Introduction
Hepatosteatosis describes the abnormal buildup of
fat within the hepatic cells in the absence of alcohol consumption.
It results from an imbalance between uptake, oxidation and
transport of fat and may be associated with metabolic dysfunction,
obesity and dyslipidemia. Hepatosteatosis may develop into
progressive hepatic inflammation (steatohepatitis), fibrosis or
cirrhosis (1,2).
The prevalence of hepatosteatosis is increasing
globally and it is one of the most common diseases of the liver
(3,4). Hepatosteatosis is associated with
metabolic syndrome, high alcohol consumption, a poor diet and
obesity (1,5,6). The
symptoms of hepatosteatosis are often nonspecific, which makes them
easy to miss and means that hepatosteatosis can be difficult to
diagnose (7). Elevated liver
transaminase levels may be a useful diagnostic marker if present,
however they often remain within a normal range (8).
Hepatosteatosis may develop into hepatic cirrhosis
or hepatocellular carcinoma and may cause the mortality of elderly
patients (9). At present the primary
method of treating hepatosteatosis/non-alcoholic steatohepatitis
(NASH) is adopting a healthy lifestyle, for example, by partaking
in regular exercise. However, lifestyle changes are often hard to
implement and patients with non-alcoholic fatty liver disease
(NAFLD) often have difficulty maintaining an improved lifestyle;
therefore, it has been suggested that such patients should be
prescribed pharmacological therapy (10).
NAFLDs cover a spectrum of liver conditions
extending from simple steatosis, severe steatosis to NASH and can
be complicated by fibrosis, cirrhosis, end-period liver failure and
hepatocellular carcinoma (3,4). In a majority of cases simple steatosis
does not affect the overall survival, while severe steatosis,
without inflammation, may affect metabolic function. NASH, an
inflammatory disorder, has been associated with increased mortality
(1,7). NASH includes steatosis, inflammation
and hepatocyte ballooning with or without fibrosis (1,7).
Hepatosteatosis increases the risk of cardiovascular
disease (CVD) and metabolic syndrome (11). The primary indicator of
hepatosteatosis is dyslipidemia, which is characterized by low
high-density lipoprotein (HDL) levels, hypertriglyceridemia and a
high level of low-density lipoprotein (LDL) (12). Currently thiazolidinedione (TZD),
which improves blood lipid profiles, glucose levels and insulin
sensitivity is the therapy of choice for type 2 diabetes mellitus
(T2DM) (13). TZD is a member of the
peroxisome proliferator-activated receptor-γ agonists family
(PPARγ), which has been used in clinical trials, either alone or in
combination with oral antidiabetic medication to treat patients
with T2DM (14).
The molecular, enzymatic and hormonal changes
associated with hepatosteatosis and other similar disorders remain
unknown. In addition, the method of pathogenesis of hepatosteatosis
has not yet been adequately described, although inflammatory
cytokines, oxidative stress, leptin and insulin resistance are
considered to be significant during its development (2). At present, TZD, an insulin sensitizer
and vitamin E, an antioxidant, in combination with lipid-depressing
medications are the most promising therapeutic strategies to treat
patients with hepatosteatosis/NASH (15). TZD is used to treat hyperglycemia,
insulin resistance and T2DM, however its mechanism of action
against hepatosteatosis remains unclear. It has been reported that
30–45 mg TZD (commercial name, pioglitazone) significantly improves
insulin sensitivity, glycated hemoglobin levels and the lipid
profile of patients with T2DM that have abnormal glycemic
regulation and slight dyslipidemia (16).
The administration of pioglitazone, which is a type
of TZD, has various effects; it increases fat deposition in several
types of muscle, perirenal and brown adipose tissue but decreases
fat accumulation in hepatic tissue (17). In adipose tissue, increased fat
deposition is associated with lipoprotein lipase (LPL) activity,
however this is not the case in the muscles (18). Therefore, further investigations into
the association between TZD and LPL activity is required.
It has been demonstrated that mice lacking
stearoyl-CoA desaturase (SCD-1) exhibit increased lipid oxidation
due to the upregulated expression of acyl-CoA oxidase and very
long-chain acyl-CoA dehydrogenase and the downregulation of lipid
synthesis (19). In addition, SCD-1
is involved in the metabolic response to leptin, a hormone of the
fat metabolism, upregulates constituents of insulin signaling and
affects the metabolism of glycogen (20). SCD-1 therefore appears to serve a key
role in metabolic control and the regulation of its activity may be
beneficial in the treatment of hepatosteatosis, obesity, T2DM,
cardiovascular disorders and other metabolic illnesses.
It was therefore hypothesized that the expression of
SCD-1 promotes lipogenesis and inhibits lipid oxidation, which may
induce the accumulation of fat in those consuming a high-calorie
diet and potentially lead to hepatosteatosis. High-carbohydrate
foods enhance de novo lipogenesis (21); however the effect of TZD on LPL and
SCD-1 remains unclear. Previous studies have investigated the
association between TZD and lipids; however, the results of such
studies are contradictory and the effect on serum triglycerides
(TGs) and total cholesterol (TC) and their role exacerbating
hepatosteatosis remain unclear (14,22,23).
Furthermore, the results of studies investigating the role of TZD
in the hormonal and enzymatic control of lipid metabolism are
varied and sometimes controversial (23–27).
Therefore the present study aimed to explore the lipid metabolism
and hormonal regulation associated with hepatosteatosis. In
addition, the role of TZD and the possible molecular mechanisms of
hepatosteatosis therapy were investigated, as well as the potential
connection between hepatosteatosis and SCD-1.
Materials and methods
Experimental animals
A total of 60 male 6 week-old albino rats, weighing
80–90 g, were obtained from the Institute for Research and Medical
Consultation (IRMC; Imam Abdulrahman Bin Faisal University, Dammam,
Saudi Arabia). Animals were kept for 1 week prior to the start of
the experiment. The rats were kept separately in elastic pens at a
temperature of 22±3°C, humidity of 40–60% and a 12-h light/dark
cycle in the animal house of the IRMC laboratories. All rats had
ad libitum access to food and water.
Diet
Two different types of diet were provided to the
rats, either a normal rat chow diet or a high-fat,
high-carbohydrate diet (HFCD; Enterprise Grain Company, Kremlin,
OK, USA). Normal rat chow pellets were composed of protein ration
(whey protein, soya bean and meat), corn, calcium phosphate,
calcium carbonate, magnesium oxide, sodium chloride and vitamins.
The standard rat food was composed of 60% carbohydrates, 8% fat,
22% basic protein, 10% dietary fiber, vitamins and minerals. The
HFCD contained 65% carbohydrates, 15% crude protein, 15% fat
[palmitate and stearate saturated fatty acids (SFAs)] and 5%
dietary fiber, vitamins and minerals. The HFCD was used to develop
the rat model of experimental hepatosteatosis and was consistent
with the formula used previously by Al-Muzafar and Amin (28).
Chemicals
TZD (pioglitazone hydrogen chloride) was purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA; cat. no.
TX75220). This was the only type of TZD used in the present study.
A total of 70 mg TZD powder was dissolved in 20 ml distilled water
and each rat received 1 ml per oral administration (os) per day at
a dose of 17.5 mg/kg body weight for 4 weeks.
Assay kits
Colorimetric assay kits obtained from HUMAN
Gesellschaft für Biochemica und Diagnostica mbH (Wiesbaden,
Germany) were used to assess the serum of rats and determine their
lipid profile (TGs, TC, LDL and HDL) and liver function [alanine
transaminase (ALT), albumin and bilirubin]. TGs were measured by
GPO-PAP method, enzymatic colorimetric test for TG with lipid
clearing factor (cat. no. 10724 kit). TC was determined by CHOD-PAP
method, enzymatic colorimetric test for cholesterol with lipid
clearing factor (cat. no. 10028 kit). LDL was determined by direct
homogenous enzymatic colorimetric test (cat. no. 10294). HDL
measured using, liquicolor direct homogenous enzymatic test (cat.
no. 10284). ALT analyzed by IFCC mod. liquiUV (cat. no. 12022).
Albumin was determined by BCG-method, photometric colorimetric test
(cat. no. 10560). Bilirubin measured by bilirubin Direct/total
liquicolor (cat. no. 10740). LDH evaluated by LDH SCE mod. liquiUV
(cat. no. 12024). Lipid hormones, leptin and resistin were measured
using ELISA kits from Bertin Bioreagent (cat. nos. A051760 and
A05179; Montigny le Bretonneux, France). SCD-1 was measured using
an ELISA kit (cat. no. MBS923022) obtained from MyBiosource, Inc.
(San Diego, CA, USA).
