Introduction
Dubin-Johnson syndrome (DJS) is an autosomal
recessive disorder, originally described in 1954 (1). The syndrome is characterized by
fluctuating predominantly conjugated hyperbilirubinemia without
hemolysis, and patients with DJS may suffer from nonspecific
symptoms, such as abdominal discomfort (1). Urinary coproporphyrin output is normal,
with the majority of the coproporphyrin fraction being
coproporphyrin I (2).
Cholescintigraphy in DJS patients displays unique delayed
visualization of the liver and biliary tract (3). In addition, liver histology in DJS
patients demonstrates distinctive deposits of melanin-like pigment
in the hepatocyte lysosomes, which gives the organ a characteristic
black color. The disorder occurs in all races and nationalities,
and in both sexes. It is considered to be a rare disorder, however,
in Sephardic Jews a higher incidence is observed of approximately
1:3,000 (4).
The adenosine triphosphate (ATP)-binding cassette
subfamily C member 2 (ABCC2) gene is located on chromosome
10q24, which encodes the human canalicular multispecific organic
anion transporter, also called the multidrug resistance-associated
protein 2 (MRP2). MRP2, comprising 1,545 amino acids, is a specific
non-bile acid organic anion transporter, which mediates the primary
active export of conjugates of lipophilic compounds with
glucuronate or glutathione from cells in an ATP-dependent manner
(5). Genetic alterations of the
ABCC2 gene have been identified in DJS patients, including
deletions, missense mutations, nonsense mutations and splice
junction mutations, such as IVS15+2T→C (6). However, no hotspot mutations have been
identified in the ABCC2 gene. The majority of the
DJS-associated mutations are considered to cause defects in MRP2
protein deletions and maturation, leading to greatly reduced
biliary secretion of organic anions (7). Certain mutations may lead to rapid
degradation of the mutant mRNA, whereas others may affect protein
maturation, protein stability, or the function of correctly
localized proteins (8,9).
ABCC2 mutations have been identified in
patients with DJS worldwide; however, little is known regarding the
mutation pattern of ABCC2 in China. In the present study,
the mutation patterns of the ABCC2 gene were investigated in
7 Chinese DJS patients, and genotype and phenotype analysis of the
ABCC2 mutations in these patients was conducted.
Patients and methods
Study population
A total of 7 DJS patients enrolled from the China
Registry of Genetic/Metabolic Liver Diseases (ClinicalTrials.gov identifier, NCT03131427) between
June 2015 and December 2017 were included in the present study. The
clinical diagnosis of DJS (7) was
based on biochemical evidence of fluctuating predominantly
conjugated hyperbilirubinemia with or without family history,
and/or histochemistry and immunohistology of the liver. Patients
did not present hemolysis, other genetic and metabolic liver
diseases, obstruction or dilation of the biliary tree, viral
hepatitis, malignant tumors, and drug-induced or autoimmune liver
diseases. A total of 4 patients with hepatocellular carcinoma who
had undergone hepatectomy in Beijing Friendship Hospital (Beijing,
China) from October 2017 to December 2017 were recruited to the
current study. Distal normal tissues from the resected hepatic
tissues of these patients were collected and used as controls.
The study was conducted in accordance with the
Declaration of Helsinki. The Ethics Committee of the Beijing
Friendship Hospital at Capital Medical University approved the
study protocol. All patients provided written informed consent.
Whole blood samples were collected from the 7 patients with DJS,
and liver biopsy samples were available and collected from 4 of the
7 patients. The whole blood was stored at −20°C prior to the
extraction of genomic DNA for the detection of ABCC2
mutations. For the liver biopsy samples, formalin-fixed and
paraffin-embedded tissue was prepared, and 4-µm sections were cut
for hematoxylin and eosin (HE) staining, Schmorl's staining and
immunohistochemical (IHC) analysis of MRP2 expression.
