Introduction
Epilepsy is a common type of neurological disorder
hallmarked by over-synchronization of neurons in the brain
(1–3). Epileptic electrical disturbances are
associated with abnormalities in behavior, awareness, sensation and
movement (4–10). According to reports, 5–8% of the
population suffers from epilepsy and 60% of cases occur in
childhood or infancy (11–13).
It is known that astrocytes provide support for
neurons via a variety of mechanisms (14). Insufficient support may lead to
alterations in neuronal function, which results in seizures.
Clearance and transport of glutamate is another neuroprotective
function of astrocytes, the loss of which serves an important role
in epilepsy development (15).
Disturbances in the ion hemostasis of astrocytes may also result in
seizures (16–18). Furthermore, astrocytes are capable of
direct communication with neurons at the synapse (19,20).
Together, these previous studies indicate that loss of astrocyte
function is associated with epilepsy development and that
maintaining astrocyte hemostasis is important for epilepsy
prevention.
Recently, the role of the cannabinoid system in
epilepsy has been explored (21).
The cannabinoid system comprises cannabinoid receptors (CBRs),
cannabinoid (endocannabinoid) and the enzymes that regulate the
synthesis and degradation of endocannabinoids (21). There are two subtypes of CBR: CB1R
and CB2R. CB2R was first isolated from human bone marrow cells and
was thought to exist in the peripheral immune system, whereas CB1R
was generally considered to exist in the central nervous system
(CNS). Recent reports have demonstrated that that CB2R also serves
an important role in the nervous system (22). It has been reported that CB2R
potentiates the antiepileptic action of CB agonists via the cGMP
signaling pathway in a model of hippocampal seizures (23). However, the biological effects and
exact mechanism of CB2R during the development of epilepsy is not
well understood. The aim of the present study was to explore the
biological effects and mechanisms of CB2R activation in astrocyte
and rat models of epilepsy.
Materials and methods
Preparation of the epileptiform cell
model
CTX TNA2 rat astrocytes were purchased from American
Type Culture Collection (Manassas, VA, USA) and cultured in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% heat-inactivated
fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in a
humidified atmosphere containing 5% CO2. Cells were
passaged every 2 days with 0.25% trypsin.
To establish epileptiform activity, cells were
placed in Mg free medium (145 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 2
mM CaCl2, 10 mM glucose, and 0.002 mM glycine, pH 7.3, adjusted to
325 mOsm with sucrose) for 3 h at 37°C in a humidified atmosphere
containing 5% CO2. CB2R agonist (JWH133, 4 µM; R & D
Systems, Inc., Minneapolis, MN, USA), CB2R antagonist (AM630, 1 µM)
and AKT inhibitor (wortmannin, 2 µM; R&D Systems, Inc.) were
administered for 24 h at 37°C in a humidified atmosphere containing
5% CO2.
Pilocarpine-induced status epilepticus
(SE) in rats
The present study was approved by the Institutional
Ethical Review Boards of the Shengjing Hospital of China Medical
University (Shenyang, China). Male Sprague-Dawley rats had access
to food and water ad libitum. All the rats were used following
university animal care and use protocols. A total of 40 male
Sprague-Dawley rats (age, ~20 days; weight, 100–120 g) were
purchased from the Department of Experimental Animals, Shengjing
Hospital of China Medical University. Rats were housed in single
cages with a 12-h light/dark cycle at 25°C and 50% humidity. Rats
were divided into four different groups: Control group, SE group,
CB2R agonist group and CB2R antagonist group. At 30 min prior to
pilocarpine injections, rats were administered with
methylscopolamine nitrate (S8502-1G; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany; 1 mg/kg i.p.) to minimize the peripheral
parasympathetic effects of pilocarpine treatment. Pilocarpine
nitrate (P6503-5G; Sigma-Aldrich; Merck KGaA; 375 mg/kg i.p.) was
subsequently administered. Onset of SE typically occurred 20–40 min
following the pilocarpine injection when the animal displayed
continuous moderate to severe behavioral seizures characterized by
forelimb clonus, rearing, and falling. The CB2R agonist (JWH133,
1.5 mg/kg) was administered every 6 h following termination of
seizure activity in the agonist group. The CB2R antagonist (AM630,
1 mg/kg) was administered every 6 h following termination of
seizure activity in the antagonist group.
SE was defined as continuous seizure activity
lasting ≥30 min or intermittent seizures without regaining
consciousness between seizures lasting ≥30 min (24). The severity of convulsions was
evaluated and only the rats that displayed behaviors consistent
with ongoing SE were used in the present study. Seizure activity
was terminated by consecutive diazepam (Shanghai Xudong Haipu
Pharmaceutical Co., Ltd., Shanghai, China) injections (5 mg/kg
i.p., solubilized in 10% ethanol, 45% propylene glycol and 45%
H2O) at 1, 3, and 5 h post onset of SE. Control groups
were composed of naive and sham control rats that received
methylscopolamine nitrate and diazepam injections only. Rats were
sacrificed at 24 h after termination of SE. Hippocampus tissue was
isolated and frozen at −80°C.
Western blotting
Total proteins were extracted from frozen
hippocampus tissue and cells using lysis buffer (P0013B; Beyotime
Institute of Biotechnology, Haimen, China) and quantified using the
Bradford method. Proteins (30 µg per lane) were separated using
SDS-PAGE (6% gels) after denaturation by mixing with the loading
buffer at a ratio of 1:4 for 5 min at 100°C. SDS-PAGE was run at
120 V until the dye reached the bottom of the separation gel, and
the proteins were then transferred to a polyvinylidene fluoride
membrane (EMD Millipore, Billerica, MA, USA) using wet
electroblotting at 120 V for 2 h. The membrane was blocked with 5%
skimmed milk powder for 2 h, then washed three times with
Tris-buffered saline plus Tween-20 (15 min/wash). Membranes were
incubated overnight at 4°C with rabbit monoclonal antibodies
against B-cell lymphoma 2 (Bcl-2; 1:1,000; cat. no. 3498),
phosphorylated Retinoblastoma (p-Rb; 1:1,000; cat. no. 8147),
phosphoinositide 3 kinase (PI3K) 110α (1:1,000; cat. no.),
p-mammalian target of rapamycin (mTOR; 1:1,000; cat. no. 2971),
mTOR (1:1,000; cat. no. 2972), glial fibrillary acidic protein
(GFAP; 1:1,000; cat. no. 3670,), p-AKT (1:1,000; cat. no. 9271) AKT
(1:1,000; cat. no. 9272) cyclin D1 (1:1,000; cat. no. 2978) cyclin
E (1:1,000; cat. no. 20808; all Cell Signaling Technology, Inc.,
Danvers, MA, USA), β-actin (1:1,000; sc-47778) and GAPDH (1:1,000;
sc-47724; both Santa Cruz Biotechnology, Dallas, TX, USA).
