Introduction
Coronary artery disease (CAD) is a leading cause of
mortality and morbidity worldwide (1). Previous studies have reported that CAD
is a complex, multi-factorial polygenic disorder that is mediated
by genetic and environmental factors (2,3). Lipid
metabolism disorders are a major factor contributing to coronary
artery disease (4).
Apolipoprotein M (apoM), which is mainly expressed
in the liver and kidney, has recently been identified (5). ApoM is considered to serve an important
role in the generation of lipid-deficient pre-β high-density
lipoprotein (HDL), an early receptor of cellular cholesterol in
reverse cholesterol transport (RCT) (6). Ye et al (7) reported that dihydrotestosterone could
downregulate apoM mRNA expression via the classical androgen
receptor, independent of protein kinase C. In addition, Su et
al (8) suggested that serum apoM
protein levels are positively correlated with total cholesterol
(TC) and serum HDL. ApoM overexpression in Ldlr−/− mice
fed with a cholesterol-enriched diet was demonstrated to protect
against atherosclerosis, indicating that apoM may exert
anti-atherosclerotic effects in vivo (9). Christoffersen et al (10) reported that apoM, as a subpopulation
of HDL, was able to protect against the oxidation of low-density
lipoprotein (LDL) and stimulate cholesterol efflux more efficiently
than apoM-deficient HDL. Together, these studies suggest that apoM
is associated with HDL-mediated RCT and serves a crucial role in
the development of CAD. However, the detailed mechanism of apoM in
RCT and the pathogenesis of CAD remain unclear.
At present, statins are used as the first-line
treatment for lowering plasma cholesterol levels (11). In addition to their inhibitory effect
on cholesterol synthesis, statins have also been reported to have
anti-oxidative (12),
anti-inflammatory (13) and
anti-thrombotic effects (14), as
well as the ability to restore endothelial function and coronary
microcirculation (15). Yang et
al (16) administered healthy
mice and HepG2 cells with simvastatin and observed that apoM mRNA
and protein expression was upregulated in vivo and in
vitro. Conversely, a study by Zhang et al (17) had contradictory results, suggesting
that simvastatin inhibits apoM expression in HepG2 cells, but had
no effect in vivo. Therefore, the role of apoM in
statin-regulated lipid metabolism requires further
investigation.
Liver X receptor (LXR) was initially identified as
an orphan member of the nuclear receptor superfamily that exhibits
a stable regulatory effect on cholesterol and lipid metabolism
(18,19). LXR is activated by synthetic
agonists, including T0901317, and by endogenous oxysterols
(20). Wong et al (21) postulated that statins inhibit the
synthesis of an oxysterol ligand for LXR in human macrophages and
decrease cholesterol efflux. They also demonstrated that
supplementing human macrophages with cholesterol reverses the
statin-mediated downregulation of ABC transporter expression,
indicating that cellular lipid levels may influence the expression
of LXR-target genes. Zhang et al (22) demonstrated that the administration of
T0901317 resulted in hepatic apoM downregulation in healthy
C57BL/6J mice and HepG2 cells. However, the association between
apoM and LXRα in the hyperlipidemic microenvironment remains
unclear. Considering the contradictory nature of previous studies,
the present study was performed to investigate whether atorvastatin
regulates apoM expression and to elucidate the potential underlying
mechanisms.
Materials and methods
Cells, animals and reagents
The human hepatoblastoma cell line (HepG2) was
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA). A total of 16 male 8-week-old ApoE−/− (weight,
19.12±0.44 g) and 8 male 8-week-old C57BL/6 (weight, 20.08±0.31 g)
mice were purchased from the Model Animal Research Center of
Nanjing University (Nanjing, China). Atorvastatin original powder
was purchased from Abcam (Cambridge, UK), LXR agonist T0901317 was
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and a
quantitative polymerase chain reaction (qPCR) kit (SYBR®
Premix Ex Taq™ II) was obtained from Takara Bio, Inc.
(Otsu, Japan). Antibodies against apoM (cat. no. ab122896) and LXRα
(cat. no. ab41902) were purchased from Abcam, while a pre-β HDL
ELISA kit (cat. no. ml001270) was obtained from Mlbio (Shanghai,
China). Reverse transcription (RT)-qPCR primers were obtained from
GENEWIZ (South Plainfield, NJ, USA).
Animal experiments
Mice received humane care according to the
Guidelines for the Care and Use of Research Animals established by
Soochow University (Suzhou, China) and the experimental protocols
were approved by the Ethics Committee of Soochow University. A
total of 12 8-week-old apoE−/− mice and 4 8-week-old
C57BL/6 mice were acclimated to housing in standard polycarbonate
cages in the Animal Facility of Soochow University under a 12 h
light/dark cycle for 1 week prior to experimentation. Mice were
housed with free access to food and water, and under the
temperature of 23°C and air humidity of 55%. The in vivo
study included four groups (16 mice total; 4 mice for each group):
C57BL/6 mice and apoE−/− mice fed with regular diet
(control and apoE−/− groups, respectively),
apoE−/− mice fed with high-fat diet treated with normal
saline (vehicle control group) and apoE−/− mice fed with
high-fat diet and treated with atorvastatin (statin group). In the
control and apoE−/− groups, mice were fed with regular
chow for 8 weeks and administered with 0.2 ml (0.9%) normal saline
by lavage every day. For the vehicle control and atorvastatin
groups, mice were provided with high-fat feed (Suzhou Shuangshi
Animal Feed Technology Co., Ltd., Suzhou, China), comprising 4%
cholesterol, 0.5% sodium cholate, 10% lard, 0.2% propylthiouracil
and 85.3% normal chow diet, for 8 weeks to induce hyperlipidemia.