Serum LPL activity was measured using an assay kit
(cat. no. 700640) purchased from Cayman Chemical Company (Ann
Arbor, MI, USA). The assay used fluorescence to identify serum LPL
activity by converting arachidonoyl-1-thioglycerol into
thioglycerol and arachidonic acid. The thioglycerol then reacted
with the thiol-fluorometric indicator to provide a fluorescent
compound, which was measured at 380–390 (excitation) and 510–520 nm
(emission).
Experimental design and animal
grouping
The 60 male rats were randomly allocated into 3
groups (n=20/group). The first group was administered a standard
chow diet throughout the study period (16 weeks). The second group
(positive control) was fed HFCD for 16 weeks to induce
hepatosteatosis and the third group was fed HFCD and received
treatment with TZD (17.5 mg/kg per os) daily between weeks 12 and
16. TZD was administered daily at 8:00 a.m. in a volume equal to 5
ml/kg at a dose of 17.5 mg/kg. Rats of the TZD-untreated groups
received equal volumes (1 ml) distilled water daily at the same
time. The total experiment period was 16 weeks, which was divided
into the hepatosteatosis induction period (weeks 1–12) and the
therapy period (weeks 13–16). Rats were weighed each week, body
weights were recorded and weight gains were calculated.
Blood and tissue sampling
Under anesthesia with diethyl ether, blood was
collected from the medial canthus of the eye of all rats following
overnight fasting at the end of week 16 by using a microhematocrit
capillary tube and stored in dry glass centrifuge tubes. The blood
was stored at 24±3°C and permitted to coagulate. Samples were
centrifuged at 1,400 × g for 20 min at a temperature 20°C and the
clear supernatant serum were stored at −80°C prior to further
biochemical analysis. Immediately after blood sampling, the rats
were sacrificed by decapitation while still under anesthesia. Part
of the liver was extracted, washed with saline and fixed in 10%
formalin at 25°C and for 48 h. The tissue samples were embedded in
paraffin and cut using a rotary microtome into 3-µm-thick sections
and transferred to slides prior to hematoxylin and eosin staining
for histopathological analysis.
Histopathological examinations
Tissue samples underwent hematoxylin and eosin
staining. Sections (3 µm) were incubated in xylene for 5 min at
37°C followed by 10×10 sec absolute alcohol and running water for 1
min at 22–25°C. Sections were incubated with Harris' Hematoxylin
for 9 min at 35°C and washed with water for 2 min at 22–25°C.
Differentiation was achieved using 1% acid alcohol (2×10 sec),
followed by 30 sec in 0.2% ammonia water and running water for 1
min at 22–25°C. Prior to microscopic examination, eosin solution
was added for 2 min and sections were washed with water for 1–5
min. and dehydrated in absolute alcohol (10×10 sec) at room
temperature. The nucleus was stained blue and the cytoplasm
appeared pink. A light microscope was used to inspect the structure
and characteristics of hepatic cells, whether fat globules or
inflammation were present and whether the hepatocytes had undergone
any degenerative alterations. The histological grading system for
hepatosteatosis, NAFLD activity score, was used to assess the
samples at low (×40) and high (×100) magnification (29). The presence of minor/moderate fat
droplets, micro- and macrovesicular hepatosteatosis, hepatolobular
inflammation and the presence of inflammatory cells were recorded
and used to determine the hepatosteatosis score, as previously
described (28,29).
Statistical analysis
The results were analyzed by one-way analysis of
variance followed by Tukey-Kramer post-hoc analysis. The data are
presented as the mean ± standard error of the mean. P<0.05 was
considered to indicate a statistically significant difference.
Statistical examination was performed using GraphPad Prism 6
software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
HFCD increases body weight and liver
function markers
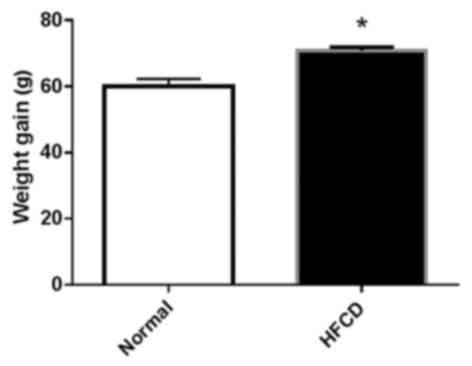
The mean body weight gain was significantly
increased in the HFCD group compared with the normal group
(P<0.05; Fig. 1). A significant
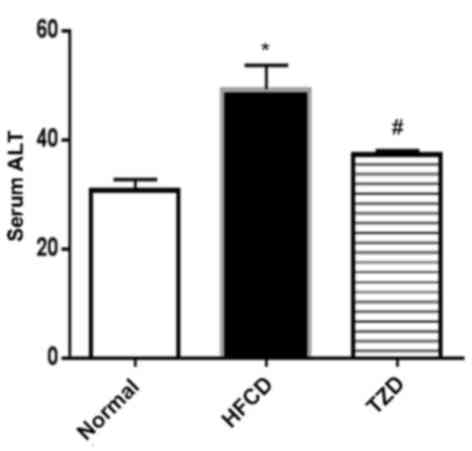
elevation in the serum total bilirubin and direct bilirubin levels,
in addition to LDH and ALT activities as liver function markers was
observed in the HFCD group compared with the control (P<0.05;
Table I and Fig. 2). The increased levels of bilirubin
and ALT activity indicated injuries to the hepatocytes however
these changes were ameliorated by the administration of TZD. No
significant changes in albumin between the different groups were
observed.
 | Table I.Effect of HFCD and TZD on liver
function in rats. |
Table I.
Effect of HFCD and TZD on liver
function in rats.
| Marker | Normal | HFCD | HFCD+TZD |
|---|
| Albumin (g/dl) | 3.12±0.20 | 2.99±1.98 | 3.62±0.30 |
| Total proteins
(g/dl) | 8.41±0.70 | 8.39±0.60 | 8.11±0.55 |
| Total bilirubin
(mg/dl) | 1.42±0.16 |
2.36±0.23a |
1.73±0.17b |
| Direct bilirubin
(mg/dl) | 0.79±0.09 |
2.08±0.10a |
1.51±0.13b |
| Lactate
dehydrogenase | 621.49±24.21 |
825.31±56.11a |
636.98±44.48b |
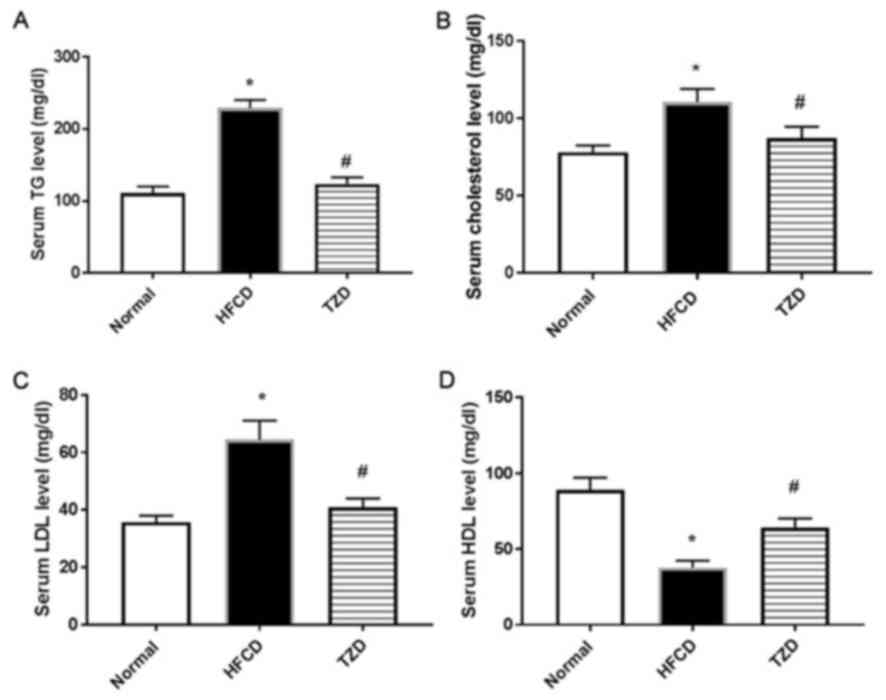
HFCD affects the blood lipid profile
of rats
A significant increase in serum TG, TC and LDL was
observed in the HFCD group compared with the normal group
(P<0.01; Fig. 3A-C), whereas
there was a significant decrease in serum HDL (P<0.01; Fig. 3D). Treatment with TZD significantly
reversed these changes (Fig. 3).
