Introduction
The renin-angiotensin-aldosterone system serves a
role in the pathogenesis of cardiovascular diseases, including
hypertension and atherosclerosis (1). Angiotensin II (Ang II) is the main
active peptide hormone of the renin-angiotensin-aldosterone system
(2). It serves a role in endothelial
dysfunction, vascular remodeling and vascular inflammation, which
are closely associated with numerous diseases, including
hypertension and atherosclerosis (3,4).
Previous studies found that Ang II triggers a large amount of
reactive oxygen species (ROS) in several human cell lines and
organic tissues (5,6). High levels of ROS can lead to
mitochondrial dysfunction, inflammation and/or autophagy (7–9). These
cellular responses are widely reported to cause disordered
homeostasis in cells, ultimately leading to cell death (10–13).
Statins typically include lipophilic (simvastatin
and atorvastatin) and hydrophilic (rosuvastatin) members (14). Atorvastatin (Ator) lowers the level
of cholesterol in the blood by inhibiting
3-hydroxy-3-methylglutaryl-coenzyme A reductase, an enzyme with a
key role in cholesterol production (15). Previous reports indicated a number of
protective functions associated with Ator in cell lines, including
the upregulation of endothelial nitric oxide expression and
antioxidant effects (16,17). Previous studies have also
demonstrated the anti-hypertensive effects of Ator, promoting the
resumption of endothelial function and via intrinsic anti-oxidant
activities (18). There is evidence
that high levels of ROS and cellular apoptosis are associated with
senescence (19). Ator was
previously reported to delay aging in endothelial progenitor cells
(20). However, few studies have
investigated the underlying mechanism by which Ator regulates
endothelial function and senescence.
The present study screened the effects of Ator on
Ang II-induced cytotoxicity in human umbilical vein endothelial
cells (HUVECs). This study examined several cellular responses
induced by Ang II, including oxidative stress, inflammation,
autophagy and cell apoptosis, and determined whether Ator could
reverse these changes. In addition, endothelial nitric oxide
synthase (eNOS) protein expression was detected following exposure
to Ang II alone and in combination with Ator, and this was shown to
be associated with HUVEC function. The tube formation assay was
performed to determine whether Ator influenced the angiogenic
damage induced by treatment with Ang II. Furthermore, this study
examined whether Ator was able to attenuate Ang II-induced
senescence in HUVECs.
Materials and methods
Materials
Ang II was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Ator was purchased from Pfizer, Inc. (New
York, NY, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT), ROS Detection kit, Senescence-associated
β-Galactosidase (SA-β-gal) staining kit, Rhodamine 123 (Rh123)
Staining kit and Terminal deoxynucleotidyl-transferase-mediated
dUTP nick-end labeling (TUNEL) Staining kit were purchased from
Beyotime Institute of Biotechnology (Jiangsu, China). Anti-B-cell
lymphoma 2 (BCL-2), anti-Bcl-2-associated X (BAX), anti-caspase-3
and anti-GAPDH antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Anti-microtubule-associated
protein 1A/1B-light chain 3 (LC3)-II, anti-LC3-I, anti-Beclin 1,
anti-sequestosome 1 (p62), anti-endothelial nitric oxide synthase
(eNOS), anti-tumor protein p53 (p53) and anti-cyclin dependent
kinase inhibitor 2A (p16) antibodies were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The
Chemicon® in vitro Angiogenesis Assay kit
was obtained from Merck KGaA. Human interleukin (IL)-12 (cat. no.
EK0421), IL-6 (cat. no. EK0410), IL-1β (cat. no. EK0932) and tumor
necrosis factor-α (TNF-α; cat. no. EK0525) ELISA kits were supplied
by Boster Biological Technology (Pleasanton, CA, USA).
Cell culture
HUVECs were obtained from the Cell Bank of the
Chinese Academy of Science (Shanghai, China; cat. no. H082). Cells
were cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen) supplemented with 10% heat-inactivated fetal bovine
serum (FBS) and 1% penicillin-streptomycin (all Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in a 5% CO2-humidified
incubator.