Following the successful establishment of hyperlipidemia, mice were
randomly divided into the vehicle control and statin groups. In the
statin group, mice were administered with 10 mg/kg/day atorvastatin
dissolved in 0.2 ml (0.9%) normal saline by oral gavage for 4
weeks. Mice in the vehicle control group were administered with 0.2
ml (0.9%) normal saline only.
In addition to the groups described above, a total
of 4 male 8-week-old apoE-deficient mice (Model Animal Research
Center of Nanjing University) were housed with free access to food
and water in standard polycarbonate cages under a 12 h light/dark
cycle, and under the temperature of 23°C and air humidity of 55%.
The mice were used as the fifth experimental (agonist) group.
Hyperlipidemia was induced in apoE-deficient mice as previously
described. Thereafter, mice were administrated with 50 mg/kg/day of
T0901317 + 10 mg/kg/day atorvastatin dissolved in 0.2 ml (0.9%)
normal saline by lavage for 4 weeks. Following treatment, mice in
all groups were anesthetized and sacrificed. Blood samples were
collected through the tail vein and centrifuged at 4°C at 5,000 × g
for 20 min to collect the serum, which was subsequently stored at
−80°C. Liver samples were harvested and flash frozen for subsequent
RT-qPCR and western blotting.
Cell culture
HepG2 cells were cultured in high-glucose Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Biological Industries, Kibbutz Beit Haemek, Israel),
1.0×105 U/l penicillin and 1.0×105 U/l
streptomycin at 37°C. The medium was replaced every 2 days and
cells were passaged. Cells were seeded in a 6-well plate at a
density of ~5×105 cells/well and grown to 50–70%
confluence. Prior to experimentation, cells were washed twice with
PBS and the medium was replaced. In vitro experiments
involved four groups: Control, dimethylsulfoxide (DMSO),
atorvastatin and agonist. Cells in the control and DMSO groups were
treated with 200 µl PBS or 200 µl DMSO, respectively. In the statin
group, cells were treated with 20 nmol atorvastatin dissolved in
200 µl DMSO. In the agonist group, cells were treated with 200 nmol
T0901317 + 20 nmol atorvastatin dissolved in 200 µl DMSO. Cells
were harvested following 24 h of treatment at 37°C for western
blotting and RT-qPCR.
Serum lipid analysis
Mice were fasted for 12 h and sacrificed, following
which blood samples were collected via the tail vein. Serum was
separated by centrifugation at 600 × g under 4°C for 20 min,
following which HDL cholesterol (HDL-C), LDL cholesterol (LDL-C),
total cholesterol (TC; all cat. no. ab65390), and triglyceride (TG;
cat. no. ab65336) levels were measured (23) with assay kits (Abcam).
Western blotting
ApoM and LXRα protein expression in liver samples
and HepG2 cells were detected using western blotting as previously
described (24,25). Briefly, 40 mg tissue or approximately
2×106 cells in each group were homogenized to obtain
lysates from which protein was extracted using
ProteoPrep® Total Extraction Sample Kit (cat. no.
PROTTOT-1KT; Sigma-Aldrich; Merck KGaA). The protein concentration
was measured using a BCA protein assay kit according to the
manufacturer's instructions. A total of 25 µg of each sample was
separated by 12% SDS-PAGE and transferred onto polyvinylidene
fluoride membranes. Blocking was then performed by overnight
incubation at 4°C in Tris-buffered saline/Tween 20 (TBST)
containing 5% non-fat dried milk. Membranes were washed with TBST
and incubated with anti-apoM (1:1,000), anti-LXRα (1:1,000), and
anti-GAPDH (cat. no. AF0006; 1:1,000; Beyotime Institute of
Biotechnology, Shanghai, China) primary antibodies for 1 h at room
temperature. Subsequent to washing with TBST, the blots were
incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. no. A0181; 1:500; Beyotime Institute of
Biotechnology) for 1 h at 37°C. A chemiluminescent substrate (cat.
no. 34580; Thermo Fisher Scientific, Inc.) was used to detect the
peroxidase-conjugated antibodies and membranes were exposed to
X-ray film using BIO-RAD Gel Doc XR+ (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The intensity of bands was evaluated with
ImageJ software (version 1.51; National Institutes of Health,
Bethesda, MD, USA) and quantified relative to GAPDH bands from the
same sample.
RT-qPCR
RT-qPCR was performed to determine the expression of
LXRα and apoM mRNA in liver samples and HepG2 cells. Total RNA was
extracted from liver tissues and HepG2 cells using TRIzol (Thermo
Fisher Scientific, Inc.) and purified using a Qiagen RNeasy Mini
kit (cat. no. 74104; Qiagen AG, Sollentuna, Sweden) as previously
described (26). cDNA was
synthesized from total RNA using the PrimeScript RT reagent kit
(Takara Bio, Inc.). The temperature and duration for RT was 25°C
for 10 min, 37°C for 60 min, 70°C for 10 min and 4°C on ∞ hold. A
total of 2 µl cDNA was used for qPCR, which was performed using
Power Syber Green and the StepOne-Plus real-time PCR system (Thermo
Fisher Scientific, Inc.). The thermocycler protocol for qPCR was
95°C for 3 min, then 40 cycles of 94°C for 40 sec, 55°C for 30 sec,
72°C for 40 sec and 72°C for 5 min, then 4°C on ∞ hold. The
endogenous housekeeping gene GAPDH was used to normalize expression
levels. The sequences of primers used in qPCR are presented in
Table I. The 2−ΔΔCq
method was used to evaluate changes in target gene expression
relative to GAPDH (27).