Sanger sequencing and functional
prediction of variants in the ABCC2 gene
Genomic DNA was extracted from whole blood using a
Genomic DNA Purification kit (Qiagen, Valencia, CA, USA). All 32
exons and their associated boundary regions (adjacent base sequence
in introns) of the ABCC2 gene were amplified by polymerase
chain reaction (PCR) using primers designed with Primer3 software
(http://bioinfo.ut.ee/primer3-0.4.0/;
Table I). PCR amplification (2×Taq
PCR MasterMix; cat. no KT201-13; Tiangen Biotech Co., Ltd.,
Beijing, China) was performed in a PCR cycler (Applied Biosystems
Veriti 96-well Thermal Cycler; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) under the following conditions: Denaturation for
30 sec at 95°C followed by 38 cycles of 15 sec at 95°C, 30 sec at
the annealing temperature of each pair of primers (56°C for exons
2, 3, 10, 14, 17, 18–19, 20–21, 31 and 32; 58°C for exons 1, 7, 9,
12–13, 15, 16, 24, 25, 26, 27–28 and 30; and 60°C for exon 4–6, 8,
11, 22–23, 29) and 70 sec at 72°C, with extension for 10 min at
72°C. PCR products were sequenced in both the forward and reverse
orientations using an automated DNA sequencer (Applied Biosystems
3730 DNA Analyzer; Thermo Fisher Scientific, Inc.).
 | Table I.Primers used for sequencing of ABCC2
gene. |
Table I.
Primers used for sequencing of ABCC2
gene.
| No. | Forward | Reverse | Exon |
|---|
| 1 |
5′-GTCTTCGTTCCAGACGCAGT-3′ |
5′-TTCTTGTTGGTGACCACCCTA-3′ | 1 |
| 2 |
5′-ACAATCCTTCCCCTTTGGTC-3′ |
5′-TGCACCAAGGAATTAGAGTTCA-3′ | 2 |
| 3 |
5′-CAGTGGTCTTTTTCCCTTCTCA-3′ |
5′-GGTAAACAGGGCAGAAGTGG-3′ | 3 |
| 4 |
5′-GTCCATGGAGATGAGGCACT-3′ |
5′-GTTGCAGTGAGCCAAGATCA-3′ | 4–6 |
| 5 |
5′-CTGTGGTTCGCTCTTGTTCC-3′ |
5′-TGATGCTGATGTACCCTTGC-3′ | 7 |
| 6 |
5′-GAGCTGCTCAGGCCAGTAAC-3′ |
5′-CCCTGAAAGGACCATCTGAG-3′ | 8 |
| 7 |
5′-TGAGGAGAGAGGCATCCTTG-3′ |
5′-TGAGGGGATTTTCTTTGGTG-3′ | 9 |
| 8 |
5′-GCTTTGTCCATGGGTCCTAA-3′ |
5′-GAAAGCTTATATTCTTCTGGGTGA-3′ | 10 |
| 9 |
5′-CCCTCTCTCATGGAAGCGTA-3′ |
5′-GAGAGCCACTGCTTCTGTCC-3′ | 11 |
| 10 |
5′-GGGCAATCATGTGAGCTGTA-3′ |
5′-GGTCAAACCATTGGTCTCCA-3′ | 12–13 |
| 11 |
5′-TCTCTCTGCTTGTGCTCGTT-3′ |
5′-GCGAATAAGTTTGGGAAGCA-3′ | 14 |
| 12 |
5′-GTCACGTGGGGACCTACATT-3′ |
5′-AATAGGCCAGGCAGTGAGAA-3′ | 15 |
| 13 |
5′-TCAATACCCAACCCCTGCTA-3′ |
5′-ATTCGGGAGTCAGAGGCTTT-3′ | 16 |
| 14 |
5′-TTTGTTTCTTCCCCTCTCCA-3′ |
5′-TCACCCTTGAAGATCCCTTG-3′ | 17 |
| 15 |
5′-TCTTCCTTTTACCCCTCCCTA-3′ |
5′-ACCCATGGCCCAAGTTCTAT-3′ | 18–19 |
| 16 |
5′-TGCTGAAACCAGCAAGATCA-3′ |
5′-TTTGCAAAGGACAGAGGACA-3′ | 20–21 |
| 17 |
5′-CTCCTTGTGGTTGGCATTCT-3′ |
5′-GGGAGCTCACAGCAGGTACT-3′ | 22–23 |
| 18 |
5′-GGAGCCTCTCATCATTCTGC-3′ |
5′-CCTCCCACCGCTAATATCAA-3′ | 24 |
| 19 |
5′-GTTCTGTGAACGCCAAGGTT-3′ |
5′-CCAGGGTTTGAACCTCAGTC-3′ | 25 |
| 20 |
5′-GTTCTGTGAACGCCAAGGTT-3′ |
5′-CCAGGGTTTGAACCTCAGTC-3′ | 26 |