Subsequently, samples were incubated with peroxidase-coupled
anti-mouse monoclonal antibodies (1:1,000; cat. no. 7076, Cell
Signaling Technology, Inc.) or rabbit IgG antibodies (1:1,000; cat.
no. 7074, Cell Signaling Technology, Inc.) at 37°C for 2 h. Target
proteins on the were visualized using a Pierce enhanced
chemiluminescence kit (Thermo Fisher Scientific, Inc.) and captured
using a DNR BioImaging system (DNR Bio-Imaging Systems, Jerusalem,
Israel). Protein densitometry was evaluated using Tecan Infinite
200 PRO NanoQuant (Tecan Group Ltd., Mannedorf, Switzerland).
Flow cytometry for cell cycle
analysis
Cells (50,000) in 6-well plates were harvested 48 h
after intervention by 0.25% trypsin. Cells were washed twice with
chilled PBS and resuspended in 250 µl of binding buffer (10010049;
Gibco; Thermo Fisher Scientific, Inc.), adjusted to
1×106 cells/ml and fixed in 1% paraformaldehyde at 4°C.
Cells were stained with 5 mg/ml propidium iodide for 30 min at room
temperature for cell cycle analysis. Following the incubation,
cells were analyzed using a FACSCalibur flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
SPSS version 16 for Windows (SPSS, Inc., Chicago,
IL, USA) was used for all statistical analyses. Student's t-test
was used to compare data between control and treatment groups.
One-way analysis of variance with Bonferroni's post hoc test was
used to compare the means of two or more groups. All P-values were
based on a two-sided statistical analysis and P<0.05 was
considered to indicate a statistically significant difference.
Results
Treatment with CB2R agonist/antagonist
changes cell cycle transition in rat astrocytes
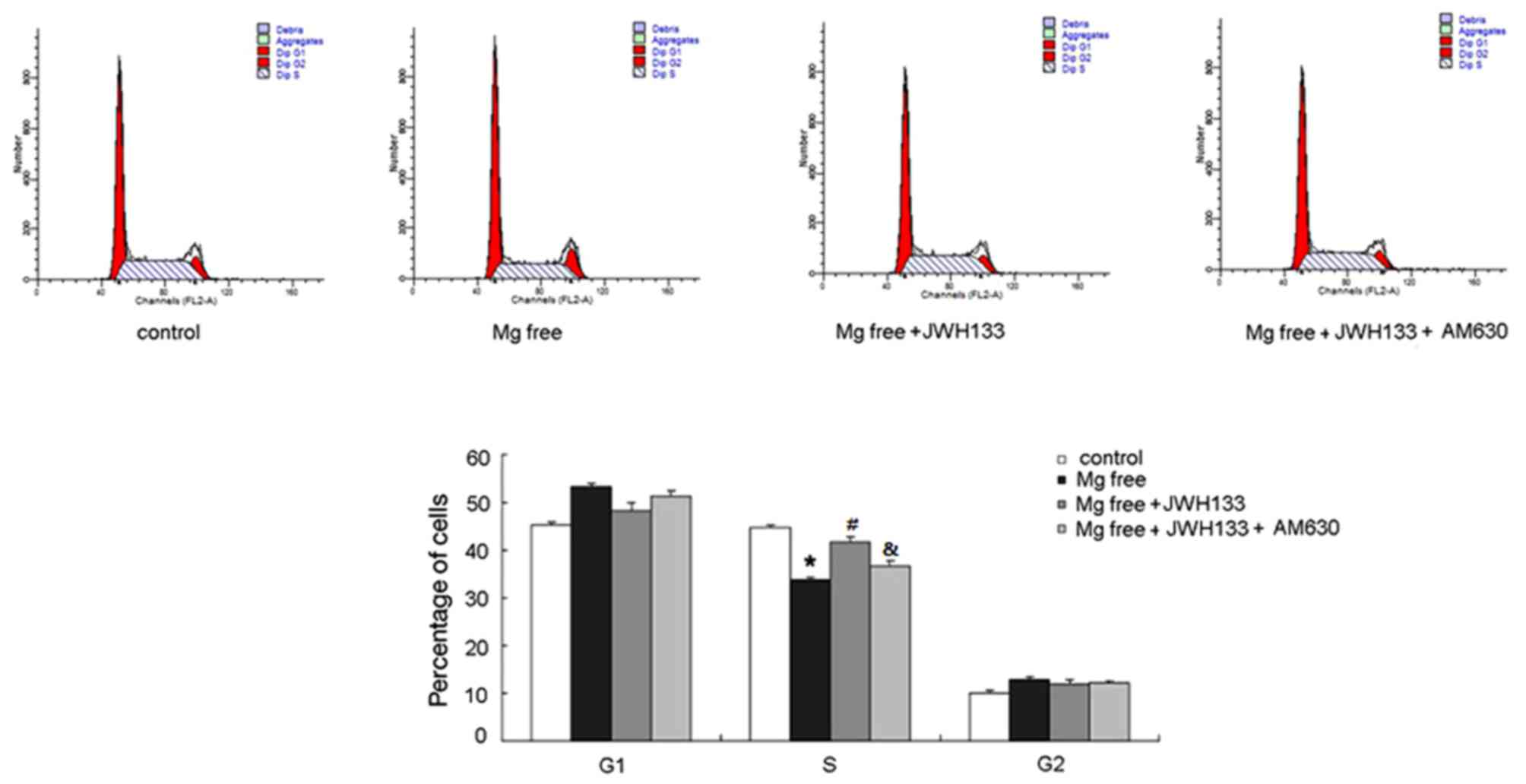
Flow cytometry was performed to examine the
biological effects of CB2R on cell cycle transition. The percentage
of cells in the S phase was decreased in astrocytes treated with Mg
free solution whereas the percentage in the G1 phase was increased
(P<0.05; Fig. 1). This suggested
that G1-S transition in astrocytes was inhibited during epilepsy.