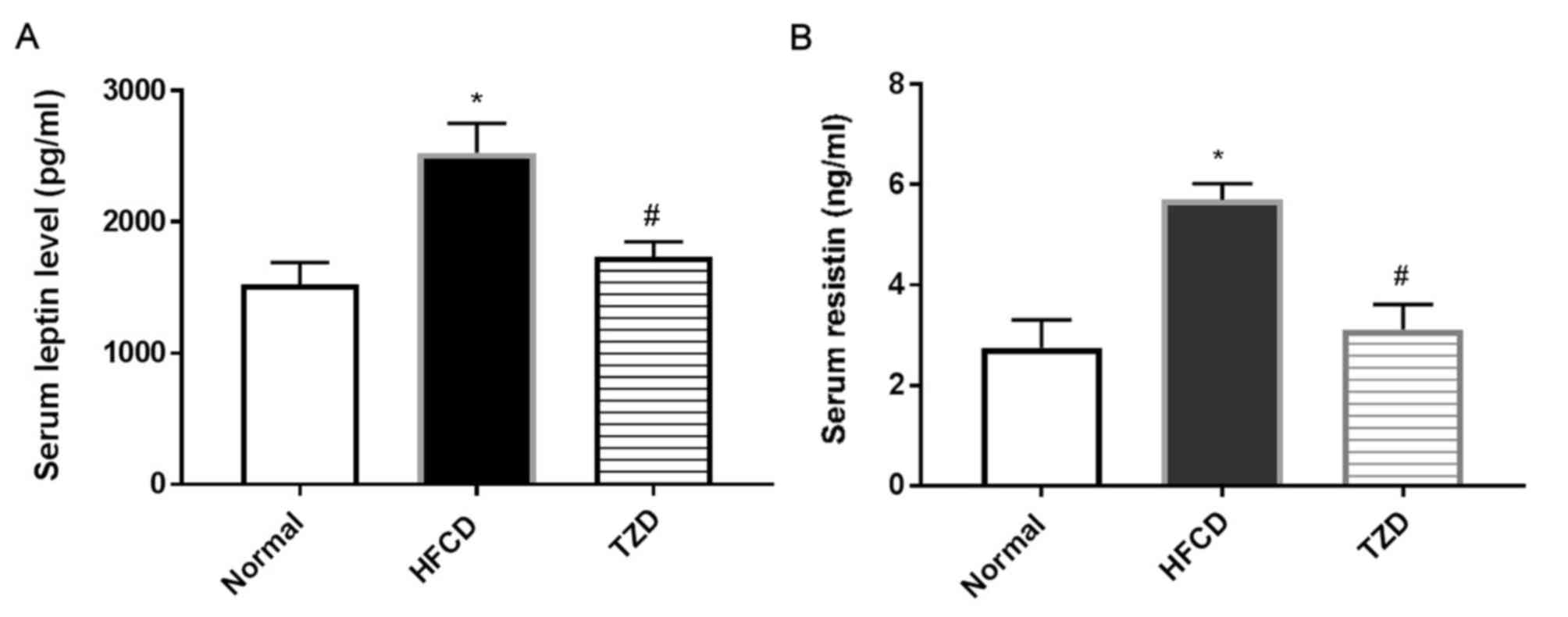
HFCD increases the level of adipose
tissue hormones
A significant increase was observed in the level of
leptin (Fig. 4A) and resistin
(Fig. 4B) in the serum of the HFCD
group compared with the normal group (P<0.05), these changes
indicated increased fat accumulation and the development of
hepatosteatosis. However, TZD administration recovered the levels
of these hormones to similar levels as the normal group.
HFCD leads to an increase in the
steatosis score
The steatosis score of the normal group was 0 for
85% of cases and 1 for 10% of cases, whereas the HFCD group
steatosis score was 1 for 65% of cases and 2 for 30% of cases
(Table II). This indicates that the
steatosis scores were markedly increased in the HFCD group compared
with the normal group. The HFCD+TZD group steatosis scores were 0
for 60% of cases and 1 for 35% of cases. These results indicate
that the administration of TZD markedly reduced the steatosis
scores to similar levels as the normal group.
| Table II.Hepatosteatosis score in the
differently treated groups of rats. |
Table II.
Hepatosteatosis score in the
differently treated groups of rats.
| Hepatosteatosis
score (%)a | Normal (%) | HFCD (%) | HFCD+TZD (%) |
|---|
| 0 (≤5) | 85 | 0 | 60 |
| 1 (6–33) | 10 | 65 | 35 |
| 2 (34–66) | 5 | 30 | 5 |
| 3 (≥67) | 0 | 5 | 0 |
TZD treatment ameliorates the increase
in lipid metabolic enzymes caused by HFCD
A notable elevation in the level of SCD-1 was
observed in the HFCD group compared with the normal group (Table III) and this was further
significantly increased by treatment with TZD. In addition, LPL
activity was notably increased in the HFCD group compared with the
normal group, however the addition of TZD significantly reversed
this increase to a lower level than the normal group.
| Table III.Effect of HFCD and TZD on lipid
metabolic enzymes in the differently treated groups of rats. |
Table III.
Effect of HFCD and TZD on lipid
metabolic enzymes in the differently treated groups of rats.
| Enzyme | Normal | HFCD | HFCD+TZD |
|---|
| Stearoyl-CoA
desaturase (ng/ml) | 9.0±1.40 | 12.0±1.39 |
18.0±1.90a,b |
| Lipoprotein lipase
(nmol/min/ml) | 39.14±3.80 | 49.61±9.60 |
24.50±2.70a,b |
Treatment with TZD restores normal
pathology following a HFCD
Histopathological examination was performed
following the administration of a normal, HFCD or TZD+HFCD diet.
Ordinary histological configuration (non-visible micro and
macrovesicular hepatosteatosis) was observed in the hepatocytes
from the normal group (Fig. 5A).
However, in the HFCD group several fat droplets, minor to moderate
micro and macrovesicular hepatosteatosis, degenerative alterations
and focal inflammation were observed in the periportal area of the
liver (Fig. 5B). In the HCFD+TZD
group, hepatocytes with an ordinary structure were observed
(Fig. 5C), indicating that treatment
with TZD restored the pathology of the hepatocytes to normal.