| 21 |
5′-AAAGTCGGCACTGGATTGTC-3′ |
5′-GTGTGATCCCTGGCTGCTAT-3′ | 27–28 |
| 22 |
5′-GCCAGTCACTGCCTCTTACC-3′ |
5′-CCGAGTAGACCGTGGAATTG-3′ | 29 |
| 23 |
5′-CAACCACAAACCAGCTTCCT-3′ |
5′-ACACGAGGAACACGAGGAGT-3′ | 30 |
| 24 |
5′-TGAAAAACGATGCTCACAGC-3′ |
5′-CCTTCTGCCATCAGGTGTTT-3′ | 31 |
| 25 |
5′-GATGTGTGTAGCTGTGGCTCA-3′ |
5′-GACAATCGAGGGGTTTCTCA-3′ | 32 |
Mutations identified in the ABCC2 gene were
uploaded to the online Human Gene Mutation Database (HGMD;
http://www.hgmd.cf.ac.uk/) to determine whether
they were known disease mutations. The online software ExAC
(http://exac.broadinstitute.org/) was
used to examine the allele frequencies of each mutation worldwide
or in specific regions. Results from the online software
MutationTaster (http://www.mutationtaster.org/), PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/) and SIFT
(http://sift.bii.a-star.edu.sg/) were
used to predict whether the novel mutations would affect the
biological function of the ABCC2 gene.
IHC analysis of MRP2 expression
Formalin-fixed and paraffin-embedded liver biopsy
tissues were prepared, and 4-µm sections (4 µm) were cut for IHC
analysis. Following deparaffinization of the slides, the endogenous
peroxidase activity was blocked with 0.3%
H2O2 in methanol for 30 min at room
temperature. Antigen retrieval was then conducted in antigen
unmasking solution (H-3300; Vector Laboratories, Inc., Burlingame,
CA, USA) by microwaving at 700 W for 15 min, keeping the solution
at boiling and then treating with 5% skimmed milk in
phosphate-buffered saline/0.1% bovine serum albumin for at least 1
h at room temperature to block non-specific staining.
Immunohistochemical staining was then performed using monoclonal
antibodies against MRP2 (cat. no. Ab3373; Abcam, Cambridge, MA,
USA) at a dilution of 1:100 at 4°C overnight, followed by addition
of secondary antibody (cat. no. MP-7402; dilution, ready-to-use;
Vector Laboratories, Inc.) at 37°C for 1 h. Visualization of
antigen-antibody reactions was achieved with 3,3′-diaminobenzidine
(DAB; SK-4100 kit; Vector Laboratories, Inc.) and examined by an
optical microscope (Eclipse E200; Nikon Corporation, Tokyo,
Japan).
Results
Patients and clinical data
In total, 7 patients with DJS and a mean age of
30±12 years (range, 13–43 years old) were enrolled into the current
study. Three of the patients were female. The median levels of
total bilirubin and direct bilirubin of these patients were 91 and
50 µmol/l (normal reference for total bilirubin range was 3.4–17.1
µmol/l and the direct bilirubin range was 0–6.8 µmol/l),
respectively. The clinical characteristics of the patients are
shown in Table II.
| Table II.Clinical characteristics and
non-synonymous variants identified in the patients with
Dubin-Johnson syndrome. |
Table II.