Treatment with the CB2R agonist JWH133 significantly upregulated
the percentage of cells in S phase (P<0.05) and reduced the
number of G1 phase cells (Fig. 1)
compared with the group treated with Mg free solution. AM630
administration ameliorated the effects of JWH133, suppressing G1 to
S transition in astrocytes compared with the Mg free solution +
JWH133 (P<0.05; Fig. 1).
CB2R agonist/antagonist alters the
expression of cell cycle related proteins
p-Rb, cyclin D1, cyclin E and Bcl-2 protein
expression was determined using western blotting. The expression of
p-Rb, cyclin D1 and cyclin E decreased significantly when cells
were treated with Mg free solution compared with the control group
(P<0.05). The expression of Bcl-2 in astrocytes treated with Mg
free solution was slightly lower than compared with the control
(Fig. 2). Administration of the CB2R
agonist JWH133 increased p-Rb, cyclin D1, cyclin E and Bcl-2
protein expression compared with Mg free solution alone
(P<0.05), whereas the addition of the CB2R antagonist (AM630)
effectively suppressed the expression of p-Rb, cyclin D1, cyclin E
and Bcl-2 compared with the Mg free solution + JWH133 (P<0.05;
Fig. 2).
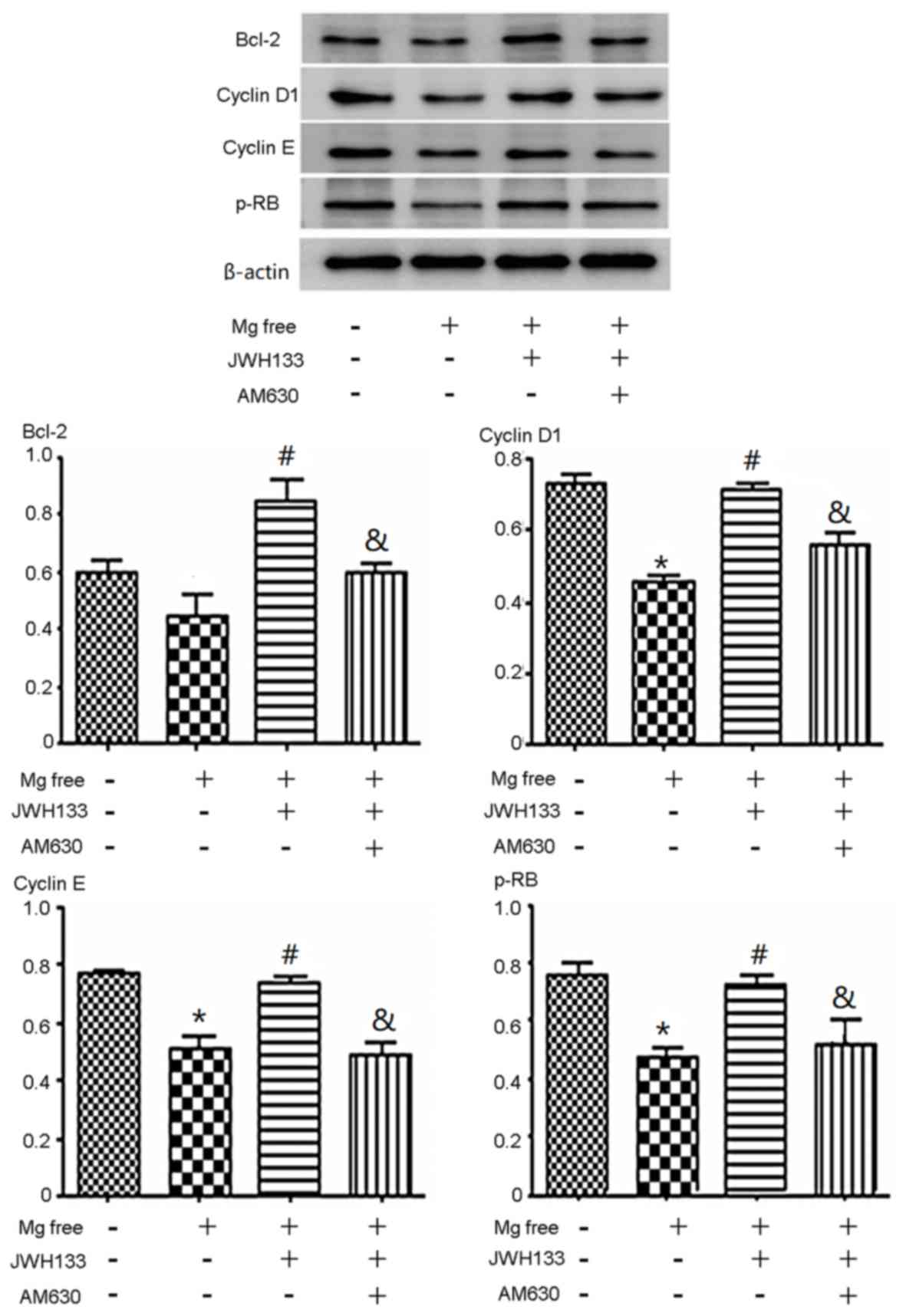 | Figure 2.Effects of CB2R agonist/antagonist on
cell cycle-associated protein expression. The expression of cyclin
D1, cyclin E and p-Rb decreased when cells were treated with Mg
free solution alone or co-administered with JWH133 and AM630. The
expression of p-Rb, cyclin D1 and cyclin E decreased significantly
when cells were treated with Mg free solution compared with the
control group. The expression of Bcl-2 in astrocytes treated with
Mg free solution was slightly lower compared with the control.