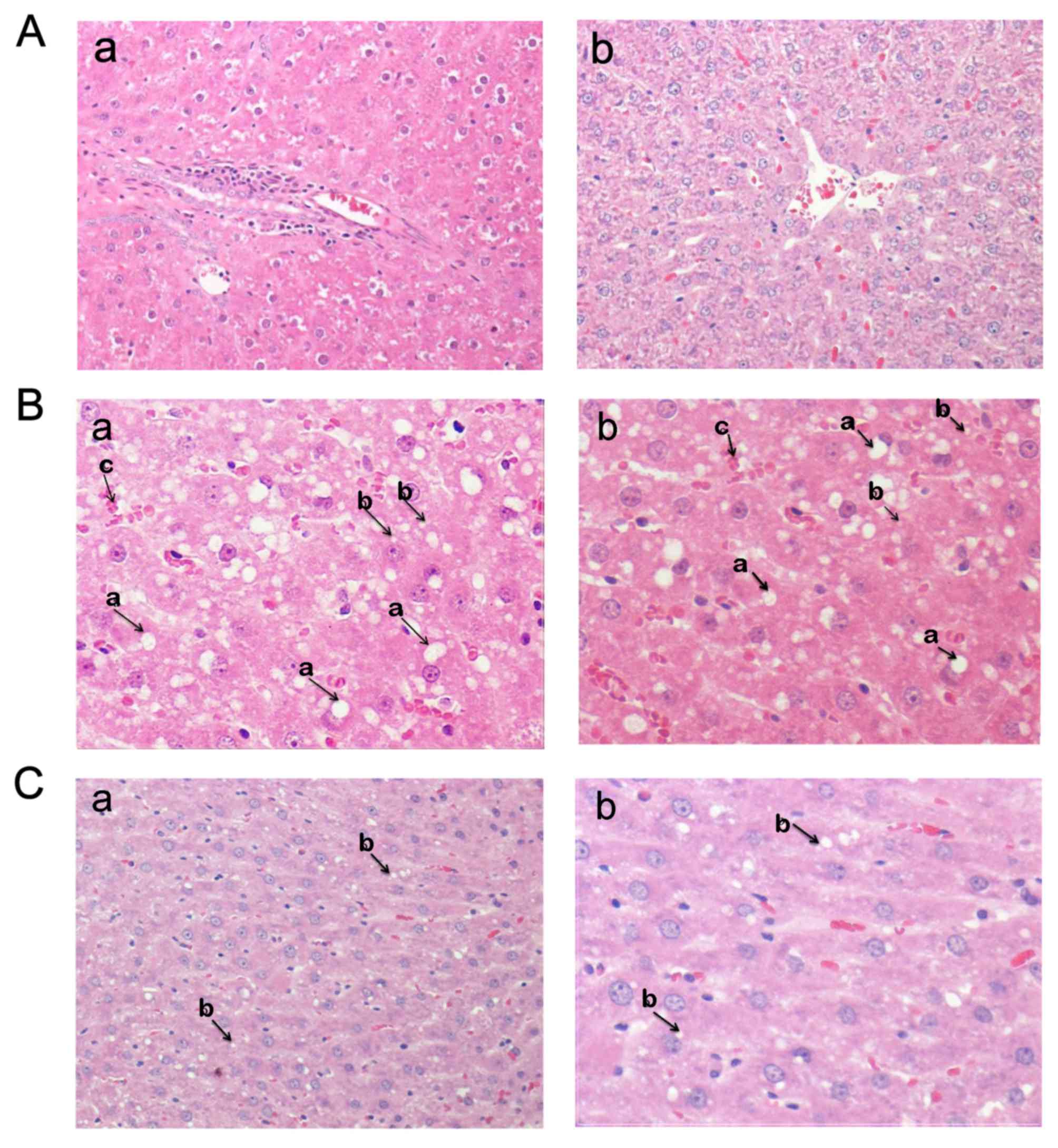 | Figure 5.Histopathological examination of
different treatment groups. Histopathological outcomes in the (A)
normal, (B) HFCD and (C) HFCD+TZD treatment groups. The normal
tissue samples (Aa) and (Ab) demonstrated characteristic
histological configuration of ordinary hepatocytes, without
inflammatory cells in the perivenular area. Several fat bubbles
were observed among and inside the hepatocytes in the (Ba) and (Bb)
HFCD liver tissue samples, which is associated with deteriorating
alterations in the hepatocytes (magnification, ×100). The HFCD
group had macrovesicular hepatosteatosis (large fat drops present
in the hepatocytes, indicated by arrow a) and microvesicular
steatosis (slight fat droplets in the hepatocytes, indicated with
arrow b). Liver focal periportal inflammation (magnification, ×100)
and accumulation (cluster) of inflammatory cells in the HFCD
group indicated with arrow c. The hepatocytes appeared normalized
and had a normal shape and minor lipid globules in the (Ca) and
(Cb) HFCD+TZD liver tissue samples. HFCD, high-fat,
high-carbohydrate diet; TZD, thiazolidinedione. |
Discussion
The present study revealed that the lipid profile
was disrupted and the liver function was impaired in the HFCD group
compared with the normal group; the enzymatic control of lipid
metabolism (SCD-1 and LPL) was also elevated. In addition, hormones
associated with lipid accumulation including, leptin and resistin
were increased in the HFCD group compared with the normal group.
These results indicate that a variety of biochemical markers are
involved in hepatosteatosis pathogenesis.
At present, a complex hypothesis suggests that
multiple parallel factors are involved in hepatosteatosis
development, these factors include: Obesity; adipose tissue
hormones; nutritional factors; gut microbiota; metabolic syndrome,
including dyslipidemia, visceral adiposity, increased lipolysis,
decreased lipogenesis, hypertriglyceridemia, hyperglycemia, insulin
resistance, high BMI and T2DM; age and genetic factors (30,31).
However, the primary regulator has yet to be identified. The
present study demonstrated that HFCD leads to elevation of the
lipid profile (TG and TC) and an increased incidence of metabolic
disturbances, which is considered to be a leading cause of
hepatosteatosis that may advance to prolonged hepatic disease and
hepatic fibrosis (32). This may
ultimately result in hepatocellular tumors and hepatic-associated
mortality (33).
Chemotherapy is considered an important treatment
option as patients suffering from obesity with hepatosteatosis
often have difficulty maintaining an improved lifestyle and diet
modifications (34). At present, the
biochemical and pathological progress of hepatosteatosis and
steatohepatitis has not been fully explained. However, lipid
metabolism, hormonal and enzymatic regulation, inflammatory markers
and oxidative stress are considered to be significant in the
initiation and progress of the disorder (30–32). The
present study indicated that hepatosteatosis may be associated with
moderate to mild lobular infiltration by fat globules, since
elevated liver indexes of serum ALT, LDH and bilirubin and an
increased lipid profile were observed compared with the control
group. The administration of TZD recovered hepatic function in
accordance with a previous study by Mazzotti et al (35).
In the present study, TZD resulted in a
significantly decreased serum TG level compared with the HFCD
group. These results are in agreement with those of Abd El-Haleim
et al (36) and also indicate
that treatment with TZD has a significant role on the serum lipid
profile, as suggested by Wang et al (37). However, it has been previously
reported that TZD administration may lead to deteriorating
microvesicular steatosis and raised hepatic TG levels, through
decreasing blood TG and free fatty acid (23). He et al (38) reported that treatment with TZD had no
significant influence on the body mass index, body fat percentage
or serum TG and TC levels.
The authors suggest that in the present study, serum
TG increased in the HFCD group and accumulated in the liver, while
TZD mobilized fat from the hepatic cells for β-oxidation and caused
normal fat deposition/lipogenesis, which led to a return of the
serum TG to normal levels. In addition, TZD as an insulin
sensitizer serves a major role in returning postprandial lipemia to
regular physiological levels and correcting fasting
hypertriglyceridemia; thereby protecting against hepatosteatosis
and atherogenic risk in T2DM (39).
TZD therapy has a favorable effect on numerous lipid types, leading
to decreases in TG, TC and LDL serum levels and an increase in HDL
levels (40).
The present study revealed a significantly decreased
serum cholesterol level in the TZD group compared with the HFCD
group, which has also been reported in a number of previous studies
(40,41). TZD reduces cholesterol by lowering
the key enzyme in cholesterol biosynthesis
hydroxymethylglutaryl-CoA reductase mRNA levels and decreasing the
intracellular content of cholesterol, which indicates a change in
the uptake of cholesterol as well as the inhibition of cholesterol
biosynthesis (42). The results of
the present study supported this theory as the activity of hepatic
LPL was inhibited and the uptake of HDL cholesterol was reduced
leading to increase in HDL level in the TZD group. TZD may function
as link between lipid and carbohydrate pathways acting as activator
for adenosine monophosphate-activated protein kinase, inducing an
activation of fatty acid oxidation and decreasing in TG synthesis
associated with a state of cellular energy change (43,44). In
summary, TZD exhibited a direct effect on the liver and confirming
these findings in humans may clarify the significant progresses in
blood lipids that detected with TZD treatment in diabetic patients
(42).
At present, TZD as an insulin sensitizer and vitamin
E as an antioxidant are the most favorable treatment options for
NAFLD and NASH as fat-lowering medications (15). Each has an optimal result on
aminotransferase, inflammation and fat accumulation. Vitamin E may
recover normal histology in hepatosteatosis but safety issues limit
its use (15,45). However, other medications, including
ursodeoxycholic acid, statins, metformin, orlistat and
pentoxifylline only have moderately positive results (6,10,15).
The current study reported that hormones and enzymes
regulating lipid metabolism serve a role in the pathogenesis and
progression of hepatosteatosis in rats and that TZD may act as a
modifier of the lipid profile, hormones and enzymatic control for
lipid metabolism for the prevention and treatment of fatty liver
induced by a HFCD. This result is supported by Xu et al
(5) who used TZD at 4 mg for 8 weeks
in HFCD rats. TZD may mitigate insulin resistance as well as
histological injury and biochemical markers in high fat-induced
fatty livers in rats as reported by Mohan et al (46) and Peng et al (23).
The present study revealed a significantly elevated
resistin level in the HFCD group, while the administration of TZD
reversed this increase. The resistin hormone (resistance to
insulin) was initially identified in mice in 2001 and was named for
its capacity to interfere with insulin action (47). Resistin was originally defined as an
adipocyte-specific hormone and it has been suggested as a link
between obesity, insulin resistance, diabetes and NAFLD (48).
Novel pharmacological agents that decrease the level
of resistin have been identified, which prevent the adverse effect
of resistin on the lipid metabolism and provide a therapeutic
strategy for the treatment of hepatic injuries (49,50). The
mechanism of action of TZD against lipid metabolism is considered
to be through decreasing resistin, which reduces any interference
with the action of insulin and stimulates lipogenesis. The reducing
effect of TZD in blood resistin was associated with the reduction
in liver fat content and improvement in sensitivity of insulin
(51). In addition TZD attenuates
insulin resistance through adiponectin-dependent and -independent
signaling pathways (49,50). The use of TZD to control leptin and
liver enzymes represents a novel approach for hepatosteatosis
therapy as presented in the current study.