Clinical characteristics and
non-synonymous variants identified in the patients with
Dubin-Johnson syndrome.
| Patient no. | Age (years) | Sex | Total bilirubin
(µmol/l) | Direct bilirubin
(µmol/l) | Variants |
|---|
| 1 | 20 | Male | 91 | 56 | p.E647X
(c.2052G>T) |
| 2 | 43 | Male | 120 | 79 | p.R393W
(c.1290C>T) |
| 3 | 21 | Female | 94 | 54 | p.G693R
(c.2190G>A) |
| 4 | 39 | Male | 97 | 49 | p.G693R
(c.2190G>A), |
|
|
|
|
|
| p.G808V
(c.2536G>T) |
| 5 | 39 | Female | 61 | 31 | p.E647X
(c.2052G>T) |
| 6 | 13 | Male | 77 | 50 | p.R393W
(c.1290C>T), |
|
|
|
|
|
| p.V417I
(c.1362G>A) |
| 7 | 40 | Female | 50 | 30 | p.Y1275X
(c.3938C>G) |
Known mutations reported by previous
studies
Six different ABCC2 gene variants including
three known variants [p.R393W (c.1290C>T) (10), p.Y1275X (c.3938C>G) (11) and p.V417I (c.1362G>A) (12)] were identified across the 7 patients,
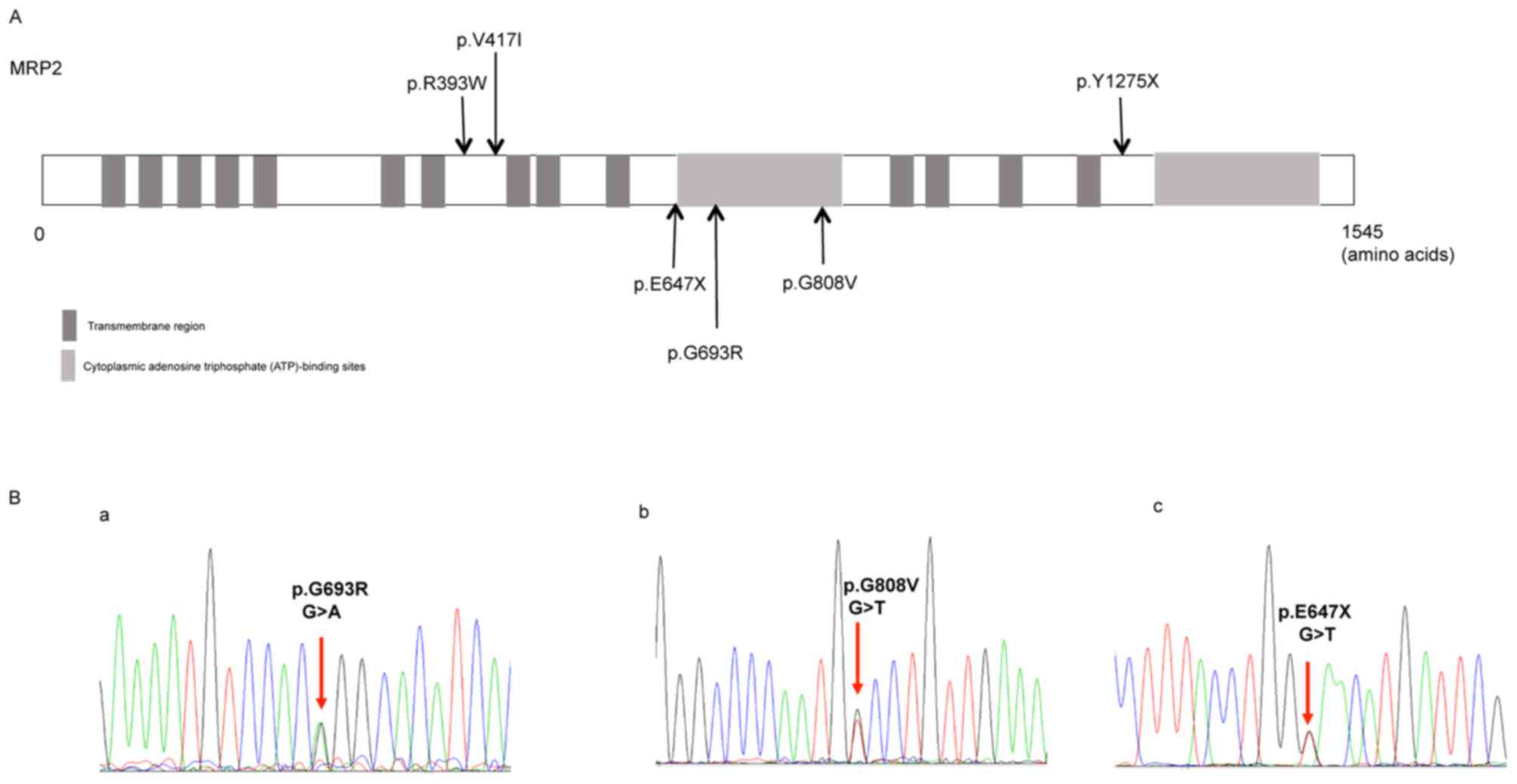
and the distribution of these variants is shown in Fig. 1A. The missense mutation p.R393W
(c.1290C>T) and nonsense mutation p.Y1275X (c.3938C>G) are
known disease mutations according to the database HGMD. In
addition, the missense variant p.V417I (c.1362G>A) in exon 10
harbored by the case no. 6 has previously been reported as a
single-nucleotide polymorphism (SNP) (12). Further details on the variants
identified in the ABCC2 gene are shown in Table III.
