Introduction
Alzheimer's disease (AD) is a chronic
neurodegenerative disease associated with aging (1). The pathogenic factors of AD are
complex. In addition to cholinergic dysfunction, β-amyloid
deposition resulting in inflammatory response is one of the most
important pathogenic mechanisms of AD (1). Treatment of AD using
acetylcholinesterase inhibitors (AchEIs), which inhibit the
acetylcholine degradation in synapses, achieved limited results
(1). However, epidemiological data
suggested that non-steroid anti-inflammatory drugs (NSAIDs), which
inhibit both cyclooxygenase 2 (COX-2) and interleukin (IL)-1β, may
reduce the incidence of AD (2).
Therefore, anti-inflammatory drugs that reduce the secretion of
inflammatory factors in the peripheral or central nervous system
have become the focus of research for the treatment of AD (3).
Berberine (BBR) is the active ingredient of the
extract from Coptis chinensis. It exhibits a variety of
biological activities, including anti-inflammatory, antidiabetic,
anticancer and antiarrhythmic effects, prevention of intestinal
bacterial infection, dilation of coronary blood vessels, and lowing
blood lipids (4). A previous study
indicated that BBR can effectively inhibit expression of
inflammatory factors, including high sensitivity c-reactive
protein, tumor necrosis factor-α (TNF-α) and IL-6 (5). However, it remains to be elucidated
whether BBR can inhibit the abnormally expressed inflammatory
factors resulting from pathological alterations in the central
nervous system.
In a preliminary study, the authors of the present
study have established a mouse model of AD (unpublished). Aberrant
expression levels of COX-2, prostaglandin E2 (PGE2), IL-1β, TNF-α
and TNF-α type 1 receptor were observed in neurons of mice with AD
exhibiting abnormal behavior. In the present study amyloid
β25–35 (Aβ25–35) was used to induce
inflammatory response in the neuroblastoma SH-SY5Y and SK-N-SH
cells to model the inflammatory process of patients with AD. The
viability of nerve cells and the alterations of mRNA and protein
expression levels of inflammatory factors COX-2, PGE2, IL-1β, TNF-α
and TNF-α type 1 receptor were observed prior to and following the
treatment. The results of the present study revealed that BBR
alleviated the inflammatory response induced by Aβ25–35.
The mechanism of action of BBR may be associated with the
inhibition of proinflammatory mediators.
Materials and methods
Cells and reagents
Human neuroblastoma SH-SY5Y and SK-N-SH cell lines
were purchased from Shanghai Institutes for Biological Sciences
(Shanghai, China). Fetal bovine serum (FBS) and Dulbecco's modified
Eagle's medium (DMEM) were purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). RNA extraction reagent TRIzol
was purchased from Thermo Fisher Scientific Inc. (Waltham, MA,
USA), reverse transcriptase and Taq polymerase were obtained from
Promega Corporation (Madison, WI, USA). COX-2 (cat. no. Sc-166475),
GAPDH (cat. no. Sc-69778), IL-1β (cat. no. Sc-32294) and tumor
necrosis factor receptor 1 (TNFR1; cat. no. Sc-8436) antibodies
were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). PGE2 and TNF-α ELISA kits were purchased from Nanjing
Jiancheng Bioengineering Institute, Nanjing, China. BBR was
purchased from Nanjing Dulai Biotechnology Co., Ltd. (Nanjing,
China).
Cell culture
SH-SY5Y and SK-N-SH cells were maintained in DMEM
supplemented with 10% fetal bovine serum (Thermo Fisher Scientific
Inc.), 100 U/ml penicillin and 100 U/ml streptomycin in a
humidified atmosphere containing 5% CO2 at 37°C.
Treatment of SH-SY5Y and SK-N-SH
cells
SH-SY5Y and SK-N-SH cells were seeded in 6-well
plates with 1×106 cells/well. When the cells adhered to
the plates, the cell culture medium was discarded and the cells
were pre-treated with DMEM medium containing different
concentrations of BBR (0, 1 and 10 mol/l) or indomethacin (200
mol/l; Zhuxi Zhongxin Biotech Co., Ltd., Nanjing, China). Cells
were incubated for 2 h at 37°C. Subsequently, cells were incubated
in DMEM medium with or without Aβ25–35 (5 mol/l) for
another 24 h at 37°C. There were five experimental groups: i)
Normal control without any treatment; ii) Aβ25–35 model
group treated with Aβ25–35 only; iii) Aβ25–35
model + indomethacin group treated with Aβ25–35 +
indomethacin (200 mol/l); iv) Aβ25–35 + low-dose BBR
group treated with Aβ25–35 + BBR (1 mol/l); and v)
Aβ25–35 + high-dose BBR group treated with
Aβ25–35 + BBR (10 mol/l). Following the treatment, cells
and culture media were collected and centrifuged at 300 × g at room
temperature for 2 min for subsequent analysis.
MTT assay on viability of SH-SY5Y and
SK-N-SH cells
The SH-SY5Y and SK-N-SH cells at their logarithmic
growth phase were collected following 0.125% trypsin digestion and
adjusted to a concentration of 1×108 cells/l. Cells were
seeded into 96-well plates at a concentration of 10,000 cells/well.
After serum starvation for 24 h, the wells were divided into
different groups in sextuplicate as mentioned above and 20 µl MTT
(5 g/l) was added to each well. The plates were incubated at 37°C
for another 4 h. Subsequently, MTT solution was removed and 150 µl
DMSO was added to each well. The plates were shook for 10 min to
dissolve the purple formazan crystals. The light absorbance was
measured at a wavelength of 490 nm using a microplate reader. Cell
survival rate was calculated using the following equation: Survival
rate (%)=A490 nm (experiment)/A490 nm
(control) ×100%.
Lactate dehydrogenase (LDH) activity
in the cell culture media
Cell culture media were centrifuged at 300 × g at
room temperature for 2 min and transferred to the 96-well
enzyme-analyzing plates using the LDH kit (Zhuxi Zhongxin Biotech
Co., Ltd. Buffer with substrate (50 µl) was added into each well.