The present study revealed that serum leptin was
significantly increased in the HFCD group compared with the normal
group. The recognition of the leptin hormone provided a molecular
association to obesity and NAFLD similar to effect observed for
resistin. Leptin regulates energy homeostasis and is known as the
essential mediator within the endocrine system (52). The data demonstrating increases in
serum leptin levels agree with previous findings (53–55).
Administration of leptin resulted in elevated energy expenditure,
hypophagia and loss of weight, whereas leptin deficiency exhibited
decreased energy expenditure, hyperphagia and neuroendocrine axis
suppression (54,55). The majority of obese individuals and
those with NAFLD are leptin resistant, which presents as high serum
leptin levels without controlling energy hemostasis (56). However Canbakan et al
(57) reported that leptin levels
were elevated in NAFLD and exhibited a preventive effect of leptin
against progressive liver damage Further understanding of the
molecular mechanisms of the leptin signaling pathway is required to
improve the therapeutic options for hepatosteatosis and metabolic
syndrome. In the normal rat group, the effects of leptin may be due
to its anorectic role and facilitate specific metabolic responses,
comprising effective depletion of triacylglycerol from hepatic and
other peripheral tissues thereby maintaining healthy liver tissue
(58).
Leptin is a lipid metabolic hormone that regulates
energy homeostasis and is considered as the principal mediator in
the negative feedback loop (52). In
addition, leptin depletes fat from hepatic and other peripheral
tissues under normal conditions (52,59). In
the current study, HFCD induced elevated leptin and resistin
levels, associated with insulin-resistance, and a non-significant
elevation in SCD. SCD levels were not sufficient to induce
significant conversion of SFA to mono unsaturated fatty acids
(MUSFA), causing elevated TGs, accumulation of fat globules in the
hepatocytes and the development of hepatosteatosis. However, STZ
reverted these changes by decreasing leptin and resistin levels and
inducing a significant increase in SCD-1, consequently mobilizing
fat from the liver.
During the development of hepatosteatosis,
hypertriglyceridemia raises the free fatty acids (FFA) in the
blood, which induces insulin resistance; this frequently occurs in
obese patients (38,39). The pharmacological basis for the
effects of TZD depends on its ability to reduce
hypertriglyceridemia and serum FFAs by inhibition of lipolysis and
enhancing oxidation, re-esterification and uptake from the blood
and hepatic cells (60). The present
study investigated whether TZDs regulate lipolysis (the process of
monitoring the hydrolysis of TG and efflux of FFA to the blood
stream). Inhibition of FFA efflux and lipolysis in the HFCD+TZD
group may describe the underlying mechanism for hepatosteatosis
treatment by TZDs. He et al (60) proposed that TZD stimulated Akt
signaling to lower the cyclic adenosine monophosphate (cAMP) level
and thereby decrease the activity of LPL, consequently preventing
lipolysis and resulting in reduced hypertriglyceridemia. In the
current study, the response of hepatosteatosis to TZD therapy was
associated with inhibiting LPL activity; therefore, TZD treatment
may have potentially beneficial properties for the blood lipid
profile (TG, TC and LDL).
In recent years the frequency of metabolic syndrome
has increased and it is typically linked with hepatosteatosis,
particularly diabetes, hypertension and cardiovascular disease
(61). The results of the present
study demonstrated that serum leptin and SCD-1 concentration
increased in the HFCD group compared with the control group. SCD-1
is a vital metabolic regulator for body weight and is considered as
the rate-limiting lipogenic enzyme catalyzing the anabolism of
MUSFAs) (20,62), primarily palmitoleate (C16:1) and
oleate (C18:1), which describe the majority of fatty acids in TGs,
esters, phospholipids and cholesterol esters (20). In addition, SCD-1 is predominantly
located in the endoplasmic reticulum, where it undergoes a quick
turnover in response to diverse hormonal and nutritional signals
(63).
Chong et al (64) reported that activation of SCD-1 and
de novo lipogenesis in hepatic and adipose tissue was
initiated after high carbohydrate feeding for a short period (3
days). The enzymatic activity of SCD-1 was raised >7-fold in
livers of non-treated ob/ob mice compared with the wild-type
(65). Concentrations of hepatic
MUSFAs (C16:1 and C18:1), which are SCD-1 products, were increased
in ob/ob mice and normalized by treatment with leptin for 12
days (65). Changes in SCD-1 levels
in obese mice decreased body adiposity and increased energy
expenditure and insulin sensitivity and induced resistance to a
obesity induced by diet (66).
Elevated levels of SCD-1 have further been established in
insulin-resistant animals and humans (67), which are conditions associated with
hepatosteatosis.
The present study suggests that non-significant
increases of SCD-1 compared with normal levels affected the
MUFA/SFA ratio by increasing lipid profile, high lipogenesis and
inducing of steatosis. In addition, palmitate, contained in HFCD,
may induce lipogenesis by activating X-Box binding
protein-1-mediated endoplasmic reticulum stress, demonstrated by
the increase in steatosis score ≥2, associated with blood palmitate
(68,69). In the normal rat group, the
physiological level of SCD-1 maintained a balanced ratio of
MUSFA/SFA to keep the hepatic tissue healthy without fat
accumulation. However, in the HFCD group the activity of SCD-1 did
not significantly alter and subsequent changes in MUSFA may not
meet the threshold target for observing further effects. TZD
therapy may elevate the activity of SCD-1 and maintain threshold
levels of MUSFA to treat NAFLD.
In recent years, evidence has indicated that SCD-1
overexpression is associated with liver fibrosis and hepatocellular
carcinoma (68,70). In addition, a high fructose diet
prolonged hepatic SCD-1 activation and promoted TG accumulation and
steatosis (71,72). It has also been determined that
elevated SCD-1 is associated with obesity (73). However, other studies have identified
no significant correlation between muscle or adipose SCD-1 mRNA and
insulin sensitivity or body mass index (24).
In downregulated SCD-1 mice a number of genes
associated with lipid synthesis are downregulated, whereas genes
associated with lipid oxidation are upregulated (25). This suggests that a consequence of
reduced SCD-1 is the activation of fat oxidation and decreased
triacylglycol biosynthesis loading and storage (25). Consistent with this observation, a
lower SCD-1 index protects against diet-induced obesity (53), which appears to be consistent with
the level of the normal group compared with the HFCD and TZD groups
in the present study.
The non-significant notable elevated SCD-1 observed
in the HFCD group compared with the normal group may be due to the
conversion of SFA into unsaturated (U)SFA, which is important for
the clearance of solid fat by conversion into miscible fatty acids.
The activity of SCD-1 prevents the accumulation of palmitate via
the conversion into USFA (19,74). The
authors suggest that when the SFA/MUSFA ratio is increased, it
favors the saturated type. As the fatty component of HFCD was
primarily composed of palmitate and stearate SFA, it resulted in
the accumulation of TG in hepatic tissues. The ratio of SFA to
MUSFA also affects the composition of phospholipids (19). In addition, variation in this ratio
has been involved in several disease states involving obesity,
cardiovascular disease, diabetes, neurological illness and cancer.
Therefore SCD-1 expression is of physiological importance in
disease and normal conditions (19).
The high leptin and SCD-1 levels observed in HFCD group of the
current study may link to protection against steatosis in response
to HFCD or induction of steatosis and the development of steatosis.
Furthermore, the occurrence of leptin-resistance in HFCD or obese
cases may affect the results.
Elevated hepatic de novo lipogenesis due to
dietary sugar is involved in the pathophysiology of hepatosteatosis
(75,76). SFA are the end product of de
novo lipogenesis and perform lipotoxic roles that stimulate the
accumulation of fat in the liver. Desaturation of these SFA by
SCD-1 can avoid these harmful effects (74). De novo lipogenesis is
associated with hepatosteatosis with a nutritional status rich in
monosaccharides and it is proposed that individual hepatic SCD-1
activity is negatively correlated with hepatic lipid aggregation
under lipogenic nutritional situations (77). The current study indicated that SCD-1
may be an essential factor in the pathogenesis of HFCD induced
steatosis.
TZD may control SCD-1, which catalyzes a
rate-regulating step in the synthesis of unsaturated oleic acid, by
catalyzing the desaturation of stearic acid (20). The ratio of stearic to oleic acid is
associated with the control of cell growth and differentiation via
effects on signal transduction and cell membrane flexibility
(18,24).