| Table III.Functional analysis of the variants
identified in the ABCC2 gene. |
Table III.
Functional analysis of the variants
identified in the ABCC2 gene.
| Location | Base change | Amino acid
change | Variation
types | Patient
frequency | Allele frequency
(Asian) | Allele frequency
(total) | Polyphen-2
prediction | SIFT
prediction | HGMD gene
result |
|---|
| Exon 9 | c.1290C>T | p.R393W | Missense | 2/7 |
1.817×10−4 |
3.295×10−5 | Probably
damaging | Deleterious | Known disease
mutation |
| Exon 10 | c.1362G>A | p.V417I | Missense | 1/7 |
2.785×10−1 |
1.953×10−1 | Benign | Tolerated | rs2273697 |
| Exon 15 | c.2052G>T | p.E647X | Nonsense | 2/7 |
6.061×10−5 |
8.248×10−6 | NA | NA | NA |
| Exon 16 | c.2190G>A | p.G693R | Missense | 2/7 |
2.442×10−4 |
4.139×10−5 | Probably
damaging | Deleterious | NA |
| Exon 18 | c.2536G>T | p.G808V | Missense | 1/7 | NA | NA | NA | NA | NA |
| Exon 27 | c.3938C>G | p.Y1275X | Nonsense | 1/7 |
4.594×10−4 |
6.59×10−5 | NA | NA | Known disease
mutation |
Novel variants identified in the
present study
Of the six variants identified, three were novel
variants, including the missense variants p.G693R (c.2190G>A)
and p.G808V (c.2536G>T), and the nonsense variant p.E647X
(c.2052G>T; Fig. 1B). Software
prediction demonstrated that the novel missense mutation p.G693R
(c.2190G>A) is probably damaging, while p.E647X is a typical
deleterious mutation (Table III).
The reports of allele frequency and functional prediction were
absent for p.G808V (Table
III).
Correlation between the ABCC2
genotypes and histology of liver biopsy
All the identified variants were heterozygous. Among
the cases enrolled, 1 patient (case no. 4) presented with compound
heterozygous mutations, namely p.G693R/p.G808V, while in another
patient (case no. 6), p.R393W/p.V417I was identified, with the
p.V417I considered as an SNP (12).
Other cases carried only one single mutation.