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| Liver X receptor
α |
CTGTGCCTGACATTCCTCCT |
CATCCTGGCTTCCTCTCTGA |
| Apolipoprotein
M |
GCGCCCAGACATGAAAACAG |
AGGCCTCTTGATTCCTGGGA |
| GAPDH |
AATCCCATCACCATCTTCCA |
TGGACTCCACGACGTACTCA |
ELISA for pre-β HDL
quantification
Serum pre-β HLD levels were measured using a
sandwich ELISA kit according to the manufacturer's protocol.
Optical density (OD) values were measured at 450 nm (background
reading at 620 nm) with an absorbance reader (BioTek Instruments,
Inc., Winooski, VT, USA). The serum concentration of pre-β HLD
(pg/ml) in each group was calculated using a standard curve.
Statistical analysis
Data are presented as the mean ± standard deviation.
All data were analyzed using GraphPad Prism 6.0 software (GraphPad
Software, Inc., La Jolla, CA, USA) using one-way analysis of
variance with Bonferroni's correction for multiple group
comparisons and Student's t-test for the comparisons between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Serum lipid profile in high-fat diet
treated mice and the effects of atorvastatin
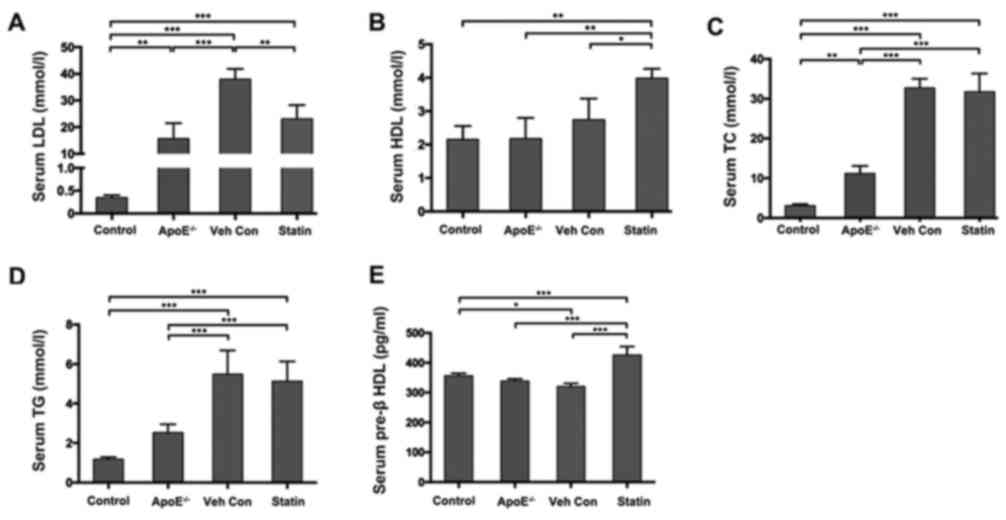
Hyperlipidemia was successfully induced in
apoE−/− mice following 8 weeks of high-fat diet
administration. As presented in Table
II, TC, LDL-C and TG were significantly increased in
apoE−/− mice fed with a high-fat diet compared with
C57BL/6 mice or apoE−/− mice fed with regular chow for 8
weeks. Following 4 weeks of statin or vehicle treatment, the serum
concentration of LDL-C was significantly decreased in the statin
group compared with the vehicle control group, while HDL-C levels
were significantly increased (Fig. 1A
and B). Serum TC and TG levels were decreased by 2.99 and
6.38%, respectively, in the statin group compared with the vehicle
control group (Fig. 1C and D).
However, no statistically significant differences were observed
between the statin group and the vehicle control group. Serum pre-β
HLD levels were reduced in the apoE−/− mice compared
with the vehicle control group (Fig.
1E). However, reduced pre-β HLD levels were reversed in the
statin group, resulting in significant pre-β HLD upregulation
compared with the control group (Fig.
1E).
| Table II.Mouse serum lipid following 8 weeks
of regular or high-fat diet. |
Table II.
Mouse serum lipid following 8 weeks
of regular or high-fat diet.