A previous study reported that SCD-1 was activated
in hepatosteatosis and consequently its suppression was a potential
treatment option (78). SCD-1
deficiency and its inhibitor were considered important for the
prevention of steatosis and other metabolic diseases (79). The present study investigated the
treatment of hepatosteatosis with TZD and identified an alternative
mechanism, which was supported by a number of studies using TZD
in vitro and in vivo (18,24).
The results revealed that SCD-1 concentration
notably increased in the HFCD group compared with the control
group. However, treatment with TZD increased its level
significantly compared with HFCD group. The authors suggest that
the ratio of SFA/USFF is an important factor in the effect of TZD.
SCD-1 is associated with pioglitazone (TZD) administration in
humans and a 2-fold elevation of SCD-1 mRNA and protein is observed
in fat tissue (24). Pioglitazone
also raised the level of SCD-1 in vitro (24). These changes suggest that PPARγ
upregulation of SCD-1 resulted in elevated lipogenesis and
strengthened adiponectin signaling. Further studies indicated that
PPARγ agonists weaken palmitate-induced endoplasmic reticulum
stress and apoptosis by SCD-1 generation (80–82).
SCD-1 upregulation may lead to the lessening of cardiovascular
disturbances through therapy with PPARγ agonists (81).
TZD significantly upregulated the activity of SDC-1,
leading to the conversion of inert saturated palmitate into
reactive monounsaturated acid, which consequently increased its
ability to be incorporated in cell membrane phospholipids (24,81,83). In
this respect, the degree of unsaturation in membrane phospholipids
serves a vital role in numerous membrane functions and irregular
incorporation of saturated phospholipids may lead to stiffening and
loss function (84). In addition,
the conversion increased the likelihood of fatty acid movement and
its reaction with cholesterol to form cholesterol esters (85). These events prevent the formation of
inert fat globules and mobilize the accumulated fat droplets in the
liver compared with the HFCD group. In the TZD group, higher
activity of SCD1 favor the MUSFA/SFA ratio. TZD raised the
composition of delta 9-cis USFA as (C14:1) myristoleic acid,
(C16:1) palmitoleic acid, (C18:1) oleic acid, and (C18:2) linoleic
acid, however, TZD decreased the composition of SFA, (C14:0)
myristic acid, (C16:0) palmitic acid and (C18:0) stearic acid.
These changes resulted in the formation of phospholipids and the
mobilization of TG from the accumulated fat in hepatosteatosis and
fatty liver, however these findings require further research.
Toyama et al (26)
demonstrated that hepatic or serum SCD-1 activity and the mRNA
level of SCD-1 were not altered by TZD treatment and no variations
have been detected for the activity index of SCD-1 in cells treated
with TZD (86). TZD may maintain the
leptin level resulting in the activation and promotion of
SCD-1.
In conclusion, HFCD induced-steatosis resulted in
lipid metabolism disorders including hypertriglyceridemia and
hypercholesterolemia, which are hormonally controlled by leptin and
resistin. Enzymatic changes were also observed in the activity of
SCD-1 and lipase. The administration of TZD served a beneficial
role in lipid metabolism by normalizing the level of serum TG, TC,
leptin and resistin by increasing the level of SCD-1 and
suppressing LPL activity. TZD appeared to increase the lipogenesis
of oleic USFA and thereby prevented the effect of
hepatosteatosis.
The mechanism of action of TZD was inhibition of
lipolysis (blood TG hydrolysis and FFA efflux), which occurred by
the inhibition of LPL activity. This mechanism may present a
pharmacological basis for using TZDs as a hepatosteatosis therapy.
The results of the present study suggest that SCD-1 is an essential
component in the control of fat metabolism and the clinical use of
SCD-1 may be an effective therapy against hepatosteatosis and other
constituents of metabolic syndrome, including diabetes and
obesity.
Acknowledgements
The authors thank the histopathological unit of King
Fahd Hospital for their support and Dr Awadia S. Awadalla from the
College of Medicine and Prof. Dr. Ebtesam A. Al-Suhaimi from the
Institute for Research and Medical Consultations at the Imam
Abdulrahman bin Faisal University for their cooperation in this
study.
Funding
The present study was financed by a grant from the
Deanship of Scientific Research at the University of Imam
Abdulrahman Bin Faisal (grant no. 2014082).
Availability of data and materials
All data produced or analyzed during this study are
included in this published article.
Authors' contributions
KA and HA designed the study, performed the
experiments, analyzed and interpreted the data. KA and HA drafted
and revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal experiments were performed in accordance
with the National Institutes of Health guidelines for the care and
use of laboratory animals and with local ethical guidelines. The
study was approved by Imam Abdulrahman Bin Faisal University's
Institutional Review Board (approval no. IRB-b-2014-9066).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Noureddin M, Mato JM and Lu SC:
Nonalcoholic fatty liver disease: Update on pathogenesis,
diagnosis, treatment and the role of S-adenosylmethionine. Exp Biol
Med (Maywood). 240:809–820. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tarantino G, Savastano S and Colao A:
Hepatic steatosis, low-grade chronic inflammation and
hormone/growth factor/adipokine imbalance. World J Gastroenterol.
16:4773–8783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Younossi ZM, Koenig AB, Abdelatif D, Fazel
Y, Henry L and Wymer M: Global epidemiology of nonalcoholic fatty
liver disease-Meta-analytic assessment of prevalence, incidence,
and outcomes. Hepatology. 64:73–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Araújo AR, Rosso N, Bedogni G, Tiribelli C
and Bellentani S: Global epidemiology of non-alcoholic fatty liver
disease/non-alcoholic steatohepatitis: What we need in the future.
Liver Int. 38 Suppl 1:S47–S51. 2018. View Article : Google Scholar
|
|
5
|
Xu P, Zhang XG, Li YM, Yu CH, Xu L and Xu
GY: Research on the protection effect of pioglitazone for
non-alcoholic fatty liver disease (NAFLD) in rats. J Zhejiang Univ
Sci B. 7:627–633. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hardy T, Anstee QM and Day CP:
Nonalcoholic fatty liver disease: New treatments. Curr Opin
Gastroenterol. 31:175–183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
El-Kader Abd SM and El-Den Ashmawy EM:
Non-alcoholic fatty liver disease: The diagnosis and management.
World J Hepatol. 7:846–858. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lomonaco R, Chen J and Cusi K: An
endocrine perspective of nonalcoholic fatty liver disease (NAFLD).
Ther Adv Endocrinol Metab. 2:211–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Starley BQ, Calcagno CJ and Harrison SA:
Nonalcoholic fatty liver disease and hepatocellular carcinoma: A
weighty connection. Hepatology. 51:1820–1832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gitto S, Vitale G, Villa E and Andreone P:
Treatment of nonalcoholic steatohepatitis in adults: Present and
future. Gastroenterol Res Pract. 2015:7328702015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wójcik-Cichy K, Koślińska-Berkan E and
Piekarska A: The influence of NAFLD on the risk of atherosclerosis
and cardiovascular diseases. Clin Exp Hepatol. 4:1–6. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun DQ, Liu WY, Wu SJ, Zhu GQ, Braddock M,
Zhang DC, Shi KQ, Song D and Zhen MH: Increased levels of
low-density lipoprotein cholesterol within the normal range as a
risk factor for nonalcoholic fatty liver disease. Oncotarget.
7:5728–5737. 2016.PubMed/NCBI
|
|
13
|
Davidson MA, Mattison DR, Azoulay L and
Krewski D: Thiazolidinedione drugs in the treatment of type 2
diabetes mellitus: Past, present and future. Crit Rev Toxicol.
48:52–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Betteridge DJ: Effects of pioglitazone on
lipid and lipoprotein metabolism. Diabetes Obes Metab. 9:640–647.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takahashi Y, Sugimoto K, Inui H and
Fukusato T: Current pharmacological therapies for nonalcoholic
fatty liver disease/nonalcoholic steatohepatitis. World J
Gastroenterol. 21:3777–3785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Herz M, Johns D, Reviriego J, Grossman LD,
Godin C, Duran S, Hawkins F, Lochnan H, Escobar-Jiménez F, Hardin
PA, et al: A randomized, double-blind, placebo-controlled, clinical
trial of the effects of pioglitazone on glycemic control and
dyslipidemia in oral antihyperglycemic medication-naive patients
with type 2 diabetes mellitus. Clin Ther. 25:1074–1095. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
de Souza CJ, Eckhardt M, Gagen K, Dong M,
Chen W, Laurent D and Burkey BF: Effects of pioglitazone on adipose
tissue remodeling within the setting of obesity and insulin
resistance. Diabetes. 50:1863–1871. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ochiai M and Matsuo T:
Pioglitazone-induced increase in the stearoyl-CoA desaturation
index and fat accumulation in rat muscles are not related to
lipoprotein lipase activity. J Oleo Sci. 62:745–754. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ntambi JM and Miyazaki M: Regulation of
stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res.