MTT assay
HUVECs were seeded onto 96-well plates at a density
of 1×104 cells/well and grown for 24 h. In the first stage, the
medium was replaced with medium containing different concentrations
(0.1, 1, 10 or 100 µM) of Ang II (Sigma-Aldrich; Merck KGaA), and
further incubated at 37°C for 24, 48 and 72 h. In the second stage,
cells were incubated with fresh medium containing Ang II (1 µM)
alone or combined with various concentrations of atorvastatin (1,
10, 20 or 50 µM) at 37°C for 24 h. The effects of atorvastatin (1,
10, 20 or 50 µM) on cell viability were also examined. Cells were
washed twice with phosphate-buffered saline and MTT was added to a
final concentration of 0.5 mg/ml in each well and incubated for 4 h
at 37°C. Following MTT incubation, dimethylsulfoxide solution was
used to dissolve the formazan crystals and absorbance was measured
at a wavelength of 490 nm on a microplate reader (Thermo Multiskan
MK3; Thermo Fisher Scientific, Inc.). The viability of control
cells was set as 100%.
ROS detection assay
ROS levels were determined by
2′,7′-dichlorofluorescein diacetate assay (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. Cells
were seeded onto six-well plates at a density of 2×105 cells per
well and grown for 24 h. Subsequently, cells were treated with
fresh DMEM containing 10 µM Ator alone, 1-µM Ang II alone or
combined with 10 µM Ator at 37°C for 24 h. A single treatment of 10
µM Ator was also applied. Flow cytometry was performed to quantify
the levels of ROS using a Cyan-LX instrument (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA) and analyzed by FlowJo
V10 CL software (FlowJo LLC, Ashland, OR, USA). The mean
fluorescence was determined by counting 10,000 events.
Cytokine quantification assay
HUVECs were treated with 10 µM Ator alone and 1 µM
Ang II alone or combined with 10 µM Ator, and incubated at 37°C for
24 h. Following incubation, the supernatant was pipetted into
96-well plates and analyzed for IL-12, IL-6, IL-1β and TNF-α
expression using the Human IL-12, IL-6, IL-1β and TNF-α ELISA kits,
according to the manufacturer's protocol. Absorbance was measured
at 450 nm with a correction wavelength at 570 nm. The bicinchoninic
acid (BCA) protein assay was used to measure the total protein
concentration in the supernatant. Cytokine levels were calculated
from a standard curve for each sample.
Mitochondrial membrane potential (MMP)
assay
MMP assay was performed using the Rh123 Staining
kit, according to the manufacturer's protocol. HUVECs were seeded
onto six-well plates at a density of 2×105 cells/well and grown for
24 h. Cells were treated with 10 µM Ator alone and 1 µM Ang II
alone or combined with 10 µM Ator and incubated for 24 h at 37°C.
Cells were washed twice with PBS and stained with 10 µg/ml Rh123
for 30 min at 37°C. Subsequently, stained cells were collected and
analyzed using a flow cytometer (DakoCytomation; Dako; Agilent
Technologies, Inc.) to quantify the fluorescence intensities of the
cells. Data were analyzed with FlowJo V10 CL software.
TUNEL assay
HUVECs were seeded onto six-well plates at a density
of 2×105 cells/well and grown for 24 h. Subsequently, cells were
treated with 10 µM Ator alone and 1 µM Ang II alone or combined
with 10 µM Ator for 24 h at 37°C. TUNEL staining was then performed
according to the manufacturer's protocol.
SA-β-gal assay
HUVECs were seeded onto six-well plates at a density
of 2×105 cells/well and grown for 24 h. Subsequently, cells were
treated with 10 µM Ator alone and 1 µM Ang II alone or combined
with 10 µM Ator for 24 h at 37°C. Cells were stained using the
SA-β-gal staining kit, according to the manufacturer's protocol.
Images were obtained using an optical microscope (SOPTOP ICX41;
Ningbo Sunny Instruments Co., Ltd., Ningbo, China) at a
magnification of ×200 and analyzed using ImageJ software (version
1.4.9; National Institute of Health, Bethesda, MD, USA).