| Group | n | TC (mmol/l) | LDL-C (mmol/l) | HDL-C (mmol/l) | TG (mmol/l) |
|---|
| C57BL/6 mice with
regular chow | 4 | 3.07±4.13 | 0.33±0.07 | 2.12±0.39 | 1.25±0.20 |
| ApoE−/−
mice with regular chow | 4 |
11.18±1.65a |
17.39±4.39a | 2.19±0.67 | 2.54±0.54 |
| ApoE−/−
mice with high-fat diet | 8 |
38.00±3.63a,b |
37.97±3.98a,b | 2.73±0.59 |
5.49±1.12a,b |
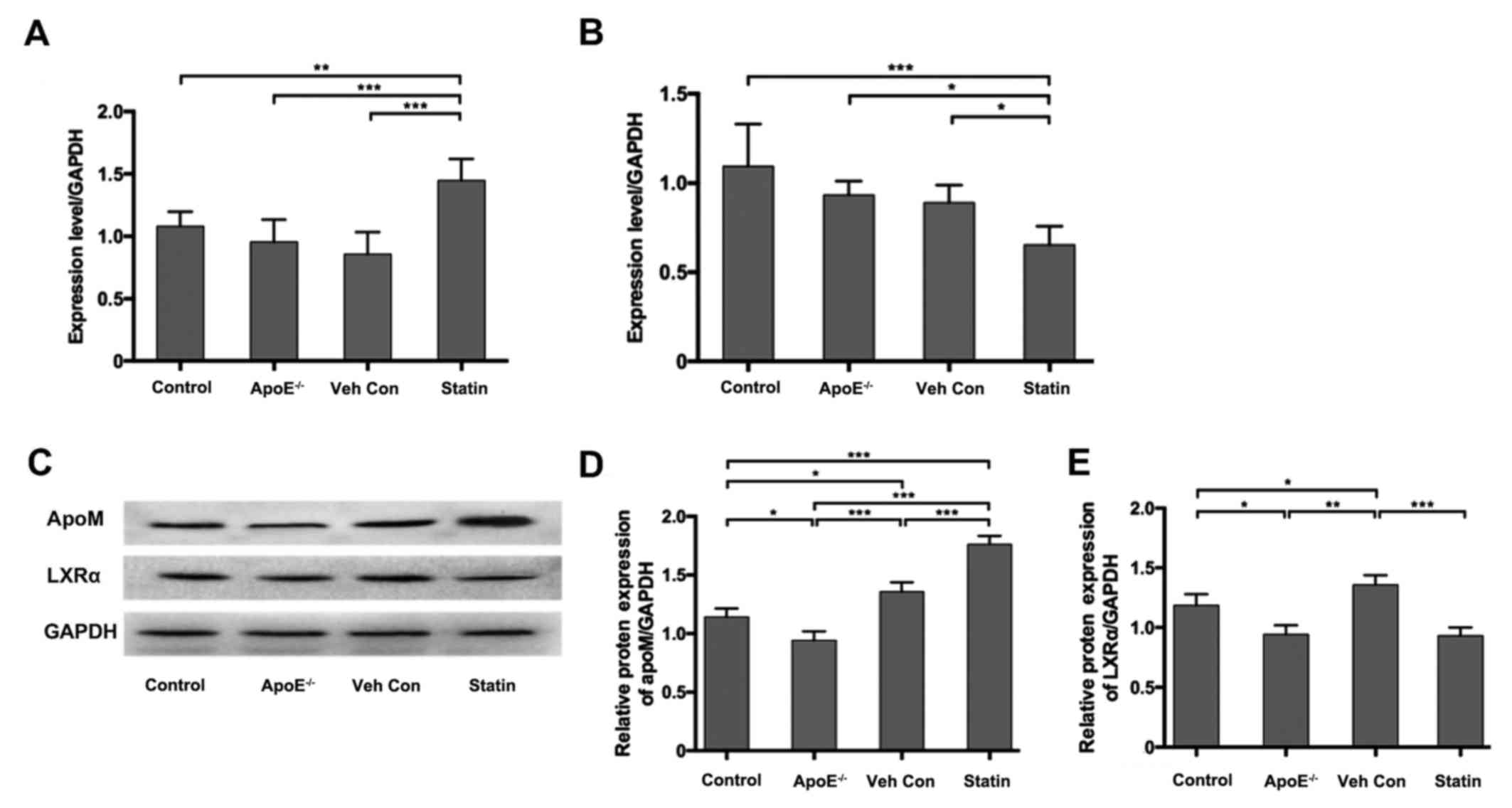
Effects of atorvastatin on apoM and
LXRα expression levels in mouse liver tissues
ApoM and LXRα expression was assessed in mouse liver
samples using RT-qPCR and western blotting. No significant
differences in apoM mRNA expression were observed between the
control, apoE−/− and vehicle control groups; however,
apoM mRNA expression levels were significantly increased in the
statin group compared with all other groups (Fig. 2A). LXRα mRNA expression was
downregulated in apoE−/− and vehicle control mice
compared with the control group, however no statistical
significance was recorded (Fig. 2B).
Mice in the statin group exhibited a significant decrease in LXRα
mRNA expression compared with the control, apoE−/− and
vehicle control groups (Fig. 2B).
The expression of apoM was significantly increased in the statin
group compared with the vehicle control group (Fig. 2C and D), consistent with the results
of RT-qPCR. However, LXRα protein expression decreased in the
statin group compared with the vehicle control group (Fig. 2C and E). These results suggest that
atorvastatin is able to upregulate apoM expression in the liver of
hyperlipidemic mice while simultaneously downregulating LXRα.