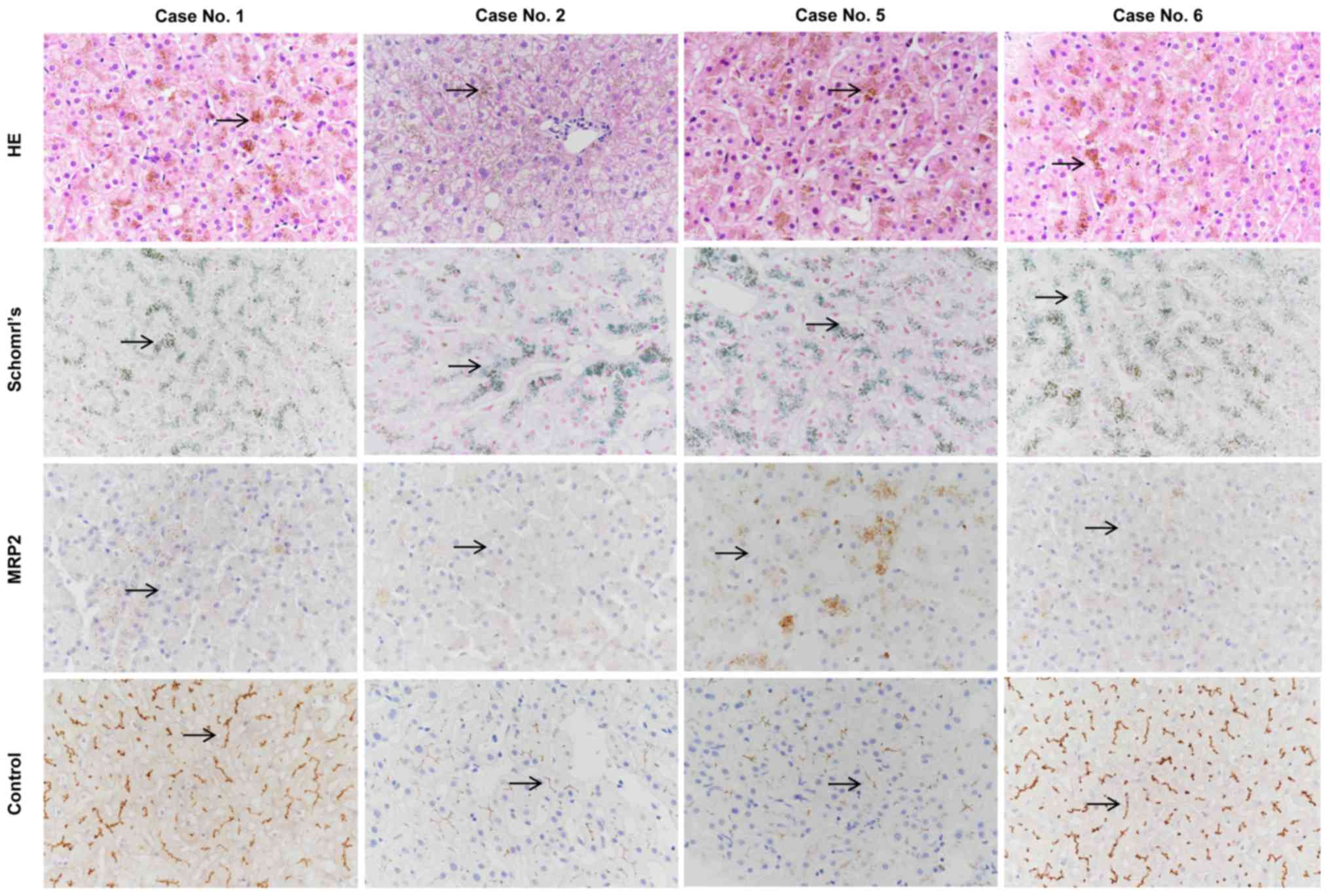
Liver biopsies and immunohistochemical analysis were
performed in cases no. 1 and 5 presenting with the novel nonsense
variant p.E647X, and in cases no. 2 and 6 presenting with the known
missense mutation p.R393W. HE and Schmorl's staining of the liver
biopsies demonstrated distinctive melanin-like, dark brown pigment
accumulation in the hepatocytes in all 4 cases (Fig. 2).
Immunohistochemical analysis of the MRP2 protein was
negative in all 4 cases compared with the normal liver tissues,
which were used as positive controls. Thus, the loss of membrane
expression of MRP2 caused by the novel nonsense variant p.E647X and
the known missense variant p.R393W was confirmed by the
immunohistochemical analysis of liver biopsy (Fig. 2).