JWH133 increased p-Rb, cyclin D1, cyclin E and Bcl-2 protein
expression compared with with Mg free solution alone. AM630
decreased the expression of p-Rb, cyclin D1, cyclin E and Bcl-2
compared with the Mg free solution + JWH133. *P<0.05 vs. control
group; #P<0.05 vs. Mg free solution;
&P<0.05 vs. Mg free solution + JWH133. CB2R,
cannabinoid 2 receptor; p-, phosphorylated; Rb, retinoblastoma;
Bcl-2, B-cell lymphoma 2. |
CB2R regulates the AKT signaling
pathway in rat astrocytes
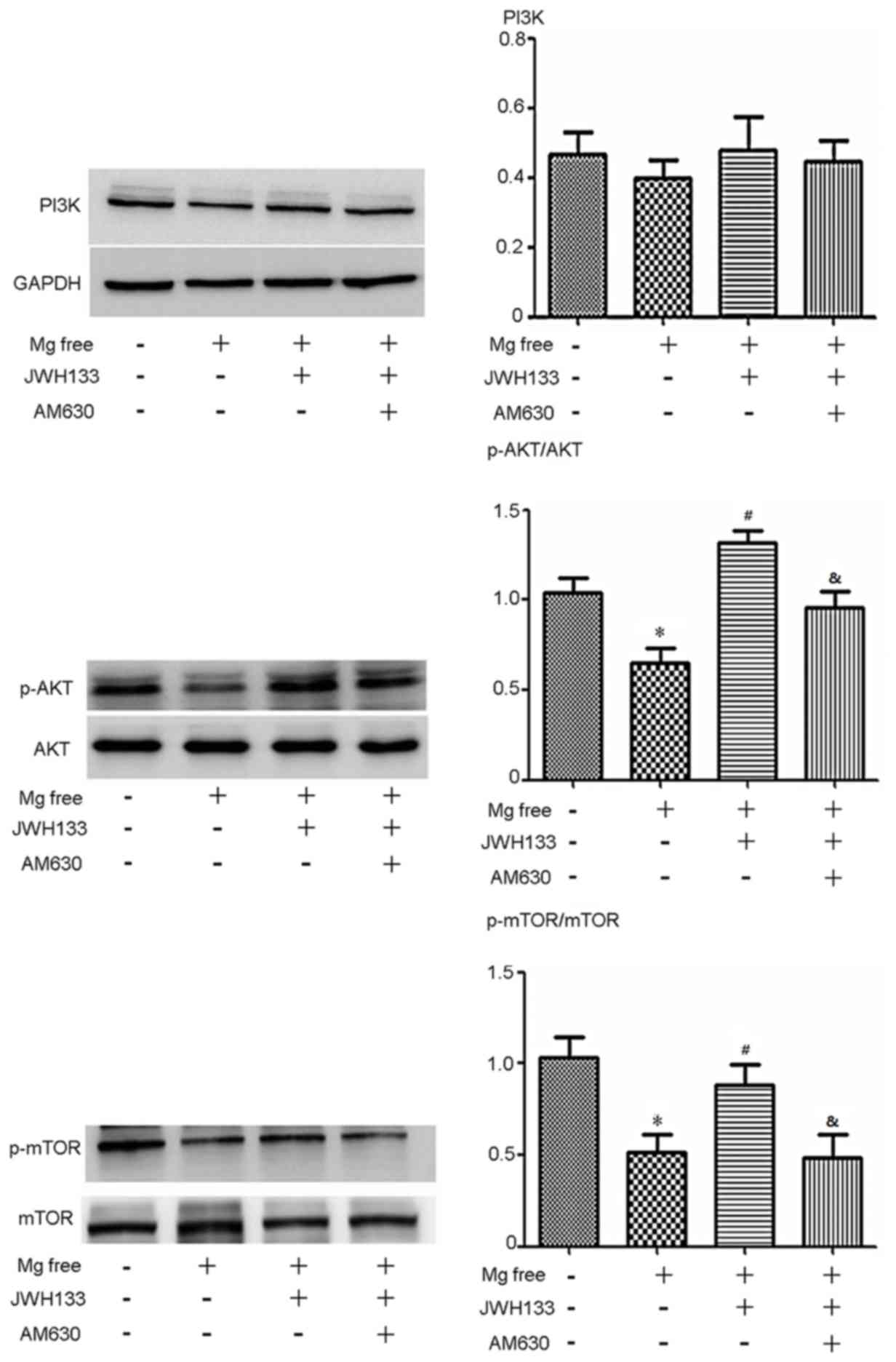
To clarify the potential mechanism of the CB2R
agonist/antagonist, several signaling pathways were assessed and it
was observed that changes in PI3K 110α-AKT activity were associated
with changes in cell cycle protein expression. The expression of
p-AKT and p-mTOR was significantly decreased in astrocytes treated
with Mg free serum compared with control astrocytes (P<0.05;
Fig. 3). Administration of the CB2R
activator JWH133 caused a significant increase in p-AKT/AKT and
p-mTOR/mTOR compared with the Mg free solution alone (P<0.05;
Fig. 3). When treated with the CB2R
inhibitor AM630, levels of p-AKT/AKT and p-mTOR/mTOR declined
significantly compared with the Mg free solution + JWH133 group
(P<0.05; Fig. 3). The expression
of PI3K 110α p110 was also assessed and followed a similar trend to
p-AKT; however, no statistically significant differences were
observed (Fig. 3).
Blocking the AKT pathway abolishes the
effects of the CB2R agonist/antagonist in rat astrocytes
To confirm that AKT signaling serves a role in
altering cell cycle distribution, the AKT inhibitor wortmannin was
used. Wortmannin treatment significantly downregulated p-AKT
expression, indicating that the AKT pathway was successfully
blocked (P<0.05; Fig. 4). The
level of p-AKT, p-Rb, cyclin D1 and Bcl-2 expression was
upregulated by activation of CB2R compared with the control group
(JWH133 treatment; P<0.05; Fig.
4). However, wortmannin blocked the effects of JWH133 on these
proteins. In the group treated with Mg free + JWH133 + wortmannin,
p-AKT, p-Rb, cyclin D1 and Bcl-2 expression decreased significantly
compared with the Mg free + JWH133 group (P<0.05; Fig. 4). In the group treated with Mg +
wortmannin, p-AKT, p-Rb, cyclin D1 and Bcl-2 expression was
significantly decreased compared with the Mg free group (P<0.05;
Fig. 4). No significant difference
in the expression of p-AKT, p-Rb, cyclin D1 and Bcl-2 was observed
between the Mg free + JWH133 + wortmannin and Mg + wortmannin
groups (Fig. 4). These results
suggest that CB2R activation by JWH133 upregulates p-Rb, cyclin D1
and Bcl-2 via the AKT signaling pathway.