The plates were incubated for 30 min at room temperature in the
dark and, subsequently, stop buffer (50 µl) was added into each
well. The light absorbance was measured at a wavelength of 490
nm.
PGE2 and TNF-α expression levels
determined by ELISA
The microplate was coated with specific antibodies
provided in the ELISA kit (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China), incubated for 1 h at 37°C and
subsequently at 4°C overnight. The plate was washed three times and
200 µl blocking buffer was added and incubated for 1 h at 37°C.
After washing 3 times, cell medium of each group was added and
incubated at 37°C for 2 h, and washed three times again. Anti-human
immunoglobulin G labeled with horseradish peroxidase was added and
incubated at 37°C for 1 h. Stop buffer was added following 3 washes
with PBS and 2 with distilled water. The optical density was
measured at a wavelength of 450 nm with a microplate reader (model
ELX800; BioTek Instrument Inc., Winooski, VT, USA.
COX-2, IL-1β and TNFR1 mRNA expression
levels detected by reverse transcription-quantitative polymerase
chain reaction
Total RNA from each group of cells was extracted
using TRIzol reagent (Thermo Fisher Scientific, Inc.). Subsequently
RNA with A260/280 value of 1.80–2.00 was used for
reverse transcription. Reverse transcription was performed using 2
µg of RNA, 1 µl of random Examer, 1 µl of RT Superscript at 200
U/l, 10 µM dNTP and 4% of MMLV reverse transcriptase. Temperature
protocol of reverse transcription was as follows: 65°C for 10 min
followed by 50°C for 60 min and 85°C for 5 min. The primer
sequences were designed using Primer Premier 5.0 (Premier Biosoft
International, Palo Alto, CA, USA) based on the gene sequences from
GenBank (6) (Table I). All primers were synthesized by
NanJing SunShine Biotechnology Co., Ltd., Nanjing, China. The
following thermocycling conditions were used for SYBR Green-Based
Real-Time PCR amplification (Invitrogen; Thermo Fisher Scientific,
Inc.): Initial denaturation at 95°C for 30 sec; 40 cycles at 94°C
for 30 sec, 60°C for 40 sec. The PCR was performed using a thermal
cycler (model TP600; Takara Bio, Inc., Otsu, Japan). The Ct values
of target genes COX-2, IL-1β, TNFR1 and control gene GAPDH were
calculated. The expression levels of COX-2, IL-1β and TNFR1 mRNA
were calculated using the 2−ΔΔCq method (7).
 | Table I.Primer sequences used for the
PCR. |
Table I.
Primer sequences used for the
PCR.
|
| Primer sequence
(5′→3′) |
|
|---|
|
|
|
|
|---|
| Gene | Forward | Reverse | Length of the PCR
product (base pair) |
|---|
| COX-2 |
CTGCGCCTTTTCAAGGATGG |
CCCCACAGCAAACCGTAGAT | 135 |
| IL-1β |
CAGAAGTACCTGAGCTCGCC |
AGATTCGTAGCTGGATGCCG | 153 |
| TNFR1 |
CTTCAATTGCAGCCTCTGCC |
CTTCCACCGTTGGTAGCGAT | 292 |
| GAPDH |
GGGAGCCAAAAGGGTCATCA |
TGATGGCATGGACTGTGGTC | 203 |
COX-2, IL-1β and TNFR1 protein
expression detected by western blotting
Cellular proteins were extracted with the RIPA lysis
buffer with an EDTA-free protease and a phosphatase inhibitor
cocktail tablet (Roche Applied Science, Penzburg, Germany).
Proteins were also determined using a BCA assay (NanJing SunShine
Biotechnology Co., Ltd.), equal amounts protein/lane (50 µg) were
resolved by 10% SDS-PAGE and electrophoretically transferred onto
PVDF membranes. Following incubation in a blocking solution (10%
milk) for 2 h at room temperature, the membranes were incubated for
5 h with polyclonal antibodies against COX-2, IL-1β, TNFR1 and
GAPDH at 1:200 dilution at room temperature. The membranes were
subsequently washed 3 times in PBS and incubated with horseradish
peroxidase-conjugated secondary antibody for 2 h at room
temperature. Horseradish peroxidase labeled goat anti-mouse
lmmunoglobulin G secondary antibodies (1:200; cat. no. A25012;
Abbkine Scientific Co., Ltd., Wuhan, China) and an enhanced
chemiluminescence (ECL) kit were used. The protein-antibody
complexes were detected using the ECL detection system. The protein
band intensities were evaluated using Scion Image software (Version
alpha 4.0.3.2; Scion Corporation, Frederick, MD, USA).