Discussion
ABCC2 mutations have been identified in
patients with DJS worldwide; however, little is known regarding the
mutation pattern of ABCC2 in Chinese patients (13). In the present study, through the
mutation analysis of 7 clinically confirmed cases of DJS, it was
observed that all cases harbored at least one non-synonymous
variant in the ABCC2 gene. All the identified mutations were
heterozygous, with only 2 cases presenting with compound
heterozygous mutations. Thus, to the best of our knowledge, the
present study is the first to report the mutation patterns of the
ABCC2 gene in Chinese patients with DJS, as well as their
clinical association with DJS.
DJS is an autosomal recessive disorder, and the
majority of mutations in this syndrome are truncation mutations,
including frame shift mutations, nonsense mutations and splice site
mutations. To date, 55 ABCC2 gene mutations have been
reported. Most of the reported mutations are predicted to cause
defects in MRP2 protein maturation and localization, leading to
greatly reduced biliary secretion of organic anions (5). The missense mutation p.R393W
(c.1290C>T) (10) and the
nonsense mutation p.Y1275X (c.3938C>G) (11) are known disease mutations for DJS
previously identified in Asian patients. In the present study,
three novel ABCC2 mutations we reported in cases with DJS,
including missense mutations p.G693R and p.G808V, and nonsense
mutation p.E647X. Software predictions of the biological effects of
these mutations revealed that p.G693R is probably damaging, while
the p.E647X mutation may lead to truncated MRP2 protein. For
p.G808V, the reports of allele frequency and functional prediction
were absent.
Notably, DJS is an autosomal recessive disorder;
therefore, only homozygous variants in target genes are
traditionally considered to cause disease phenotypes. In the
present study, although all cases presented phenotypes, the
identified genetic variants were all heterozygous, and 2 of the 7
cases harbored compound heterozygous mutations. Recently, studies
have demonstrated that autosomal recessive diseases can also be
caused by compound heterozygous genotypes. For instance,
combinations of missense mutations of MRP2 have been reported in 2
Japanese patients, including p.R393W/p.R768W (10) and p.W709R/p.R1310X (14). In the current study, one novel
combination of missense mutations, p.G693R/p.G808V, was reported in
the ABCC2 gene. However, in the remaining 6 cases only
single heterozygous mutations were identified, suggesting that
variants in other associated, yet currently unknown, genes may
exist, or that other non-genetic factors may serve a synergistic
role. This highlights the importance of future studies involving
mutation analysis of the whole genome using next generation
sequencing. In our future study with a larger sample size, whole
exome sequencing will be conducted for the case with only one
heterozygous mutation, and the synergistic role of the non-genetic
factors will be explored.
However, there were several limitations in the
current study. Firstly, certain novel variants identified in the
present study were only predicted by software as deleterious.
However, the biological functional analysis of the novel mutations
was not conducted with a cell line model, and further functional
studies are required to understand the real effect of these
mutations. Functional analysis with cell line model will be
performed in the future. Furthermore, the number of cases was
limited, and whole exon sequencing of cases with single mutations
would be helpful to further understand their role in the
development of DJS. A greater number of patients with DJS will be
enrolled to enlarge the sample size, and whole exon sequencing of
cases with single mutations will be conducted in the future.
In conclusion, to the best of our knowledge, the
present study is the first to report the mutation patterns of the
ABCC2 gene in Chinese patients with DJS and their clinical
association with this disorder. The present study may provide
information that is essential for understanding of the role of
ABCC2 mutations in the development of DJS and may provide a
genetic basis for the diagnosis of DJS in China.
Acknowledgements
The authors kindly acknowledge Dr. Yun Zhang
(Biobank of Clinical Resources, Beijing Friendship Hospital,
Beijing, China) for blood sample storage.
Funding
The present study was supported by grants from the
National Key Technologies R&D Program of China (grant no.
2015BAI13B09).
Availability of data and materials
All the data were collected from the hospital
information system and can be available from the corresponding
author upon reasonable request.
Authors' contributions
LW collected and analyzed the clinical data, and
drafted the article. JH and XO conceived the paper and revised the
manuscript. JJ analyzed the data in the manuscript and supervised
the study. SJ performed the polymerase chain reaction amplification
analysis of the ABCC2 gene. XZ performed histopathological
analysis of the liver tissues of patients. WZ, DZ, AX, WD, ZW, HL,
SZ and YN collected the clinical data. All the authors approved the
final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Beijing Friendship Hospital at Capital Medical
University (Beijing, China).