43:91–104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Paton CM and Ntambi JM: Biochemical and
physiological function of stearoyl-CoA desaturase. Am J Physiol
Endocrinol Metab. 297:E28–E37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marques-Lopes I, Ansorena D, Astiasaran I,
Forga L and Martínez JA: Postprandial de novo lipogenesis and
metabolic changes induced by a high-carbohydrate, low-fat meal in
lean and overweight men. Am J Clin Nutr. 73:253–261. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jia C, Huan Y, Liu S, Hou S, Sun S, Li C,
Liu Q, Jiang Q, Wang Y and Shen Z: Effect of chronic pioglitazone
treatment on hepatic gene expression profile in obese C57BL/6J
Mice. Int J Mol Sci. 16:12213–1229. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peng J, Huan Y, Jiang Q, Sun SJ, Jia CM
and Shen ZF: Effects and potential mechanisms of pioglitazone on
lipid metabolism in obese diabetic KKAy Mice. PPAR Res.
2014:5381832014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yao-Borengasser A, Rassouli N, Varma V,
Bodles AM, Rasouli N, Unal R, Phanavanh B, Ranganathan G, McGehee
RE Jr and Kern PA: Stearoyl-coenzyme A desaturase 1 gene expression
increases after pioglitazone treatment and is associated with
peroxisomal proliferator-activated receptor-gamma responsiveness. J
Clin Endocrinol Metab. 93:4431–4439. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ntambi JM, Miyazaki M, Stoehr JP, Lan H,
Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM and Attie
AD: Loss of stearoyl-CoA desaturase-1 function protects mice
against adiposity. Proc Natl Acad Sci USA. 99:11482–18486. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toyama T, Kudo N, Hibino Y, Mitsumoto A,
Nishikawa M and Kawashima Y: Effects of pioglitazone on
stearoyl-CoA desaturase in obese Zucker fa/fa rats. J Pharmacol
Sci. 104:137–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boutari C, Perakakis N and Mantzoros CS:
Association of adipokines with development and progression of
nonalcoholic fatty liver disease. Endocrinol Metab (Seoul).
33:33–43. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al-Muzafar HM and Amin KA: Probiotic
improves fatty liver disease by virtue of its action on lipid
profiles, leptin, and inflammatory biomarkers. BMC Complement
Altern Med. 17:432017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang W, Menke AL, Driessen A, Koek GH,
Lindeman JH, Stoop R, Havekes LM, Kleemann R and van den Hoek AM:
Establishment of a general NAFLD scoring system for rodent models
and comparison to human liver pathology. PLoS One. 9:e1159222014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mikolasevic I, Milic S, Wensveen Turk T,
Grgic I, Jakopcic I, Stimac D, Wensveen F and Orlic L: Nonalcoholic
fatty liver disease-A multisystem disease? World J Gastroenterol.
22:9488–9505. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marino L and Jornayvaz FR: Endocrine
causes of nonalcoholic fatty liver disease. World J Gastroenterol.
1:11053–11076. 2015. View Article : Google Scholar
|
|
32
|
Tilg H and Moschen AR: Evolution of
inflammation in nonalcoholic fatty liver disease: The multiple
parallel hits hypothesis. Hepatology. 52:1836–1846. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pang Y, Kartsonaki C, Turnbull I, Guo Y,
Clarke R, Chen Y, Bragg F, Yang L, Bian Z, Millwood IY, et al:
Diabetes, plasma glucose and incidence of fatty liver, cirrhosis
and liver cancer: A prospective study of 0.5 million people.
Hepatology. 2018.(Epub ahead of print). View Article : Google Scholar
|
|
34
|
Zelber-Sagi S, Godos J and Salomone F:
Lifestyle changes for the treatment of nonalcoholic fatty liver
disease: A review of observational studies and intervention trials.
Therap Adv Gastroenterol. 9:392–407. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mazzotti A, Caletti MT, Marchignoli F,
Forlani G and Marchesini G: Which treatment for type 2 diabetes
associated with non-alcoholic fatty liver disease? Dig Liver Dis.
49:235–240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
El-Haleim Abd EA, Bahgat AK and Saleh S:
Effects of combined PPAR-γ and PPAR-α agonist therapy on fructose
induced NASH in rats: Modulation of gene expression. Eur J
Pharmacol. 773:59–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang G, Wang X, Zhang Q and Ma Z: Response
to pioglitazone treatment is associated with the lipoprotein lipase
S447X variant in subjects with type 2 diabetes mellitus. Int J Clin
Pract. 61:552–557. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
He L, Liu X, Wang L and Yang Z:
Thiazolidinediones for nonalcoholic steatohepatitis: A
meta-analysis of randomized clinical trials. Medicine (Baltimore).
95:e49472016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Al Majali K, Cooper MB, Staels B, Luc G,
Taskinen MR and Betteridge DJ: The effect of sensitisation to
insulin with pioglitazone on fasting and postprandial lipid
metabolism, lipoprotein modification by lipases, and lipid transfer
activities in type 2 diabetic patients. Diabetologia. 49:527–537.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Corey KE, Vuppalanchi R, Wilson LA,
Cummings OW and Chalasani N: NASH resolution is associated with
improvements in HDL and triglyceride levels but not improvement in
LDL or non-HDL-C levels. Aliment Pharmacol Ther. 41:301–309. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zaitone SA, Barakat BM, Bilasy SE, Fawzy
MS, Abdelaziz EZ and Farag NE: Protective effect of boswellic acids
versus pioglitazone in a rat model of diet-induced non-alcoholic
fatty liver disease: Influence on insulin resistance and energy
expenditure. Naunyn Schmiedebergs Arch Pharmacol. 388:587–600.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Djaouti L, Jourdan T, Demizieux L, Chevrot
M, Gresti J, Vergès B and Degrace P: Different effects of
pioglitazone and rosiglitazone on lipid metabolism in mouse
cultured liver explants. Diabetes Metab Res Rev. 26:297–305. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
LeBrasseur NK, Kelly M, Tsao TS, Farmer
SR, Saha AK, Ruderman NB and Tomas E: Thiazolidinediones can
rapidly activate AMP-activated protein kinase in mammalian tissues.
75-E181. Am J Physiol Endocrinol Metab. 291:E175–E181. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saha AK, Avilucea PR, Ye JM, Assifi MM,
Kraegen EW and Ruderman NB: Pioglitazone treatment activates
AMP-activated protein kinase in rat liver and adipose tissue in
vivo. Biochem Biophys Res Commun. 314:580–585. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Banini BA and Sanyal AJ: Current and
future pharmacologic treatment of nonalcoholic steatohepatitis.
Curr Opin Gastroenterol. 33:134–141. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mohan SK, Veeraraghavan VP and Jainu M:
Effect of pioglitazone, quercetin and hydroxy citric acid on
extracellular matrix components in experimentally induced
non-alcoholic steatohepatitis. Iran J Basic Med Sci. 18:832–836.
2015.PubMed/NCBI
|
|
47
|
Steppan CM, Bailey ST, Bhat S, Brown EJ,
Banerjee RR, Wright CM, Patel HR, Ahima RS and Lazar MA: The human
Resistin links obesity to diabetes. Nature. 409:307–312. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jamaluddin MS, Weakley SM, Yao Q and Chen
C: Resistin: functional roles and therapeutic considerations for
cardiovascular disease. Br J Pharmacol. 165:622–632. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kubota N, Terauchi Y, Kubota T, Kumagai H,
Itoh S, Satoh H, Yano W, Ogata H, Tokuyama K, Takamoto I, et al:
Pioglitazone ameliorates insulin resistance and diabetes by both
adiponectin-dependent and -independent pathways. J Biol Chem.
281:8748–8755. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dongiovanni P, Rametta R, Meroni M and
Valenti L: The role of insulin resistance in nonalcoholic
steatohepatitis and liver disease development-a potential
therapeutic target? Expert Rev Gastroenterol Hepatol. 10:229–242.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bajaj M, Suraamornku S, Hardies LJ,
Pratipanawatr T and DeFronzo RA: Plasma resistin concentration,
hepatic fat content, and hepatic and peripheral insulin resistance
in pioglitazone-treated type II diabetic patients. Int J Obes Relat
Metab Disord. 28:783–789. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Park HK and Ahima RS: Physiology of
leptin: Energy homeostasis, neuroendocrine function and metabolism.