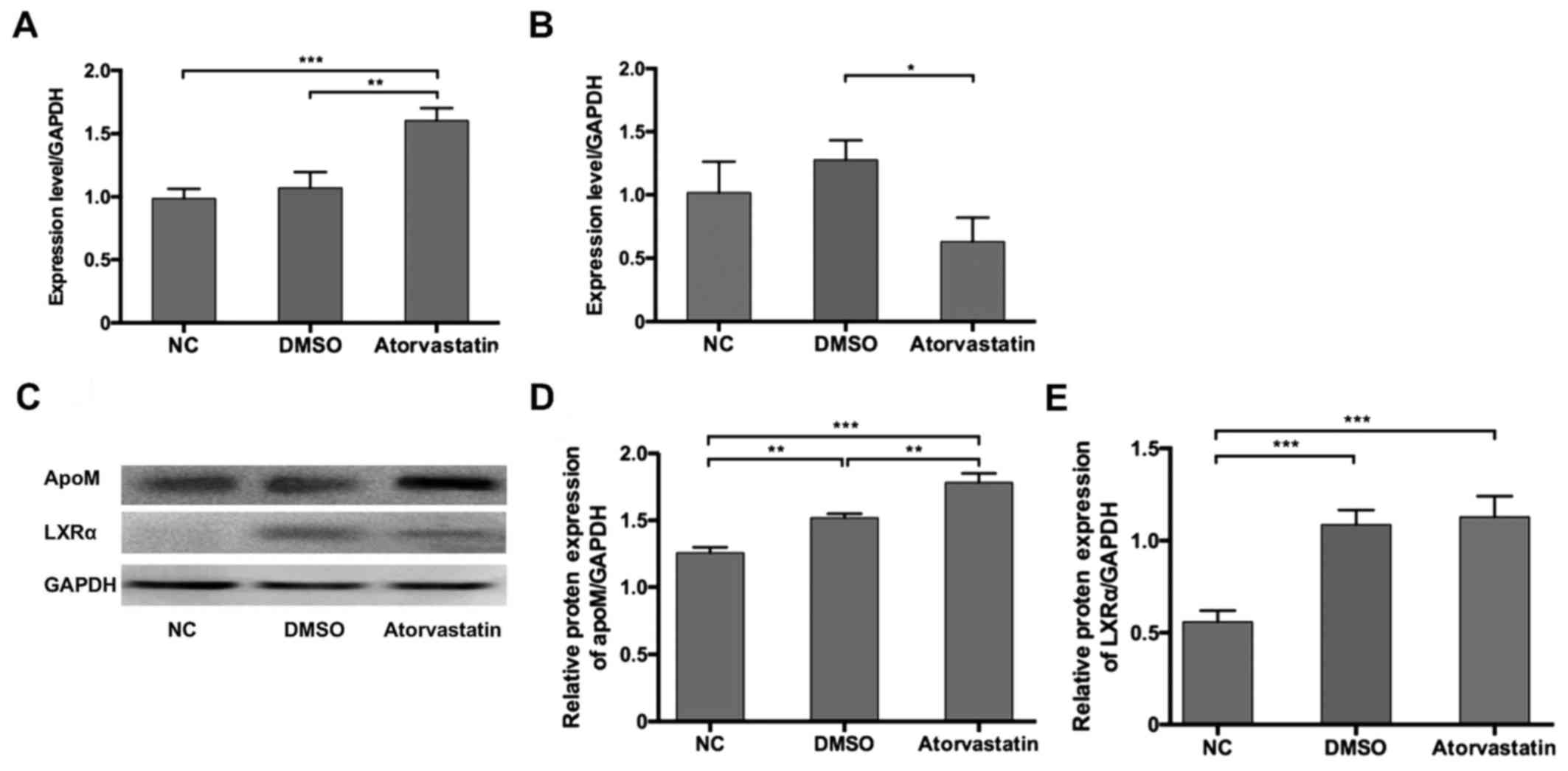
Effects of atorvastatin on apoM and
LXRα expression in HepG2 cells
The in vivo study indicated that atorvastatin
was able to downregulate serum lipid levels in hyperlipidemic mice
by regulating liver apoM and LXRα expression at the mRNA and
protein levels. In order to further study the mechanism underlying
mechanism, an in vitro cell model was employed. RT-qPCR
results suggested that apoM mRNA expression was significantly
increased 1.6-fold compared with the DMSO group following statin
treatment (Fig. 3A). Furthermore,
LXRα was significantly downregulated at the mRNA level in response
to statin treatment compared with the DMSO group (Fig. 3B). Western blotting results revealed
that apoM protein was overexpressed following statin treatment
compared with the DMSO group (Fig. 3C
and D). Although LXRα mRNA expression levels were reduced
following statin treatment, no significant differences were
observed in LXRα protein expression between the DMSO and
atorvastatin groups (Fig. 3C and
E).