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analysis was performed using SPSS software
(version 18.0; SPSS, Inc., Chicago, IL, USA). Data were analyzed
using one-way analysis of variance followed by Least Significant
Difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
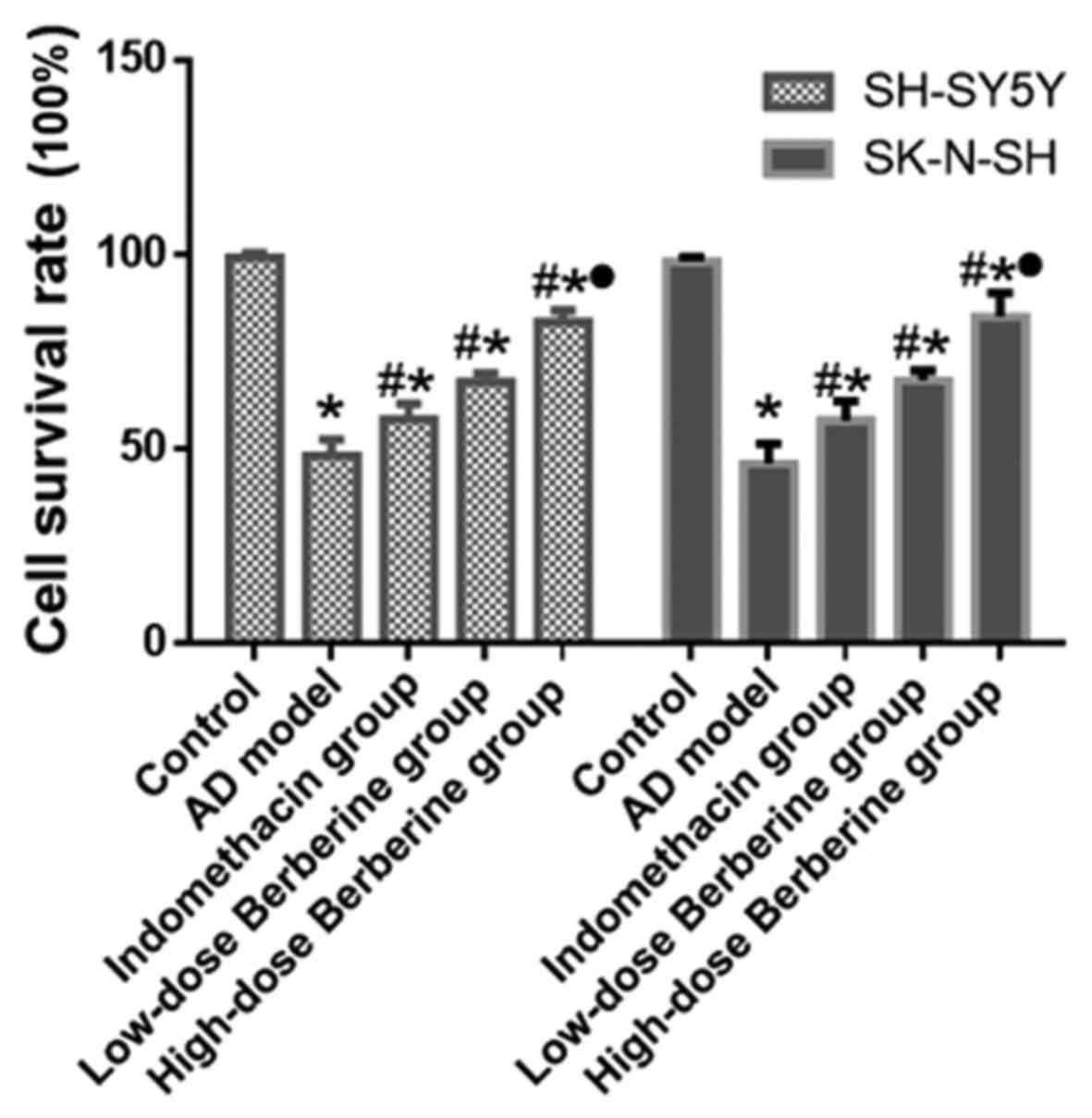
Effect of BBR on viability of SH-SY5Y
and SK-N-SH cells treated with Aβ25–35
Following treatment with Aβ25-35, cell
viability of the AD model (Aβ25–35) significantly
decreased compared with the normal control (SH-SY5Y and SK-N-SH
cells; both P<0.05). Cell viability increased following
treatment with BBR or indomethacin, especially in the high-dose BBR
group (SH-SY5Y and SK-N-SH cells; all P<0.05). The results
indicated that Aβ25–35 induced cellular damage in
SH-SY5Y and SK-N-SH cells, and BBR could protect against this
damage. Among the experimental groups, high-dose BBR could better
protect the damaged cells compared with the indomethacin and
low-dose BBR groups (Fig. 1).
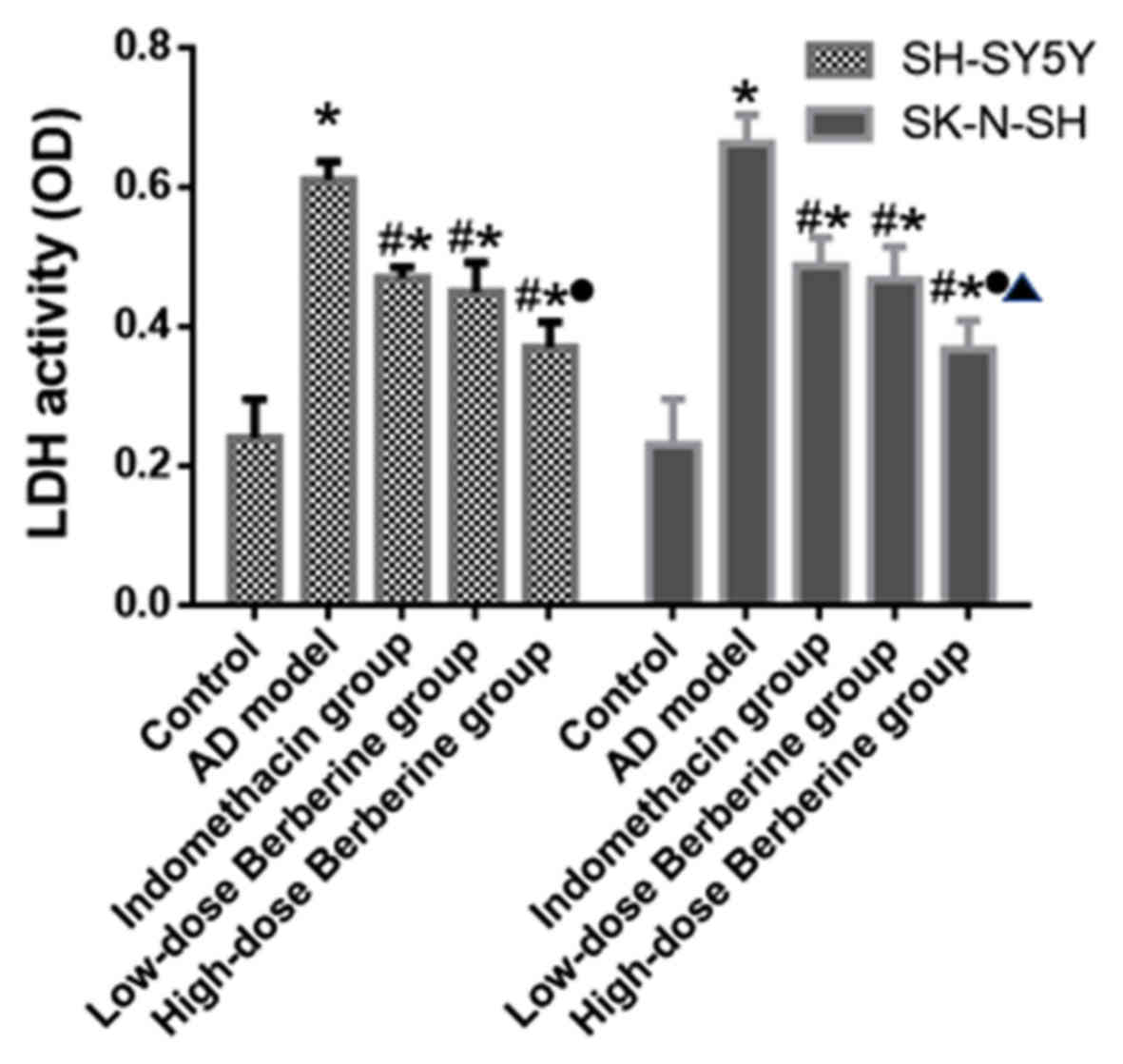
Effect of BBR on LDH activity of
SH-SY5Y and SK-N-SH cells
The LDH activity of SH-SY5Y and SK-N-SH cells was
evaluated in cell culture media. Compared with the respective
control groups, the LDH activity of cells induced by
Aβ25–35 increased significantly (SH-SY5Y and SK-N-SH
cells; all P<0.05). Both indomethacin and BBR reduced the LDH
activity of cells compared with the model group (SH-SY5Y and
SK-N-SH cells; all P<0.05). High-dose BBR decreased the LDH
activity by 39.3% in SH-SY5Y cells and 44.6% in SK-N-SH cells,
which was a greater difference compared with that of the low-dose
BBR or indomethacin groups (P<0.05; Fig. 2).