Metabolism. 64:24–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cedernaes J, Alsiö J, Västermark A,
Risérus U and Schiöth HB: Adipose tissue stearoyl-CoA desaturase 1
index is increased and linoleic acid is decreased in obesity-prone
rats fed a high-fat diet. Lipids Health Dis. 12:22013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Amitani M, Asakawa A, Amitani H and Inui
A: The role of leptin in the control of insulin-glucose axis. Front
Neurosci. 7:512013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou Y and Rui L: Leptin signaling and
leptin resistance: Front Med. 7:207–222. 2013.
|
|
56
|
Huang XD, Fan Y, Zhang H, Wang P, Yuan JP,
Li MJ and Zhan XY: Serum leptin and soluble leptin receptor in
non-alcoholic fatty liver disease. World J Gastroenterol.
14:2888–2893. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Canbakan B, Tahan V, Balci H, Hatemi I,
Erer B, Ozbay G, Sut N, Hacibekiroglu M, Imeryuz N and Senturk H:
Leptin in nonalcoholic fatty liver disease. Ann Hepatol. 7:249–254.
2008.PubMed/NCBI
|
|
58
|
Cohen P, Ntambi JM and Friedman JM:
Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr Drug
Targets Immune Endocr Metabol Disord. 3:271–280. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
D'souza AM, Neumann UH, Glavas MM and
Kieffer TJ: The glucoregulatory actions of leptin. Mol Metab.
6:1052–1065. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
He J, Xu C, Kuang J, Liu Q, Jiang H, Mo L,
Geng B and Xu G: Thiazolidinediones attenuate lipolysis and
ameliorate dexamethasone-induced insulin resistance. Metabolism.
64:826–836. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Adams LA, Anstee QM, Tilg H and Targher G:
Non-alcoholic fatty liver disease and its relationship with
cardiovascular disease and other extrahepatic diseases. Gut.
66:1138–1153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dobrzyn P, Ntambi JM and Dobrzyn A:
Stearoyl-CoA desaturase: A novel control point of lipid metabolism
and insulin sensitivity. Eur J Lipid Sci Technol. 110:93–100. 2008.
View Article : Google Scholar
|
|
63
|
Heinemann FS and Ozols J: Stearoyl-CoA
desaturase, a short-lived protein of endoplasmic reticulum with
multiple control mechanisms. Prostaglandins Leukot. Essent Fatty
Acids. 68:122–133. 2003. View Article : Google Scholar
|
|
64
|
Chong MF, Hodson L, Bickerton AS, Roberts
R, Neville M, Karpe F, Frayn KN and Fielding BA: Parallel
activation of de novo lipogenesis and stearoyl-CoA desaturase
activity after 3 d of high-carbohydrate feeding. Am J Clin Nutr.
87:817–823. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cohen P and Friedman JM: Leptin and the
control of metabolism: Role for stearoyl-CoA desaturase-1(SCD-1). J
Nutr. 134:2455S–2463S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dobrzyn A and Ntambi JM: The role of
stearoyl-CoA desaturase in body weight regulation. Trends
Cardiovasc Med. 14:77–81. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dobrzyn P, Jazurek M and Dobrzyn A:
Stearoyl-CoA desaturase and insulin signaling-what is the molecular
switch? Biochim Biophys Acta. 1797:1189–1194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Von Loeffelholz C, Döcke S, Lock JF,
Lieske S, Horn P, Kriebel J, Wahl S, Singmann P, de Las Heras Gala
T, Grallert H, et al: Increased lipogenesis in spite of upregulated
hepatic 5′AMP-activated protein kinase in human non-alcoholic fatty
liver. Hepatol Res. 47:890–901. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lee JJ, Lambert JE, Hovhannisyan Y,
Ramos-Roman MA, Trombold JR, Wagner DA and Parks EJ: Palmitoleic
acid is elevated in fatty liver disease and reflects hepatic
lipogenesis. Am J Clin Nutr. 101:34–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gaggini M, Cabiati M, Del Turco S, Navarra
T, De Simone P, Filipponi F, Del Ry S, Gastaldelli A and Basta G:
Increased FNDC5/Irisin expression in human hepatocellular
carcinoma. Peptides. 88:62–66. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mock K, Lateef S, Benedito VA and Tou JC:
High-fructose corn syrup-55 consumption alters hepatic lipid
metabolism and promotes triglyceride accumulation. J Nutr Biochem.
39:32–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu L, Wang S, Yao L, Li JX, Ma P, Jiang
LR, Ke DZ, Pan YQ and Wang JW: Long-term fructose consumption
prolongs hepatic stearoyl-CoA desaturase 1 activity independent of
upstream regulation in rats. Biochem Biophys Res Commun.
479:643–648. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hulver MW, Berggren JR, Carper MJ,
Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm GL,
Houmard JA, et al: Elevated stearoyl-CoA desaturase-1 expression in
skeletal muscle contributes to abnormal fatty acid partitioning in
obese humans. Cell Metabolism. 2:251–261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hellemans KH, Hannaert JC, Denys B,
Steffensen KR, Raemdonck C, Martens GA, Van Veldhoven PP,
Gustafsson JA and Pipeleers D: Susceptibility of pancreatic beta
cells to fatty acids is regulated by LXR/PPARα-dependent
stearoyl-coenzyme a desaturase. PLoS One. 4:e72662009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chiu S, Mulligan K and Schwarz JM: Dietary
carbohydrates and atty liver disease: De novo lipogenesis. Curr
Opin Clin Nutr Metab Care. 21:277–282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Softic S, Cohen DE and Kahn CR: Role of
dietary fructose and hepatic de novo Lipogenesis in fatty liver
disease. Dig Dis Sci. 61:1282–1293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Silbernagel G, Kovarova M, Cegan A,
Machann J, Schick F, Lehmann R, Häring HU, Stefan N, Schleicher E,
Fritsche A and Peter A: High hepatic SCD1 activity is associated
with low liver fat content in healthy subjects under a lipogenic
diet. J Clin Endocrinol Metab. 97:E2288–E2292. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kurebayashi S, Hirose T, Miyashita Y,
Kasayama S and Kishimoto T: Thiazolidinediones downregulate
stearoyl-CoA desaturase 1 gene expression in 3T3-L1 adipocytes.
Diabetes. 46:2115–2118. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sun S, Zhang Z, Pokrovskaia N, Chowdhury
S, Jia Q, Chang E, Khakh K, Kwan R, McLaren DG, Radomski CC, et al:
Discovery of triazolone derivatives as novel, potent stearoyl-CoA
desaturase-1 (SCD1) inhibitors. Bioorg Med Chem. 23:455–465. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Meshkani R, Sadeghi A, Taheripak G,
Zarghooni M, Gerayesh-Nejad S and Bakhtiyari S: Rosiglitazone, a
PPARγ agonist, ameliorates palmitate-induced insulin resistance and
apoptosis in skeletal muscle cells. Cell Biochem Funct. 32:683–691.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ikeda J, Ichiki T, Takahara Y, Kojima H,
Sankoda C, Kitamoto S, Tokunou T and Sunagawa K: PPARγ agonists
attenuate palmitate-induced ER stress through up-regulation of
SCD-1 in macrophages. PLoS One. 10:e01285462015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Risérus U, Tan GD, Fielding BA, Neville
MJ, Currie J, Savage DB, Chatterjee VK, Frayn KN, O'Rahilly S and
Karpe F: Rosiglitazone increases indexes of stearoyl-CoA desaturase
activity in humans: Link to insulin sensitization and the role of
dominant-negative mutation in peroxisome proliferator-activated
receptor-gamma. Diabetes. 54:1379–1384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Leekumjorn S, Cho HJ, Wu Y, Wright NT, Sum
AK and Chan C: The role of fatty acid unsaturation in minimizing
biophysical changes on the structure and local effects of bilayer
membranes. Biochim Biophys Acta. 1788:1508–1516. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Leamy AK, Egnatchik RA and Young JD:
Molecular mechanisms and the role of saturated fatty acids in the
progression of non-alcoholic fatty liver disease. Prog Lipid Res.
52:165–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Arisqueta L, Navarro-Imaz H, Rueda Y and
Fresnedo O: Cholesterol mobilization from hepatic lipid droplets
during endotoxemia is altered in obese ob/ob mice. J Biochem.
158:321–329. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Saliani N, Darabi M, Yousefi B, Baradaran
B, Khaniani MS, Darabi M, Shaaker M, Mehdizadeh A, Naji T and
Hashemi M: PPARγ agonist-induced alterations in Δ6-desaturase and
stearoyl-CoA desaturase 1: Role of MEK/ERK1/2 pathway. World J
Hepatol. 5:220–225. 2013. View Article : Google Scholar : PubMed/NCBI
|