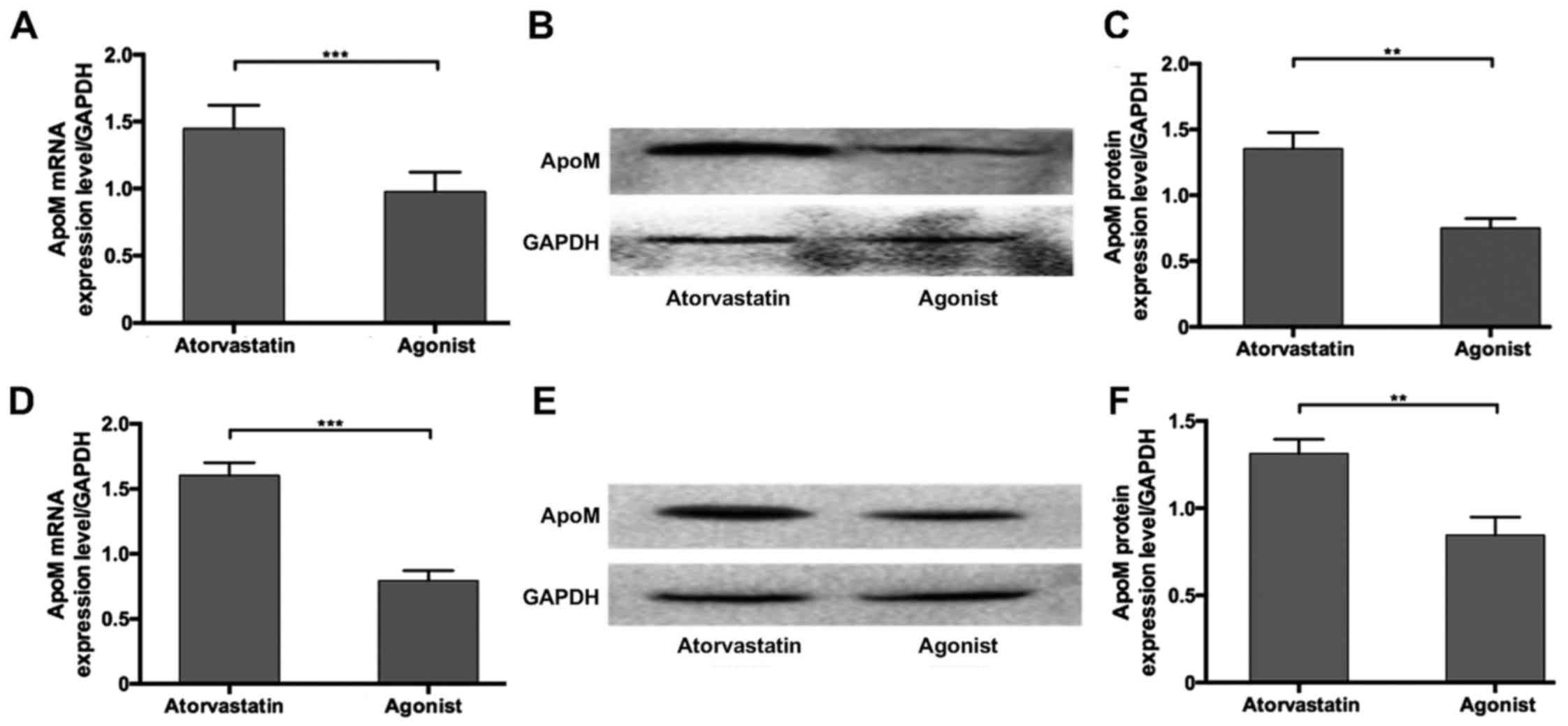
Effects of T0901317 and atorvastatin
on apoM expression in vivo and in vitro
To investigate whether apoM upregulation may be
mediated by the attenuation of LXRα, the LXR agonist T0901317 was
used. T0901317 was administered in combination with atorvastatin to
hyperlipidemic mice and HepG2 cells. The results revealed that apoM
mRNA and protein expression was significantly decreased by combined
treatment with the agonist compared with atorvastatin alone in
vivo and in vitro (Fig.
4). These results suggest that T0901317 is able to inhibit
atorvastatin-induced apoM upregulation.
Discussion
Statins are a class of drugs that inhibit the enzyme
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (28). Previous investigations into statins
have revealed a number of mechanisms underlying its
anti-atherosclerotic effects: i) Inhibition of HMG-CoA conversion
to mevalonic acid with consequential decreases in cholesterol
biosynthesis and reductions in serum TC and LDL-C (29); ii) increased biosynthesis of nitric
oxide (NO) and amelioration of endothelial function (30); iii) inhibition of the inflammatory
reaction and the formation of foam cells in atheromatous plaques
(31,32); and iv) regulation of the platelet
membrane composition and inhibition of platelet aggregation
(33).
ApoM was first identified as a novel apolipoprotein
by Xu et al (34) in 1999.
ApoM is primarily synthesized in the liver and secreted into the
plasma where it participates in the formation of HDL and serves a
role in lipid metabolism (35).
Richter et al (36)
determined that apoM serves a pivotal role in the formation of
pre-β HDL. They reported that both pre-β HDL and normal HDL
expression was increased following the recovery of hepatocyte
nuclear factor-1α (HNF-1α) and apoM expression in an
HNF-1α-deficient mouse model (36).
The current hypothesis is that apoM is not essential for HDL to
mobilize cholesterol, however it facilitates this action via
enhancing pre-β HDL formation (37).
In brief, apoM may increase the formation of pre-β HDL and
facilitate cholesterol mobilization of pre-β HDL from macrophages
via its interaction with ATP-binding cassette transporter member 1
(38). ApoM expression may be
regulated by multiple factors in vivo and ex vivo,
including HNF-1α 4α, liver receptor homolog-1, forkhead box A2 and
platelet activating factor, which are able to upregulate apoM
expression (5). Furthermore, LXR,
retinoid X receptor, farnesoid X receptor and small heterodimer
partner may downregulate apoM (5). A
number of studies have investigated the effects of statins on apoM;
however, the results are controversial (6,16,17).
Thus, whether statins are able to regulate apoM expression and its
underlying mechanisms remains unclear.
In the present study, hyperlipidemic apoE-deficient
mice were treated with atorvastatin and it was demonstrated that
serum HDL and pre-β HDL levels were elevated, while LDL expression
was decreased. Considering that of apoM overexpression in mice
increases serum HDL-C concentrations, apoM deficiency may be
associated with reduced serum HDL-C concentrations (39). As apoM is associated with the
formation of pre-β HDL (37), it was
next investigated whether the cholesterol-lowering effects of
statins are associated with its effect on apoM and other
cholesterol efflux-associated genes. ApoM upregulation in the liver
was revealed to be accompanied by LXRα downregulation, while serum
pre-β HDL expression was also increased in the statin treated
group. These results indicate that statin treatment enhances
cholesterol efflux, which may be mediated by pre-β HDL and
facilitated by apoM. Based on this, the effect of statin treatment
in vitro was investigated to further explore the mechanism
responsible. ApoM and LXRα expression were measured in HepG2 cells
and the results were similar to those of in vivo analysis;
statin-induced apoM upregulation in HepG2 cells was accompanied by
LXRα downregulation. Importantly, co-treatment with atorvastatin
and T0901317 in vivo and in vitro significantly
inhibited statin-induced apoM overexpression.