Patient consent for publication
Written informed consent was obtained from all
patients for the publication of this article and any accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviation
Abbreviations:
|
DJS
|
Dubin-Johnson syndrome
|
|
ABCC2
|
adenosine triphosphate-binding
cassette subfamily C member 2
|
|
MRP2
|
multidrug resistance-associated
protein 2
|
References
|
1
|
Dubin IN and Johnson FB: Chronic
idiopathic jaundice with unidentified pigment in liver cells; A new
clinicopathologic entity with a report of 12 cases. Medicine
(Baltimore). 33:155–197. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frank M, Doss M and de Carvalho DG:
Diagnostic and pathogenetic implications of urinary coproporphyrin
excretion in the dubin-johnson syndrome. Hepatogastroenterology.
37:147–151. 1990.PubMed/NCBI
|
|
3
|
Morita M and Kihara T: Intravenous
cholecystography and metabolism of meglumine iodipamide
(biligrafin) in dubin-johnson syndrome. Radiology. 99:57–60. 1971.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shani M, Seligsohn U, Gilon E, Sheba C and
Adam A: Dubin-johnson syndrome in israel. I. Clinical, laboratory,
and genetic aspects of 101 cases. Q J Med. 39:549–567.
1970.PubMed/NCBI
|
|
5
|
Gazzin S, Masutti F, Vitek L and Tiribelli
C: The molecular basis of jaundice: An old symptom revisited. Liver
Int. 37:1094–1102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wakusawa S, Machida I, Suzuki S, Hayashi
H, Yano M and Yoshioka K: Identification of a novel 2026G->C
mutation of the MRP2 gene in a Japanese patient with Dubin-Johnson
syndrome. J Hum Genet. 48:425–429. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Erlinger S, Arias IM and Dhumeaux D:
Inherited disorders of bilirubin transport and conjugation: New
insights into molecular mechanisms and consequences.
Gastroenterology. 146:1625–1638. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Keitel V, Nies AT, Brom M,
Hummel-Eisenbeiss J, Spring H and Keppler D: A common Dubin-Johnson
syndrome mutation impairs protein maturation and transport activity
of MRP2 (ABCC2). Am J Physiol Gastrointest Liver Physiol.
284:G165–G174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hashimoto K, Uchiumi T, Konno T, Ebihara
T, Nakamura T, Wada M, Sakisaka S, Maniwa F, Amachi T, Ueda K and
Kuwano M: Trafficking and functional defects by mutations of the
ATP-binding domains in MRP2 in patients with dubin-johnson
syndrome. Hepatology. 36:1236–1245. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Machida I, Wakusawa S, Sanae F, Hayashi H,
Kusakabe A, Ninomiya H, Yano M and Yoshioka K: Mutational analysis
of the MRP2 gene and long-term follow-up of dubin-johnson syndrome
in japan. J Gastroenterol. 40:366–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JH, Chen HL, Chen HL, Ni YH, Hsu HY
and Chang MH: Neonatal dubin-johnson syndrome: Long-term follow-up
and MRP2 mutations study. Pediatr Res. 59:584–589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen C, Zhang B, Liu Z, Tang Y, Zhang Y,
Wang S, Guo Y, Ding Y, Wang S and Ding M: Effects of ABCB1, ABCC2,
UGT2B7 and HNF4α genetic polymorphisms on oxcarbazepine
concentrations and therapeutic efficacy in patients with epilepsy.
Seizure. 51:102–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiang R, Li JJ, Fan LL, Jin JY, Xia K and
Wang F: Identification of a compound heterozygous mutation of ABCC2
in a patient with hyperbilirubinemia. Mol Med Rep. 16:2830–2834.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Uchiumi T, Tanamachi H, Kuchiwaki K,
Kajita M, Matsumoto S, Yagi M, Kanki T and Kang D: Mutation and
functional analysis of ABCC2/multidrug resistance protein 2 in a
japanese patient with dubin-johnson syndrome. Hepatol Res.
43:569–575. 2013. View Article : Google Scholar : PubMed/NCBI
|