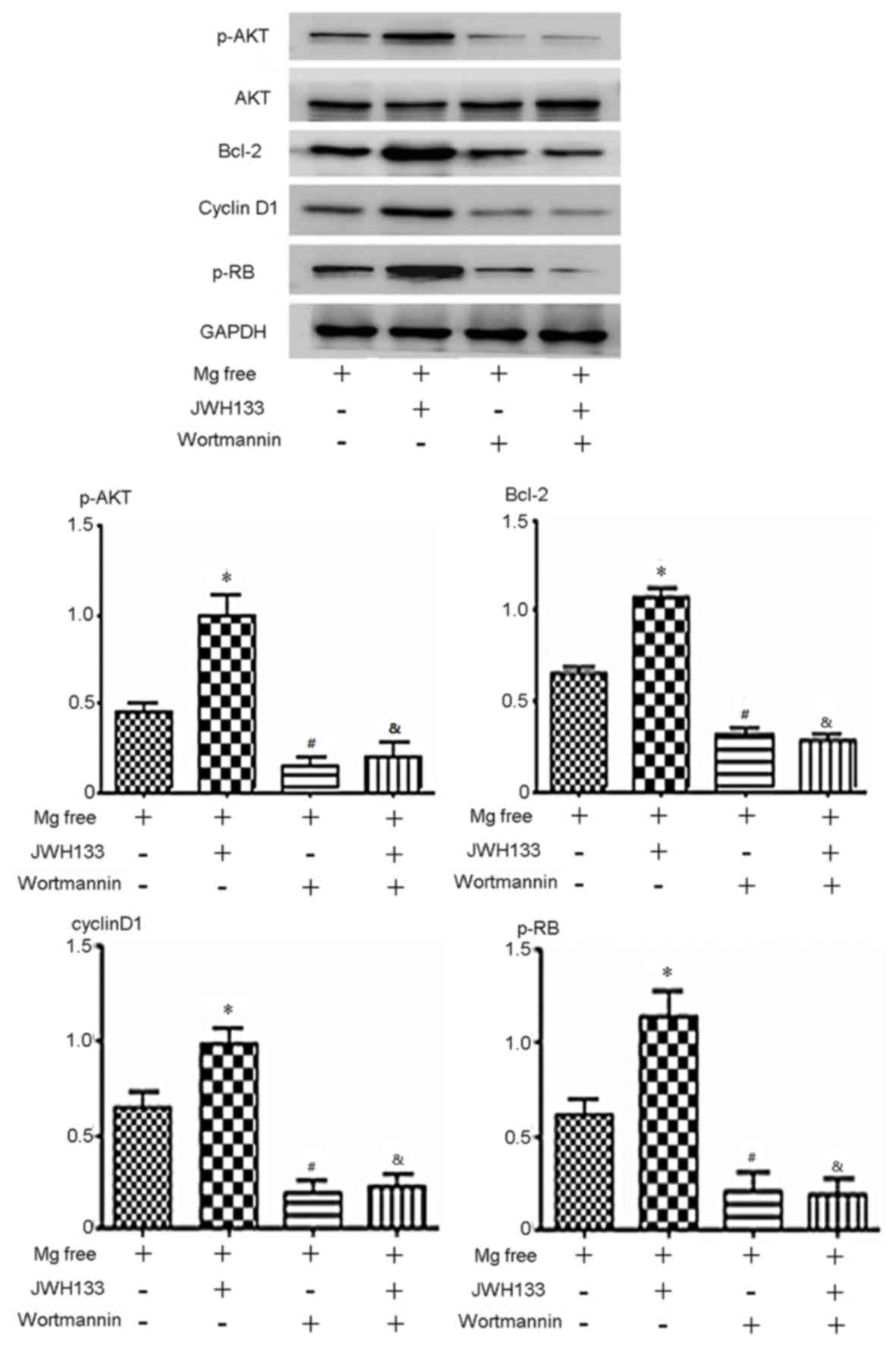 | Figure 4.Expression of p-AKT, AKT, p-Rb,
cyclin D1 and Bcl-2 protein in cells treated with the PI3K 110α-AKT
inhibitor wortmannin. p-AKT, AKT p-Rb, cyclin D1 and Bcl-2 were
downregulated when cells were treated with Mg free solution and
wortmannin, with or without JWH133. The level of p-AKT, p-Rb,
cyclin D1 and Bcl-2 expression was upregulated by JWH133 compared
with the Mg free group. In the group treated with Mg + wortmannin,
p-AKT, p-Rb, cyclin D1 and Bcl-2 expression decreased significantly
compared with Mg free. In the group treated with Mg + JWH133 +
wortmannin, p-AKT, p-Rb, cyclin D1 and Bcl-2 expression decreased
significantly compared with Mg + JWH133. *P<0.05 vs. Mg free
group; #P<0.05 vs. Mg free; &P<0.05
vs. Mg free solution + JWH133. p-, phosphorylated; AKT, protein
kinase B; Rb, retinoblastoma; Bcl-2, B-cell lymphoma 2. |
CB2R regulates the PI3K 110α-AKT
signaling pathway in a rat model of epilepsy
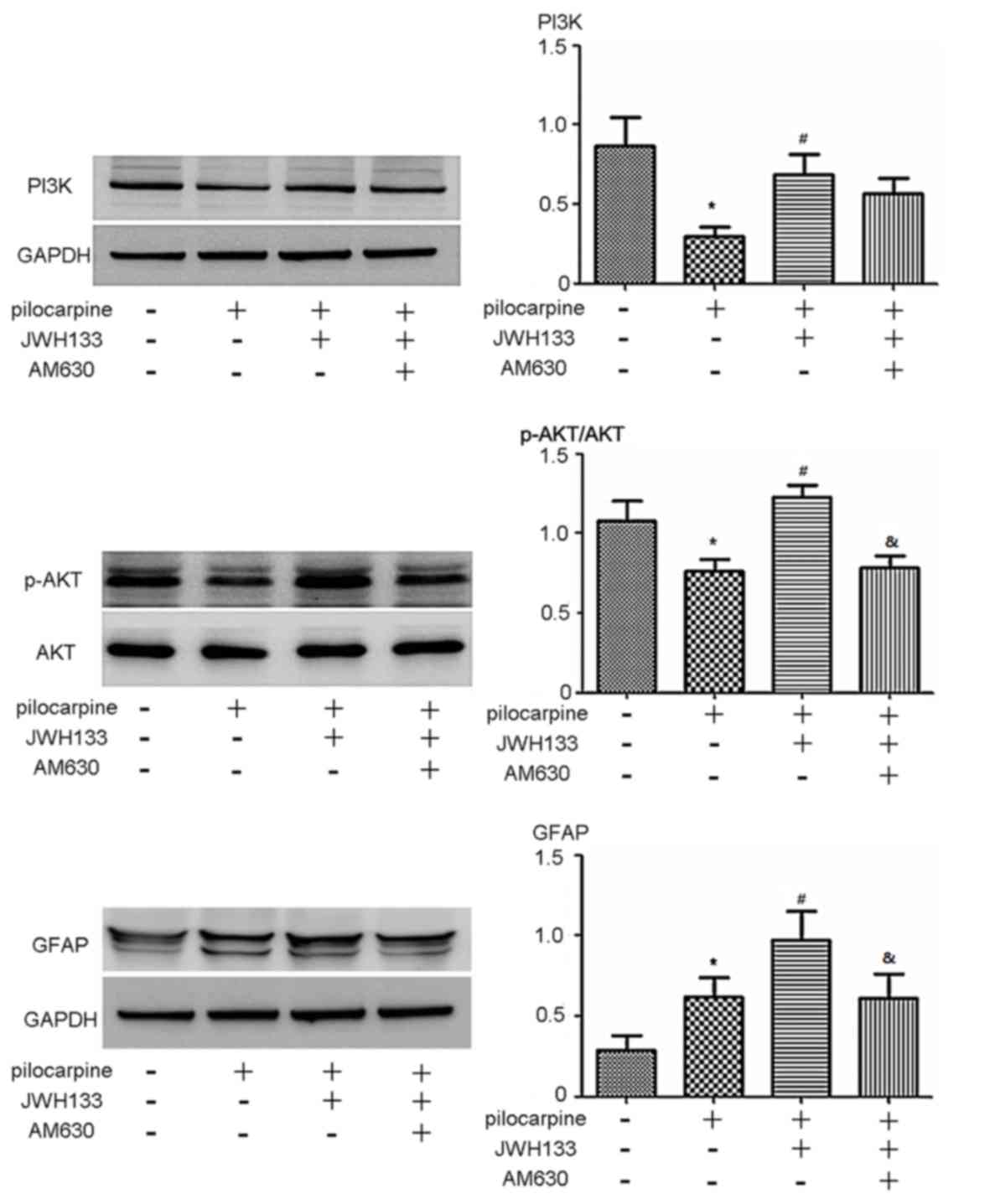
To further investigate the role of CB2R in
regulating the PI3K 110α-AKT signaling pathway, CB2R
agonist/antagonists were administered to rats with
pilocarpine-induced epilepsy. The expression of p-AKT and PI3K 110α
was significantly decreased in rats with epilepsy compared with the
control group (P<0.05; Fig. 5).
p-AKT and PI3K 110α expression was significantly increased in rats
treated with the CB2R agonist JWH133 compared with rats treated
with pilocarpine alone (P<0.05; Fig.
5). When rats with epilepsy were treated with the CB2R
antagonist AM630, p-AKT expression was significantly reduced
compared with agonist JWH133 (P<0.05; Fig. 5). The expression of PI3K 110α in the
AM630 group was markedly reduced compared with the JWH133 group
(P<0.05; Fig. 5). The expression
of GFAP, the biomarker of astrocytes (25), was also assessed. The CB2R agonist
JWH133 effectively promoted GFAP expression (P<0.05; Fig. 5). However, administration of the CB2R
antagonist AM630 reduced the expression of GFAP compared with the
JWH133 group (P<0.05: Fig.
5).
Discussion
Epilepsy is a common and complex disorder of the
CNS, the mechanism of which remains to be elucidated. The
pathogenesis of epilepsy is considered to be associated with
dysfunctional ion channels, neurotransmitters and glial cells
(26–28). The cannabinoid system comprises the
cannabinoid receptor, cannabinoids and enzymes. There are two types
of cannabinoid receptor, CB1R and CB2R, of which CB2R exists in the
CNS, including the cerebral cortex, corpus callosum, hippocampus,
basal ganglia region and opisthencephalon. Previous reports have
demonstrated that cannabinoid may relieve the symptoms of epilepsy
in animal models, suggesting a potential link between CB2R and
epilepsy (29,30).
Astrocytes are the main type of glial cell in the
CNS and they provide support and protection for neurons (31). Astrocytes supply neurons with
nutrients, oxygen and cytokines and maintain homeostasis. Astrocyte
dysfunction has been reported to be associated with the development
of epilepsy; for example, disturbances in astrocyte ion hemostasis
result in seizures (15). Astrocytes