In conclusion, the results of the present study
indicate that atorvastatin treatment is able to upregulate apoM
expression and attenuate LXRα expression, which may enhance RCT in
the mouse liver. Additionally, T0901317 may block statin-induced
apoM upregulation. These results suggest that atorvastatin may
upregulate apoM via mediating LXRα in mouse models. Future studies
may futher elucidate the mechanism behind atorvastatin-induced apoM
upregulation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shanghai
Municipal Commission of Health and Family Planning Program (grant
no. 201440293), Jiangsu Province's Key Discipline/Laboratory of
Medicine (grant no. XK201118), the National Clinical Key Specialty
of Cardiovascular Surgery and Jiangsu Clinical Research Center for
Cardiovascular Surgery (grant no. BL201451) and the National
Natural Science Foundation of China (grant nos. 81770260 and
81400199.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL, HH, SS, XW, YY, YH, JS and CR were responsible
for performance of experiment, data analysis, and manuscript
preparation. JY and ZS were responsible for manuscript writing and
revision, and experimental design.
Ethical approval and consent to
participate
The experiment protocols were approved by the Ethic
Committee of Soochow University (reference number:
SZUM2008031233).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there is no conflict of
interests regarding the publication of this paper.
References
|
1
|
Ma T, Sun J, Zhao Z, Lei W, Chen Y, Wang
X, Yang J and Shen Z: A brief review: Adipose-derived stem cells
and their therapeutic potential in cardiovascular diseases. Stem
Cell Res Ther. 8:1242017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marenberg ME, Risch N, Berkman LF,
Floderus B and de Faire U: Genetic susceptibility to death from
coronary heart disease in a study of twins. N Engl J Med.
330:1041–1046. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nora JJ, Lortscher RH, Spangler RD, Nora
AH and Kimberling WJ: Genetic-epidemiologic study of early-onset
ischemic heart disease. Circulation. 61:503–508. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manting L, Haihong Z, Jing L, Shaodong C
and Yihua L: The model of rat lipid metabolism disorder induced by
chronic stress accompanying high-fat-diet. Lipids Health Dis.
10:1532011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ren K, Tang ZL, Jiang Y, Tan YM and Yi GH:
Apolipoprotein M. Clin Chim Acta. 446:21–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kappelle PJ, Ahnstrom J, Dikkeschei BD, de
Vries R, Sluiter WJ, Wolffenbuttel BH, van Tol A, Nielsen LB,
Dahlbäck B and Dullaart RP: Plasma apolipoprotein M responses to
statin and fibrate administration in type 2 diabetes mellitus.
Atherosclerosis. 213:247–250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye YZ, Cao B, Li MQ, Wang W, Wang RX, Rui
J, Wei LY, Jing ZH, Ji Y, Jiao GQ and Zou J: Dihydrotestosterone
regulating apolipoprotein M expression mediates via protein kinase
C in HepG2 cells. Lipids Health Dis. 11:1682012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su W, Jiao G, Yang C and Ye Y: Evaluation
of apolipoprotein M as a biomarker of coronary artery disease. Clin
Biochem. 42:365–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wolfrum C, Poy MN and Stoffel M:
Apolipoprotein M is required for prebeta-HDL formation and
cholesterol efflux to HDL and protects against atherosclerosis. Nat
Med. 11:418–422. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Christoffersen C, Nielsen LB, Axler O,
Andersson A, Johnsen AH and Dahlback B: Isolation and
characterization of human apolipoprotein M-containing lipoproteins.
J Lipid Res. 47:1833–1843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Endo A: A historical perspective on the
discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci.
86:484–493. 2006. View Article : Google Scholar
|
|
12
|
Tsai NW, Lee LH, Huang CR, Chang WN, Chang
YT, Su YJ, Chiang YF, Wang HC, Cheng BC, Lin WC, et al: Statin
therapy reduces oxidized low density lipoprotein level, a risk
factor for stroke outcome. Crit Care. 18:R162014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Ghoul WM, Kim MS, Fazal N, Azim AC and
Ali A: Evidence for simvastatin anti-inflammatory actions based on
quantitative analyses of NETosis and other inflammation/oxidation
markers. Results Immunol. 4:14–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmidt M, Cannegieter SC, Johannesdottir
SA, Dekkers OM, Horváth-Puhó E and Sorensen HT: Statin use and
venous thromboembolism recurrence: A combined nationwide cohort and
nested case-control study. J Thromb Haemost. 12:1207–1215. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paraskevaidis IA, Iliodromitis EK,
Ikonomidis I, Rallidis L, Hamodraka E, Parissis J, Andoniadis A,
Tzortzis S and Anastasiou-Nana M: The effect of acute
administration of statins on coronary microcirculation during the
pre-revascularization period in patients with myocardial
infraction. Atherosclerosis. 223:184–189. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang L and Zhao S: Effect of simvastatin
on the expression and regulation mechanism of apolipoprotein M. Int
J Mol Med. 29:510–514. 2012.PubMed/NCBI
|
|
17
|
Zhang X, Mao S, Luo G, Wei J,
Berggren-Söderlund M, Nilsson-Ehle P and Xu N: Effects of
simvastatin on apolipoprotein M in vivo and in vitro. Lipids Health
Dis. 10:1122011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ulven SM, Dalen KT, Gustafsson JA and Nebb
HI: LXR is crucial in lipid metabolism. Prostaglandins Leukot
Essent Fatty Acids. 73:59–63. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y and Mangelsdorf DJ: LuXuRies of
lipid homeostasis: The unity of nuclear hormone receptors,
transcription regulation, and cholesterol sensing. Mol Interv.