Tube formation assay
Tube formation assays were performed using the
Chemicon in vitro Angiogenesis Assay kit according to
manufacturer's protocol. Tube formation assays were performed using
24-well plates precoated with Matrigel, which was stored for 24 h
at 4°C before the experiment. The Matrigel was thawed and 200 µl
was added to a 24-well plate. The plate was incubated for 30 min at
37°C to form a gel layer. Cells seeded at a density of 1×104
cells/well were treated with 10 µM Ator alone, and 1 µM Ang II
alone or a combined of the two at 37°C overnight. The cells (1×104
cells/well) were then plated onto Matrigel pre-coated 24-well
plates in DMEM supplemented with 10% FBS medium and incubated for 4
h. Images were obtained using an SOPTOP ICX41 optical microscope at
a magnification of ×200 and analyzed using ImageJ software.
Western blot analysis
HUVECs were seeded onto six-well plates at a density
of 2×105 cells/well and grown for 24 h. Total protein was extracted
from cells using radioimmunoprecipitation lysis buffer (Beyotime
Institute of Biotechnology). The resultant mixture was centrifuged
at 10,000 × g for 10 min at 4°C. Total protein was quantified using
a BCA protein assay kit (Beyotime Institute of Biotechnology),
according to the manufacturer's protocol. The proteins (20 µg/lane)
were separated by SDS-PAGE, transferred onto a polyvinylidene
difluoride membrane and blocked with 5% nonfat milk at room
temperature for 2 h. The membranes were incubated with primary
antibodies against human BCL-2 (cat. no. SC509), BAX (cat. no.
SC6236), caspase-3 (cat. no. SC271028) and GAPDH (cat. no. SC47724;
all 1:400; Santa Cruz Biotechnology, Inc.), p38 (cat. no. 8690),
LC3-II (cat. no. 2775), LC3-I (cat. no. 4108), Beclin 1 (cat. no.
3778), p62 (cat. no. 88588), eNOS (cat. no. 32027), p53 (cat. no.