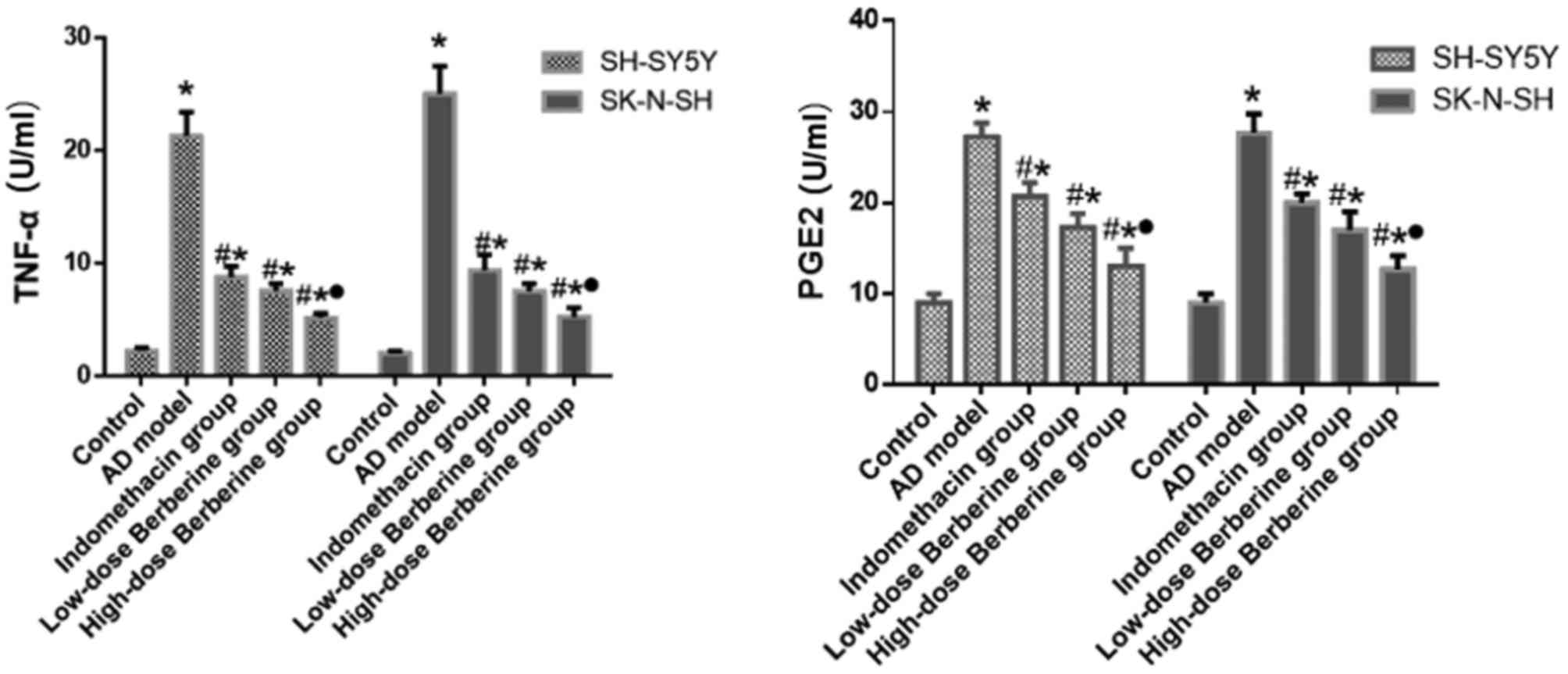
Effect of BBR on the expression levels
of PGE2 and TNF-α in SH-SY5Y and SK-N-SH cells induced by
Aβ25–35
After SH-SY5Y and SK-N-SH cells were treated with
Aβ25-35, the expression levels of PGE2 and TNF-α
secreted into the culture media were examined by ELISA. Compared
with the control group, the expression levels of PGE2 and TNF-α
increased following Aβ25–35 induction (SH-SY5Y and
SK-N-SH cells; all P<0.05). Both indomethacin and BBR reduced
the levels of PGE2 and TNF-α in cells (SH-SY5Y and SK-N-SH cells;
all P<0.05). High-dose BBR significantly decreased the
expression level of PGE2 by 52.4% in SH-SY5Y cells and 54.2% in
SK-N-SH cells, compared with the AD model group, which is a greater
difference compared with the low-dose BBR or indomethacin groups
(P<0.05; Fig. 3). Similarly,
high-dose BBR significantly decreased the expression level of TNF-α
by 75.6% in SH-SY5Y cells and 78.9% in SK-N-SH cells, which was a
greater difference compared with that of the low-dose BBR or
indomethacin groups (Fig. 3).