2:78–87. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Houck KA, Borchert KM, Hepler CD, Thomas
JS, Bramlett KS, Michael LF and Burris TP: T0901317 is a dual
LXR/FXR agonist. Mol Genet Metab. 83:184–187. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wong J, Quinn CM and Brown AJ: Statins
inhibit synthesis of an oxysterol ligand for the liver × receptor
in human macrophages with consequences for cholesterol flux.
Arterioscler Thromb Vasc Biol. 24:2365–2371. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Zhu Z, Luo G, Zheng L,
Nilsson-Ehle P and Xu N: Liver X receptor agonist downregulates
hepatic apoM expression in vivo and in vitro. Biochem Biophys Res
Commun. 371:114–117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gervois P, Fruchart JC and Staels B: Drug
Insight: Mechanisms of action and therapeutic applications for
agonists of peroxisome proliferator-activated receptors. Nat Clin
Pract Endocrinol Metab. 3:145–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Lei W, Yan W, Li X, Wang X, Zhao
Z, Hui J, Shen Z and Yang J: microRNA-206 is involved in survival
of hypoxia preconditioned mesenchymal stem cells through targeting
Pim-1 kinase. Stem Cell Res Ther. 7:612016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Z, Yang J, Yan W, Li Y, Shen Z and
Asahara T: Pretreatment of cardiac stem cells with exosomes derived
from mesenchymal stem cells enhances myocardial repair. J Am Heart
Assoc. 5:e0028562016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
You J, Sun J, Ma T, Yang Z, Wang X, Zhang
Z, Li J, Wang L, Ii M, Yang J and Shen Z: Curcumin induces
therapeutic angiogenesis in a diabetic mouse hindlimb ischemia
model via modulating the function of endothelial progenitor cells.
Stem Cell Res Ther. 8:1822017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Friesen JA and Rodwell VW: The
3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome
Biol. 5:2482004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonetti PO, Lerman LO, Napoli C and Lerman
A: Statin effects beyond lipid lowering-are they clinically
relevant? Eur Heart J. 24:225–248. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fichtlscherer S, Schmidt-Lucke C, Bojunga
S, Rössig L, Heeschen C, Dimmeler S and Zeiher AM: Differential
effects of short-term lipid lowering with ezetimibe and statins on
endothelial function in patients with CAD: Clinical evidence for
‘pleiotropic’ functions of statin therapy. Eur Heart J.
27:1182–1190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cortellaro M, Cofrancesco E, Arbustini E,
Rossi F, Negri A, Tremoli E, Gabrielli L and Camera M: Atorvastatin
and thrombogenicity of the carotid atherosclerotic plaque: The
ATROCAP study. Thromb Haemost. 88:41–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Otto C, Geiss HC, Empen K and Parhofer KG:
Long-term reduction of C-reactive protein concentration by regular
LDL apheresis. Atherosclerosis. 174:151–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luzak B, Boncler M, Rywaniak J, Wilk R,
Stanczyk L, Czyz M, Rysz J and Watala C: The effect of a platelet
cholesterol modulation on the acetylsalicylic acid-mediated blood
platelet inhibition in hypercholesterolemic patients. Eur J
Pharmacol. 658:91–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu N and Dahlbäck B: A novel human
apolipoprotein (apoM). J Biol Chem. 274:31286–31290. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang XY, Dong X, Zheng L, Luo GH, Liu YH,
Ekström U, Nilsson-Ehle P, Ye Q and Xu N: Specific tissue
expression and cellular localization of human apolipoprotein M as
determined by in situ hybridization. Acta Histochem. 105:67–72.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Richter S, Shih DQ, Pearson ER, Wolfrum C,
Fajans SS, Hattersley AT and Stoffel M: Regulation of
apolipoprotein M gene expression by MODY3 gene hepatocyte nuclear
factor-1alpha: Haploinsufficiency is associated with reduced serum
apolipoprotein M levels. Diabetes. 52:2989–2995. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Elsoe S, Christoffersen C, Luchoomun J,
Turner S and Nielsen LB: Apolipoprotein M promotes mobilization of
cellular cholesterol in vivo. Biochim Biophys Acta. 1831:1287–1292.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mulya A, Seo J, Brown AL, Gebre AK,
Boudyguina E, Shelness GS and Parks JS: Apolipoprotein M expression
increases the size of nascent pre beta HDL formed by ATP binding
cassette transporter A1. J Lipid Res. 51:514–524. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Christoffersen C, Jauhiainen M, Moser M,
Porse B, Ehnholm C, Boesl M, Dahlbäck B and Nielsen LB: Effect of
apolipoprotein M on high density lipoprotein metabolism and
atherosclerosis in low density lipoprotein receptor knock-out mice.
J Biol Chem. 283:1839–1847. 2008. View Article : Google Scholar : PubMed/NCBI
|