2527) or p16 (cat. no. 80772; all 1:400; Cell Signaling Technology,
Inc.) overnight at 4°C. Following primary incubation, membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies (1:5,000; cat. no. A0201) for 2 h at room temperature.
Protein bands were visualized with BeyoECL Plus (both Beyotime
Institute of Biotechnology). Protein expression was quantified
using ImageJ software and normalized to GAPDH.
Statistical analysis
The results are reported as the mean ± standard
error of the mean of at least three independent experiments. All
the experimental data were analyzed using one-way analysis of
variance with Bonferroni correction multiple testing. Statistical
analyses were performed using GraphPad Prism software (version 6.0;
GraphPad Software, Inc., La Jolla, CA, USA). *P<0.05 was
considered to indicate a statistically significant difference.
Results
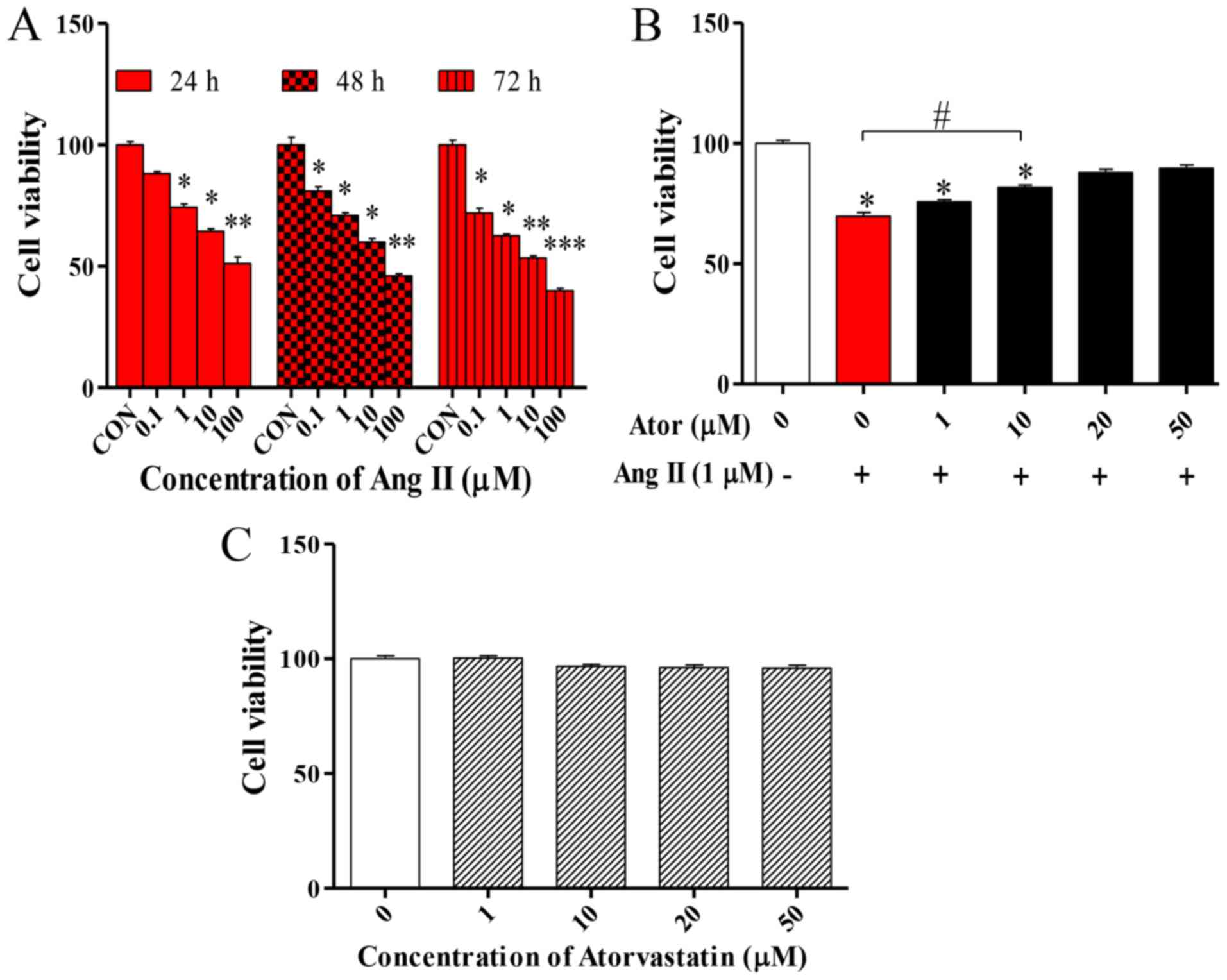
Cell viability measurement
To determine the effect of Ang II in HUVECs, cell
viability was examined using the MTT assay. The MTT assay was
performed following treatment with various concentrations (0, 0.1,
1, 10 or 100 µM) of Ang II at 24, 48 and 72 h. Ang II significantly
decreased cell viability in what appears to be a dose- and
time-dependent manner (Fig. 1A).
Based on these results, the effect of Ator on the viability of
HUVECs was analyzed. HUVECs were treated with 1 µM Ang II in
combination with various concentrations (1, 10, 20 or 50 µM) of
Ator for 24 h. Ator appeared to suppress cell death in a
dose-dependent manner (Fig. 1B).
HUVECs were treated with various concentrations (1, 10, 20 or 50
µM) of Ator alone. Ator did not affect cell viability (Fig. 1C). These results indicated that Ator
may be able to repair the cellular damage caused by Ang II and
protect cells from death.