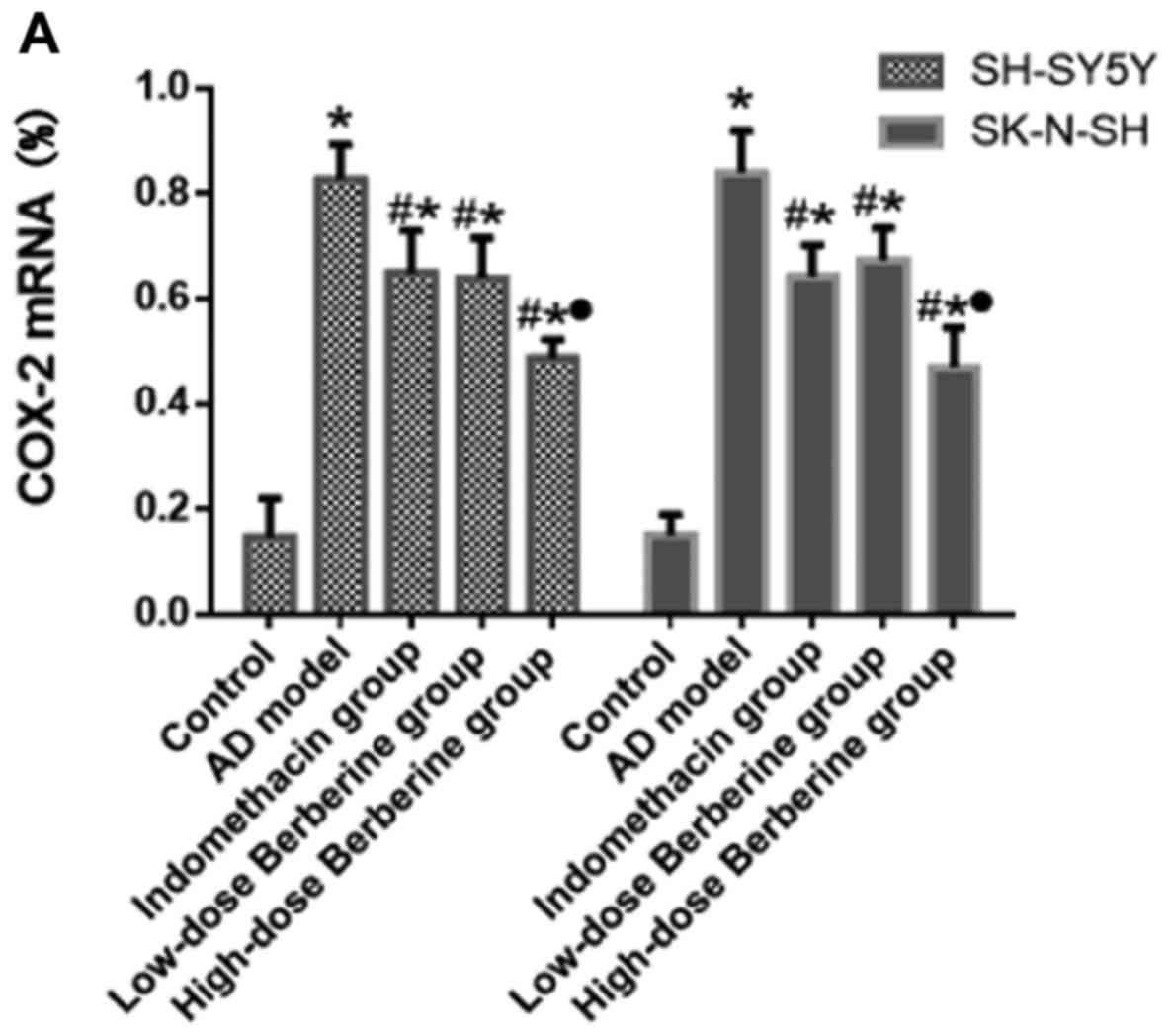
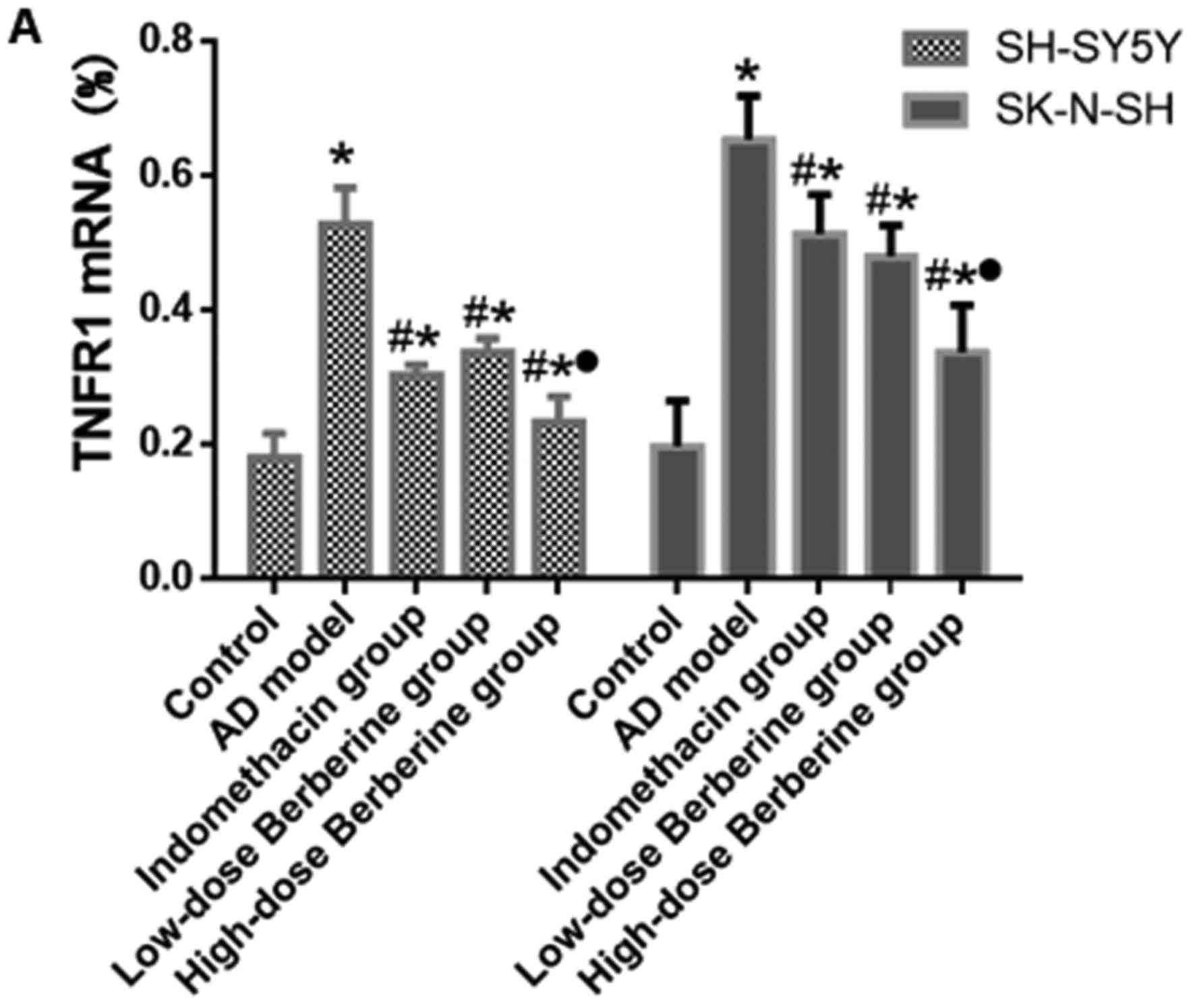
Effect of BBR on mRNA and protein
expression of COX-2, IL-1β, and TNFR1 in SH-SY5Y and SK-N-SH cells
induced by Aβ25–35
Following treatment with Aβ25–35, the
mRNA expression of IL-1β increased 3.32-fold in SH-SY5Y cells and
3.39-fold in SK-N-SH cells; the protein expression increased
5.86-fold in SH-SY5Y cells and 3.68-fold in SK-N-SH cells. COX-2
mRNA expression increased 5.86-fold in SH-SY5Y cells and 5.6-fold
in SK-N-SH cells; protein expression increased 4.04-fold in SH-SY5Y
cells and 4.64-fold in SK-N-SH cells. TNFRI mRNA expression
increased 2.93-fold in SH-SY5Y cells and 3.32-fold in SK-N-SH
cells; protein expression increased 4.06-fold in SH-SY5Y cells and
3.02-fold in SK-N-SH cells. These increases have significant
difference in AD model compared with control group (P<0.05).
Both indomethacin and BBR reduced mRNA and protein expression
levels of COX-2, IL-1β, and TNFR1 (all P<0.05). There were no
significant differences in the mRNA and protein expression levels
of COX-2, IL-1β and TNFR1 between indomethacin and low-dose BBR
groups (P>0.05), however, there was a significant difference in
the mRNA and protein expression level of COX-2, IL-1β and TNFR1
treated by high-dose BBR compared with the low-dose BBR group (all
P<0.05; Figs. 4–6).
Discussion
Previously, studies on the pathogenesis of AD
revealed that chronic inflammation of the central nervous system is
one of the important pathological features, along with the
phosphorylation of tau protein, senile plaques and amyloidosis
(8–10). It was reported that factors that
participate in the pathophysiological process of AD include
cytokines, such as IL-1, TNF-α and transforming growth factor β. It
has been hypothesized that pathogenesis of AD may be initiated by
up-regulating the expression of amyloid precursor protein by the
stimulation of its promoter (11).
Increased expression levels of amyloid precursor protein result in
up-regulated expression of acetylcholinesterase (AchE) and