are able to communicate directly with neurons at the synapse to
control the ion channel function of neurons (19,20).
Maintaining astrocyte function may therefore be important for
protecting against epilepsy.
In the present study, changes in cell cycle
distribution induced by the epileptic microenvironment and the
potential underlying mechanisms were investigated. Flow cytometry
analysis revealed that G1 to S transition was inhibited when cells
were treated with Mg free solution. The CB2R agonist JWH133
successfully upregulated the percentage of cells in S phase and
downregulated the percentage of G1 phase cells, demonstrating that
JWH133 promotes G1 to S transition. However, the CB2R antagonist
AM630 abolished these effects. These results suggest that CB2R
regulates cell cycle transition in astrocytes.
In addition, it was determined that the expression
of cell cycle-associated proteins was altered in accord with cell
cycle changes. Bcl-2, p-Rb, cyclin D1 and cyclin E expression
decreased when cells were treated with Mg free solution. The CB2R
agonist JWH133 enhanced the expression of p-Rb, cyclin D1, cyclin E
and Bcl-2, whereas the CB2R antagonist AM630 suppressed the
expression of p-Rb, cyclin D1, cyclin E and Bcl-2. It has
previously been reported that the CB2R activator JWH-015 reduced
neuron loss and cytochrome C release to improve the biological
function of nerves, an effect that was reversed using the CB2R
inhibitor SR144528 (32). The
results of the present study support those of previous studies,
suggesting CB2R activation protects astrocytes and promotes
astrocyte growth.
Several signaling pathways were assessed to clarify
the potential mechanism of biological effects induced by CB2R
agonist/antagonist. The results indicated that PI3K-AKT signaling
was responsible for the changes in cell cycle protein expression.
PI3K-AKT signaling comprises PI3K 110α, AKT and downstream
proteins. Second-messenger phosphatidylinositol (3,4,5)-trisphosphate was generated following the
activation of PI3K 110α, which in turn activated AKT
phosphorylation. PI3K-AKT activation is associated with neuronal
activities, including proliferation, apoptosis, differentiation and
metabolism (33). PI3K-AKT
dysregulation is critical in the development of CNS diseases,
including Parkinson's Disease, ischemic brain injury and epilepsy
(34–36). In the present study it was
demonstrated that p-AKT expression decreased significantly in
astrocytes in an epileptic environment. CB2R activator JWH133
promoted p-AKT expression, whereas the CB2R inhibitor AM630
inhibited it. Changes in the expression of PI3K p110 were similar
to p-AKT in these groups. mTOR is a downstream protein of AKT
signaling which has been reported to be associated with the
development of epilepsy (37–41). It
was reported that mTOR serves an important role in epileptic
seizures via adjusting the excitability of dentate gyrus gramular
cells (37). p-mTOR expression
decreased significantly in astrocytes in the epileptic
microenvironment. The CB2R activator JWH133 elevated the ratio of
p-mTOR/mTOR while CB2R inhibitor AM630 reduced it. These results
suggest that activating CB2R induces AKT signaling.