Ator attenuates Ang II-induced
cytotoxicity in HUVECs
To monitor the effects of Ator on Ang II-induced
cytotoxicity in HUVECs, several cellular processes were examined in
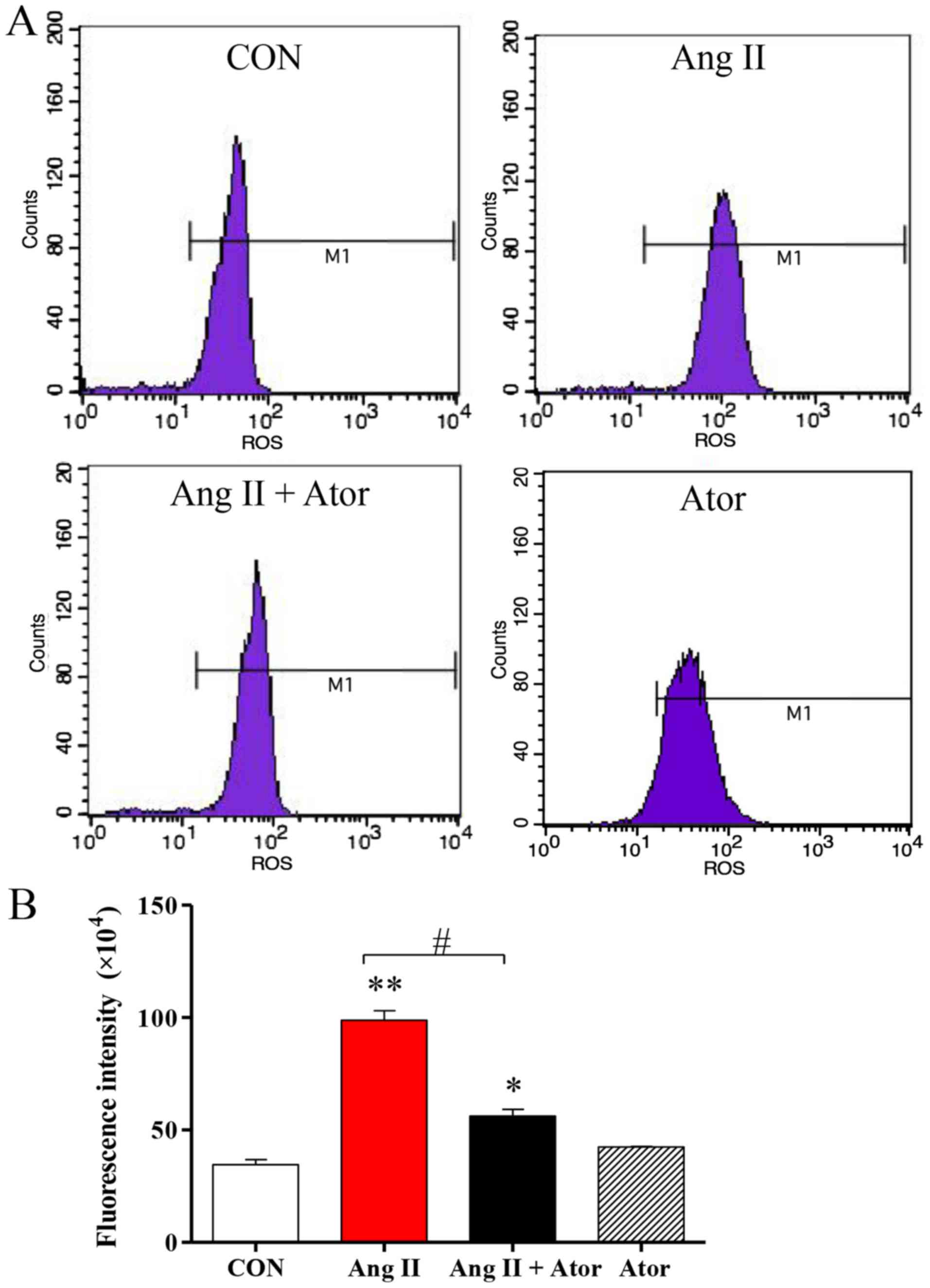
this study. Intracellular levels of ROS were assessed using flow
cytometry (Fig. 2A). ROS generation
in HUVECs was significantly elevated following treatment with Ang
II, compared with the untreated control. However, the addition of
Ator significantly decreased intracellular ROS compared with the
Ang II treatment group. Treatment with Ator alone did not cause any
significant changes in ROS generation (Fig. 2B).