increased AchE activity (12). These
alterations may further induce malnutritional axonal growth and
increase the level of phosphorylated tau protein (13). Finally, activation of astrocytes
increases the level of inflammatory factors including TNF-α
(14).
In the central nervous system, neurons and
microglial cells normally exhibit no or very low levels of
inflammatory factors (1). In chronic
neurodegenerative diseases, the high level of inflammatory factors
in the central nervous system may result from the invasion of
peripheral lymphocytes and exogenous inflammatory factors or be
induced by amyloid deposits (1).
Peripheral lymphocytes and inflammatory factors are not the primary
causes of inflammation of the central nervous system due to the
blood-brain barrier (15). The main
cause of the neurodegenerative process in AD is neuronal
inflammation induced by abnormal deposition of amyloids in the
brain (16). It has been
demonstrated that interaction between amyloid β and its receptors
resulted in intracellular signal transduction and activation of
microglial cells to generate inflammatory factors (17). NSAIDs induce a protective effect by
inhibiting inflammatory factors (18).
A small clinical trial revealed that oral
administration of low dose (0.4 g) BBR 3 times/day can inhibit the
levels of inflammatory factors (5).
BBR can cross the blood brain barrier and directly affect the
cerebral cortex and hippocampus (19). BBR exhibited a protective effect and
therapeutic potential for chronic brain injury induced by aluminum
overload and other diseases of the central nervous system in mice
(20,21). It remains to be elucidated whether
BBR could attenuate the inflammatory reaction induced by AD in the
central nervous system. In the present study, Aβ25–35
was used to induce inflammatory reaction in SH-SY5Y and SK-N-SH
cells, to model the inflammatory response of AD. The results
indicated alterations of cell viability and expression levels of
inflammatory factors including COX-2, PGE2, IL-1Β, TNF-α and TNF-α
type 1 receptor following treatment with BBR.