The AKT inhibitor wortmannin was used to investigate
the role of AKT signaling in CB2R-induced cell cycle changes and it
was demonstrated that levels of p-AKT, p-Rb, cyclin D1 and Bcl-2
were upregulated by CB2R activation in astrocytes without
wortmannin treatment. However, wortmannin blocked the effects of
JWH133 on these proteins. In wortmannin treated cells, no
significant differences in p-Rb, cyclin D1 and Bcl-2 were observed
between the Mg free and JWH133 groups. These results suggest that
JWH133-induced CB2R activation upregulates p-Rb, cyclin D1 and
Bcl-2 via the AKT signaling pathway. It has been reported that AKT
phosphorylation accelerates its degradation (42,43). In
addition, the p-AKT/AKT ratio was measured in the current study and
it was identified that JWH133 significantly upregulated AKT
phosphorylation. Wortmannin suppressed the PI3K signal pathway and
p-AKT/AKT ratio was depressed even following JWH133 treatment.
To further investigate the regulatory role of CB2R
in the PI3K/AKT signaling pathway, a rat model of epilepsy was
established and treated with CB2R agonist/antagonists. The
expression of p-AKT and PI3K 110α expression was significantly
decreased in rats with epilepsy compared with control rats and
treatment with the CB2R agonist JWH133 significantly promoted AKT
and PI3K 110α expression. Rats treated with the CB2R antagonist
AM630 had significantly lower p-AKT levels and PI3K 110α expression
was markedly downregulated compared with rats treated with JWH133.
These results are similar to the findings of the in vitro
experiments. GFAP expression was also assessed to investigate the
level of astrogliosis and astrocyte viability. The results revealed
that treatment with CB2R agonist JWH133 upregulated GFAP expression
and treatment with the CB2R antagonist AM630 downregulated GFAP
expression in comparison. These results suggest that, in a rat
model of epilepsy, CB2R activation may enhance astrocyte viability
and activate AKT signaling.
In conclusion, the results of the present study
demonstrate that CB2R activation upregulates the proliferation of
rat astrocytes in vivo and in vitro, possibly via
activation of the PI3K-AKT signaling pathway.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Liaoning
Science and Technology Department of China Project (grant. no.
2014225007).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QC performed the western blot experiments and wrote
the manuscript. XL performed the flow cytometry experiments. FY
constructed the pilocarpine-induced status epilepticus model. HW
designed the experiment.
Ethics approval and consent to
participate
The present study has been approved by the
Institutional Ethical Review Boards of the Shengjing Hospital of
China Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Richter Z, Janszky J, Sétáló G Jr, Horváth
R, Horváth Z, Dóczi T, Seress L and Ábrahám H: Characterization of
neurons in the cortical white matter in human temporal lobe
epilepsy. Neuroscience. 333:140–150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Santini E and Klann E: Genetically
dissecting cortical neurons involved in epilepsy in angelman
syndrome. Neuron. 90:1–3. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharop BR, Boldyriev OI, Batiuk MY,
Shtefan NL and Shuba YM: Compensatory reduction of Cav3.1
expression in thalamocortical neurons of juvenile rats of WAG/Rij
model of absence epilepsy. Epilepsy Res. 119:10–12. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Woodbury DM: Neurotransmitters and
epilepsy: Distinguishing characteristics and unifying precepts. Fed
Proc. 43:2529–2531. 1984.PubMed/NCBI
|
|
5
|
Werner FM and Coveñas R: Classical
neurotransmitters and neuropeptides involved in generalized
epilepsy in a multi-neurotransmitter system: How to improve the
antiepileptic effect? Epilepsy Behav. 71:124–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ievglevskyi O, Isaev D, Netsyk O, Romanov
A, Fedoriuk M, Maximyuk O, Isaeva E, Akaike N and Krishtal O:
Acid-sensing ion channels regulate spontaneous inhibitory activity
in the hippocampus: Possible implications for epilepsy. Philos
Trans R Soc Lond B Biol Sci. 371:pii: 201504312016. View Article : Google Scholar
|
|
7
|
Lerche H, Shah M, Beck H, Noebels J,
Johnston D and Vincent A: Ion channels in genetic and acquired
forms of epilepsy. J Physiol. 591:753–764. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heuser K, Szokol K and Tauboll E: The role
of glial cells in epilepsy. Tidsskr Nor Laegeforen. 134:37–41.
2014.(In English; Norwegian). View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Afawi Z, Oliver KL, Kivity S, Mazarib A,
Blatt I, Neufeld MY, Helbig KL, Goldberg-Stern H, Misk AJ,
Straussberg R, et al: Multiplex families with epilepsy: Success of
clinical and molecular genetic characterization. Neurology.
86:713–722. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vezzani A, Lang B and Aronica E: Immunity
and Inflammation in Epilepsy. Cold Spring Harb Perspect Med.
6:a0226992015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim DW, Sunwoo JS and Lee SK: Incidence
and localizing value of vertigo and dizziness in patients with
epilepsy: Video-EEG monitoring study. Epilepsy Res. 126:102–105.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim H, Thurman DJ, Durgin T, Faught E and
Helmers S: Estimating epilepsy incidence and prevalence in the US
pediatric population using nationwide health insurance claims data.
J Child Neurol. 31:743–749. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang S, Zhang J, Zhang S and Fu X:
Epilepsy in adults with supratentorial glioblastoma: Incidence and
influence factors and prophylaxis in 184 patients. PLoS One.