Nerve cells and neural glial cells normally express
low levels of COX-2 and IL-1β for basic brain function, including
synaptic plasticity and memory enhancement. The neuronal membrane
excitability is modulated by adrenergic, noradrenergic and
glutamatergic neurotransmitters (1).
Under pathological conditions nerve cells may be affected by
ischemia, hypoxemia, mitogens, cytokines and hormones. In these
cases, the levels of inflammatory factors in nerve cells increase,
leading to neuronal degeneration (16). High level of COX-2 increases the
synthesis of PGE2 which can induce cell apoptosis, damage the
sulfhydryl group of intracellular proteinsand cause
neurodegenerative disorders (22).
IL-1β is the primary factor contributing to the formation of senile
plaques in AD. Overexpression of IL-1β in microglia is a
characteristic feature of AD. It has been shown in vitro
that IL-1β does not alter cell morphology, number and activity
(23). However, the combined effect
of IL-1β and Aβ25–35 exacerbated the cytotoxicity of
Aβ25–35 in a time- and dose-dependent manner. The
mechanism of action is associated with overexpression of mRNA
induced by the regulating sequences of the 5′ promoter region of
the APP gene (24). Therefore, drugs
inhibiting COX-2 and IL-1β expression can antagonize the
inflammatory reaction in the central nervous system to protect
nerve cells.
TNF-α is a pleiotropic cytokine (25). In addition to induction of neuronal
cytotoxicity, TNF-α can increase the permeability of blood-brain
barrier, which promotes lymphocyte infiltration (26) and up-regulates the expression of
intercellular adhesion molecule 1 (ICAM-1). The biological function
of TNF-α is to transmit signals through two cell surface receptors
including TNFR1 and TNFR2 (25).
Gene knockout studies and experiments using receptor agonist
antibodies indicated that these two receptors react with different
proteins and activate different signal transduction pathways
(27–29). Following the binding of TNF-α and
TNFR1, the mitogen-activated protein kinase, protein kinase C, and
protein kinase A activate nuclear factor κB, which promotes
transcription of numerous proinflammatory cytokines (30). The binding of TNF-α and TNFR1
participates in transcriptional regulation of inflammatory genes.
It serves a role in mediating inflammation in the central nervous
system (31). A previous study
revealed that TNFR1 blocking peptide binds to TNFR1 and antagonizes
the pro-inflammatory effect of TNF-α (32). Therefore, it can be hypothesized that
inhibiting the expression of COX-2 and IL-1β can antagonize the
inflammatory reaction in the central nervous system to protect
nerve cells.
In the AD group of the present study, the decrease
in cell survival and increase in LDH activity of SH-SY5Y and
SK-N-SH cells indicated that the cells were severely damaged by
Aβ25–35. Following BBR drug intervention, cell survival
increased and LDH activity decreased, indicating that BBR reduced
the cytotoxicity of Aβ25–35. The mechanism may be
associated with the down-regulation of expression of TNF-α, COX-2,
IL-1β and TNFR1. Compared with the low-dose BBR and indomethacin
groups, high dose BBR significantly decreased the expression of
LDH, PGE2, TNF-α, COX-2, IL-1β and TNFR1. The results suggested
that BBR may regulate PGE2 through other pathways in addition to
the inhibition of the COX-2 signaling. The high expression levels
of PGE2 may mediate brain tissue damage in several ways including:
i) Increased adhesion of platelets and neutrophils to vascular
endothelial cells (33); ii)
increased permeability of the blood-brain barrier (34); iii) cytotoxicity mediated by
excitatory amino acids (35); and
iv) increased generation of oxygen free radicals and the toxic
effect of nitric oxide leading to decreased cell viability.
Taken together, the present study suggested that BBR
can protect nerve cells against inflammatory response mediated by
Aβ25–35. The mechanism of action may be associated with
down-regulation of PGE2, COX-2, IL-1β, TNF-α and TNFR1. High-dose
BBR exhibits a marked inhibitory effect on the inflammatory
response and could potentially be used for the treatment of AD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the General
Project of Jiangsu Provincial Administration of Traditional Chinese
Medicine (grant no. YB2017045).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HZ and LY conceived and designed the study. JX and
WW performed the experiments and wrote the paper. LY reviewed and
edited the manuscript. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen HZ, Lin GW and Wang JY: Practice of
Internal Medicine. 14th. People's Medical Publishing House;
Beijing: 2017, View Article : Google Scholar
|
|
2
|
Breitner JC: NSAIDs and Alzheimer's
disease: How far to generalize from trials? Lancet Neurol.
2:5272003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hung CW, Chen YC, Hsieh WL, Chiou SH and
Kao CL: Ageing and neurodegenerative diseases. Ageing Res Rev. 9
Suppl 1:S36–S46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang J, Feng Y, Tsao S, Wang N, Curtain R
and Wang Y: Berberine and Coptidis Rhizoma as novel antineoplastic
agents: A review of traditional use and biomedical investigations.
J Ethnopharmacol. 126:5–17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiang W, Huang XJ and Huang GX: Study of
two anti-flammatory drugs treating primary type 2 diabetic. J Pract
Diabetes. 7:51–52. 2011.
|
|
6
|
Kearse M, Moir R, Wilson A, Stones-Havas
S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C,
et al: Geneious basic: An integrated and extendable desktop
software platform for the organization and analysis of sequence
data. Bioinformatics. 28:1647–1649. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Halliday G, Robinson SR, Shepherd C and
Kril J: Alzheimer's disease and inflammation: A review of cellular
and therapeutic mechanisms. Clin Exp Pharmacol Physiol. 27:1–8.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zamolodchikov D and Strickland S: A
possible new role for Aβ in vascular and inflammatory dysfunction
in Alzheimer's disease. Thromb Res. 141 Suppl 2:S59–S61. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hurtado DE, Molina-Porcel L, Iba M,
Aboagye AK, Paul SM, Trojanowski JQ and Lee VM: A{beta} accelerates
the spatiotemporal progression of tau pathology and augments tau
amyloidosis in an alzheimer mouse model. Am J Pathol.
177:1977–1988. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ringheim GE, Szczepanik AM, Petko W,
Burgher KL, Zhu SZ and Chao CC: Enhancement of beta-amyloid
precursor protein transcription and expression by the soluble
interleukin-6 receptor/interleukin-6 complex. Brain Res Mol Brain
Res. 55:35–44. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Liu L, Kang J, Sheng JG, Barger SW,
Mrak RE and Griffin WS: Neuronal-glial interactions mediated by
interleukin-1 enhance neuronal acetylcholinesterase activity and
mRNA expression. J Neurosci. 20:149–155. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sheng JG, Zhu SG, Jones RA, Griffin WS and
Mrak RE: Interleukin-1 promotes expression and phosphorylation of
neurofilament and tau proteins in vivo. Exp Neurol. 163:388–391.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Farooqui AA: Chapter 6: Contribution of
neuroinflammation in the pathogenesis of Alzheimer's disease.
Neurochemical Aspects of Alzheimer's DiseaseRisk Factors,
Pathogenesis, Biomarkers, and Potential Treatment Strategies.
Academic Press; pp. 201–245. 2017
|
|
15
|
Pollak TA, Drndarski S, Stone JM, David
AS, McGuire P and Abbott NJ: The blood-brain barrier in psychosis.
Lancet Psychiatry. 5:79–92. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhi-you C and Yong Y: Pathway and
mechanism of oxidative stress in Alzheimer's disease. J Med Coll
PLA. 22:320–324. 2007. View Article : Google Scholar
|
|
17
|
Teeling JL and Perry VH: Systemic
infection and inflammation in acute CNS injury and chronic
neurodegeneration: Underlying mechanisms. Neuroscience.
158:1062–1073. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Giovannini MG, Scali C, Prosperi C,
Bellucci A, Vannucchi MG, Rosi S, Pepeu G and Casamenti F:
Beta-amyloid-induced inflammation and cholinergic hypofunction in
the rat brain in vivo: Involvement of the p38MAPK pathway.
Neurobiol Dis. 11:257–274. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Wang R, Xing D, Su H, Ma C, Ding Y
and Du L: Kinetic difference of berberine between hippocampus and
plasma in rat after intravenous administration of Coptidis rhizoma
extract. Life Sci. 77:3058–3067. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kulkarni SK and Dhir A: Berberine: A plant
alkaloid with therapeutic potential for central nervous system
disorders. Phytother Res. 24:317–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu BZ, Zhang J and Yang JQ: The
protective effect and mechanism of Berberine on chronic brain
injury in mice induced by aluminum overload. Chin Herbal Med.
39:1351–1355. 2008.(In Chinese).
|
|
22
|
Lukiw WJ, Percy ME and Kruck TP: Nanomolar
aluminum induces pro-inflammatory and pro-apoptotic gene expression
in human brain cells in primary culture. J Inorg Biochem.
99:1895–1898. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Babcock AA, Ilkjær L, Clausen BH,
Villadsen B, Dissing-Olesen L, Bendixen AT, Lyck L, Lambertsen KL
and Finsen B: Cytokine-producing microglia have an altered
beta-amyloid load in aged APP/PS1 Tg mice. Brain Behav Immun.
48:86–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nie YH: Regulation of Interleukin-1 on
β-amyloid cytotoxic and gene expression. Chin Clin Rehabilitat.
8:79–81. 2004.
|
|
25
|
Tseng WY, Huang YS, Lin HH, Luo SF, McCann
F, McNamee K, Clanchy F and Williams R: TNFR signaling and its
clinical implications. Cytokine. 101:19–25. 2018. View Article : Google Scholar
|
|
26
|
Mayhan WG: Cellular mechanisms by which
tumor necrosis factor-alpha produces disruption of the blood-brain
barrier. Brain Res. 927:144–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Keller C, Keller P, Giralt M, Hidalgo J
and Pedersen BK: Exercise normalises overexpression of TNF-alpha in
knockout mice. Biochem Biophys Res Commun. 321:179–182. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Toda K, Hayashi Y and Saibara T: Deletion
of tumor necrosis factor-alpha receptor type 1 exacerbates insulin
resistance and hepatic steatosis in aromatase knockout mice.
Biochim Biophys Acta. 1801:655–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizoguchi E, Hachiya Y, Kawada M, Nagatani
K, Ogawa A, Sugimoto K, Mizoguchi A and Podolsky DK: TNF receptor
type I-dependent activation of innate responses to reduce
intestinal damage-associated mortality. Gastroenterology.
134:470–480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schmidt N, Haydn T, Schneider I, Busch H,
Boerries M and Fulda S: Smac mimetic induces an early wave of gene
expression via NF-κB and AP-1 and a second wave via TNFR1
signaling. Cancer Lett. 421:170–185. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vilela MC, Lima GK, Rodrigues DH,
Lacerda-Queiroz N, Mansur DS, de Miranda AS, Rachid MA, Kroon EG,
Vieira LQ, Campos MA, et al: TNFR1 plays a critical role in the
control of severe HSV-1 encephalitis. Neurosci Lett. 479:58–62.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang LX, Yin BJ and Wang J: Sealing
action of TNFRI blocking peptide-hIgGFc fusion protein and TNFRI
blocking peptide on the biological effect induced by TNF-α. Chin J
Immunol. 24:776–780. 2008.
|
|
33
|
Stanimirovic D, Shapiro A, Wong J,
Hutchison J and Durkin J: The induction of ICAM-1 in human
cerebromicrovascular endothelial cells (HCEC) by ischemia-like
conditions promotes enhanced neutrophil/HCEC adhesion. J
Neuroimmunol. 76:193–205. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Candelario-Jalil E, González-Falcón A,
García-Cabrera M, León OS and Fiebich BL: Post-ischaemic treatment
with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain
barrier disruption and leukocyte infiltration following transient
focal cerebral ischaemia in rats. J Neurochem. 100:1108–1120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takadera T and Ohyashiki T: Prostaglandin
E2 deteriorates N-methyl-D-aspartate receptor-mediated cytotoxicity
possibly by activating EP2 receptors in cultured cortical neurons.
Life Sci. 78:1878–1883. 2006. View Article : Google Scholar : PubMed/NCBI
|