11:e01582062016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Loewen JL, Barker-Haliski ML, Dahle EJ,
White HS and Wilcox KS: Neuronal injury, gliosis, and glial
proliferation in two models of temporal lobe epilepsy. J
Neuropathol Exp Neurol. 75:366–378. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eid T, Lee TW, Patrylo P and Zaveri HP:
Astrocytes and glutamine synthetase in epileptogenesis. J Neurosci
Res. Jul 18–2018.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hamilton NB and Attwell D: Do astrocytes
really exocytose neurotransmitters? Nat Rev Neurosci. 11:227–238.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
James LR, Andrews S, Walker S, de Sousa
PR, Ray A, Russell NA and Bellamy TC: High-throughput analysis of
calcium signalling kinetics in astrocytes stimulated with different
neurotransmitters. PLoS One. 6:e268892011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Magistretti PJ, Sorg O, Yu N, Martin JL
and Pellerin L: Neurotransmitters regulate energy metabolism in
astrocytes: Implications for the metabolic trafficking between
neural cells. Dev Neurosci. 15:306–312. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sukigara S, Dai H, Nabatame S, Otsuki T,
Hanai S, Honda R, Saito T, Nakagawa E, Kaido T, Sato N, et al:
Expression of astrocyte-related receptors in cortical dysplasia
with intractable epilepsy. J Neuropathol Exp Neurol. 73:798–806.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carmignoto G and Haydon PG: Astrocyte
calcium signaling and epilepsy. Glia. 60:1227–1233. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mecha M, Carrillo-Salinas FJ, Feliú A,
Mestre L and Guaza C: Microglia activation states and cannabinoid
system: Therapeutic implications. Pharmacol Ther. 166:40–55. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ni R, Mu L and Ametamey S: Positron
emission tomography of type 2 cannabinoid receptors for detecting
inflammation in thecentral nervous system. Acta Pharmacol Sin. Jun
19–2018.(Epub ahead of print). View Article : Google Scholar
|
|
23
|
Rizzo V, Carletti F, Gambino G, Schiera G,
Cannizzaro C, Ferraro G and Sardo P: Role of CB2 receptors and cGMP
pathway on the cannabinoid-dependent antiepileptic effects in an in
vivo model of partial epilepsy. Epilepsy Res. 108:1711–1718. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hocker SE: Status epilepticus. Continuum
(Minneap Minn) 21 (5 Neurocritical Care). 1–1383. 2015.
|
|
25
|
Xu J: New insights into GFAP negative
astrocytes in calbindin D28k immunoreactive astrocytes. Brain Sci.
8(pii): E1432018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu JJ, Diaz P, Bie B, Astruc-Diaz F, Wu J,
Yang H, Brown DL and Naguib M: Spinal gene expression profiling and
pathways analysis of a CB2 agonist (MDA7)-targeted prevention of
paclitaxel-induced neuropathy. Neuroscience. 260:185–194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Molina-Holgado E, Vela JM, Arévalo-Martín
A, Almazán G, Molina-Holgado F, Borrell J and Guaza C: Cannabinoids
promote oligodendrocyte progenitor survival: Involvement of
cannabinoid receptors and phosphatidylinositol-3 kinase/Akt
signaling. J Neurosci. 22:9742–9753. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim J and Li Y: Chronic activation of CB2
cannabinoid receptors in the hippocampus increases excitatory
synaptic transmission. J Physiol. 593:871–886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Devane WA, Dysarz FA III, Johnson MR,
Melvin LS and Howlett AC: Determination and characterization of
cannabinoid receptor in rat brain. Mol Pharmacol. 34:605–613.
1988.PubMed/NCBI
|
|
30
|
Jing N, Fang B, Wang ZL and Ma H: Remote
ischemia preconditioning attenuates blood-spinal cord barrier
breakdown in rats undergoing spinal cord ischemia reperfusion
injury: Associated with activation and upregulation of CB1 and CB2
receptors. Cell Physiol Biochem. 43:2516–2524. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seifert G and Steinhäuser C:
Neuron-astrocyte signaling and epilepsy. Exp Neurol. 244:4–10.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Viscomi MT, Oddi S, Latini L, Pasquariello
N, Florenzano F, Bernardi G, Molinari M and Maccarrone M: Selective
CB2 receptor agonism protects central neurons from remote
axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci.
29:4564–4570. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duan W, Chen Y and Wang XR: MicroRNA-155
contributes to the occurrence of epilepsy through the PI3K/Akt/mTOR
signaling pathway. Int J Mol Med. 42:1577–1584. 2018.PubMed/NCBI
|
|
34
|
Khwanraj K, Madlah S, Grataitong K and
Dharmasaroja P: Comparative mRNA expression of eEF1A isoforms and a
PI3K/Akt/mTOR pathway in a cellular model of parkinson's disease.
Parkinsons Dis. 2016:87160162016.PubMed/NCBI
|
|
35
|
Zhang W, Liu J, Hu X, Li P, Leak RK, Gao Y
and Chen J: n-3 polyunsaturated fatty acids reduce neonatal
hypoxic/ischemic brain injury by promoting phosphatidylserine
formation and Akt signaling. Stroke. 46:2943–2950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Roy A, Skibo J, Kalume F, Ni J, Rankin S,
Lu Y, Dobyns WB, Mills GB, Zhao JJ, Baker SJ and Millen KJ: Mouse
models of human PIK3CA-related brain overgrowth have acutely
treatable epilepsy. Elife. 4:pii: e127032015. View Article : Google Scholar
|
|
37
|
Hester MS, Hosford BE, Santos VR, Singh
SP, Rolle IJ, LaSarge CL, Liska JP, Garcia-Cairasco N and Danzer
SC: Impact of rapamycin on status epilepticus induced hippocampal
pathology and weight gain. Exp Neurol. 280:1–12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lasarge CL and Danzer SC: Mechanisms
regulating neuronal excitability and seizure development following
mTOR pathway hyperactivation. Front Mol Neurosci. 7:182014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bateup HS, Denefrio CL, Johnson CA,
Saulnier JL and Sabatini BL: Temporal dynamics of a homeostatic
pathway controlling neural network activity. Front Mol Neurosci.
6:282013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Berdichevsky Y, Dryer AM, Saponjian Y,
Mahoney MM, Pimentel CA, Lucini CA, Usenovic M and Staley KJ:
PI3K-Akt signaling activates mTOR-mediated epileptogenesis in
organotypic hippocampal culture model of post-traumatic epilepsy. J
Neurosci. 33:9056–9067. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Groszer M, Erickson R, Scripture-Adams DD,
Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X and Wu H: Negative
regulation of neural stem/progenitor cell proliferation by the Pten
tumor suppressor gene in vivo. Science. 294:2186–2189. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Su CH, Lan KH, Li CP, Chao Y, Lin HC, Lee
SD and Lee WP: Phosphorylation accelerates geldanamycin-induced Akt
degradation. Arch Biochem Biophys. 536:6–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liao Y and Hung MC: Physiological
regulation of Akt activity and stability. Am J Transl Res. 2:19–42.
2010.PubMed/NCBI
|