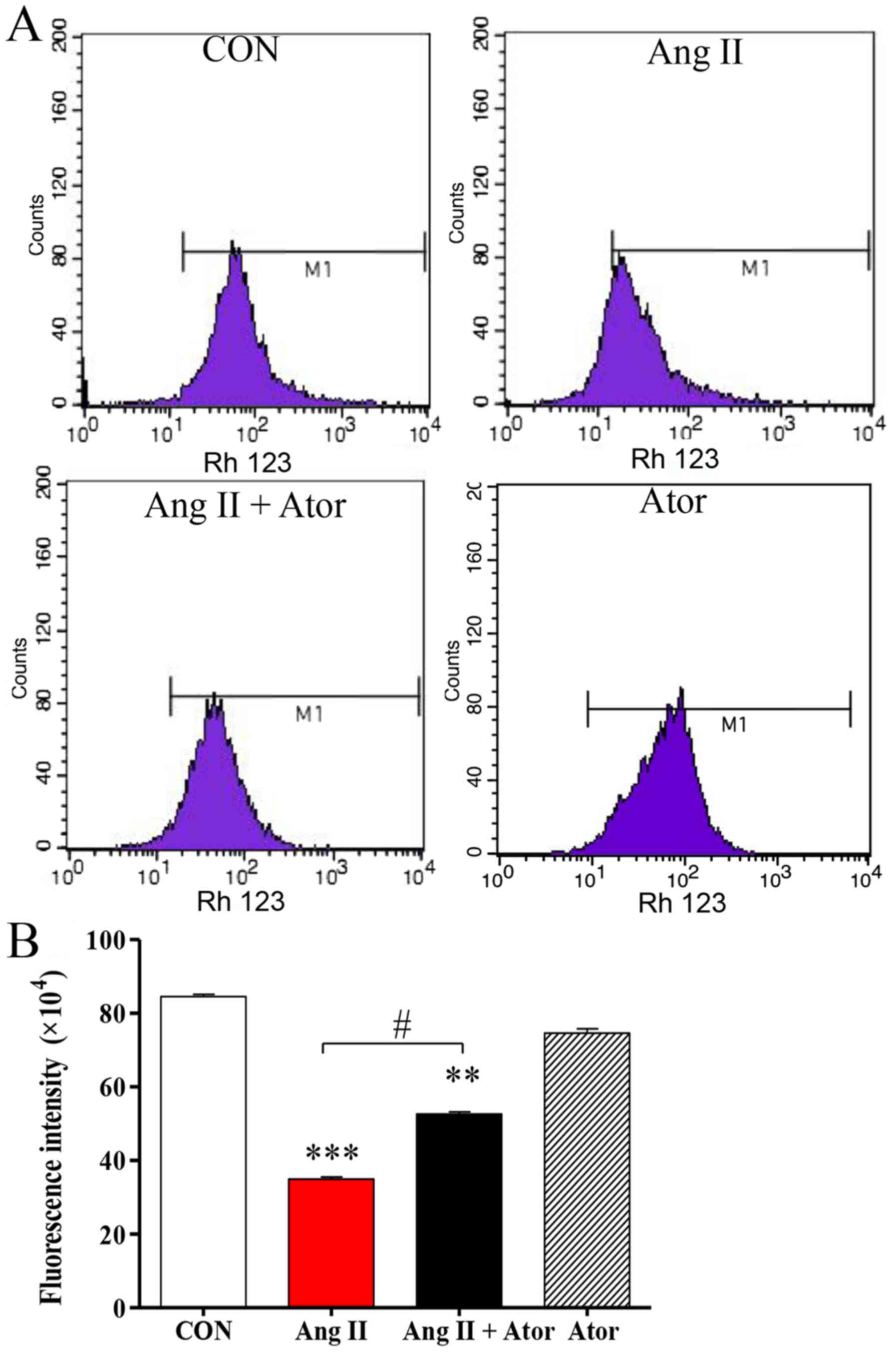
Mitochondria are one of the main sources of ROS
under conditions of oxidative stress; elevated intracellular ROS
may lead to mitochondrial dysfunction, an early indication of cell
death (21). The present study
analyzed the mitochondrial membrane potential by Rh123 staining.
Rh123 is a fluorescent dye that accumulates in normal mitochondria.
Intracellular Rh123 fluorescence was assessed using flow cytometry
(Fig. 3A). Rh123 fluorescence was
significantly weaker following treatment with Ang II, compared with
the untreated control group. However, the fluorescent signal was
attenuated in the presence of Ator compared with the Ang II alone
treatment group. Treatment with Ator alone did not cause any
obvious alterations compared with the untreated control (Fig. 3B). These results indicated that Ator
suppressed cell death and prevented mitochondrial damage caused by
Ang II.
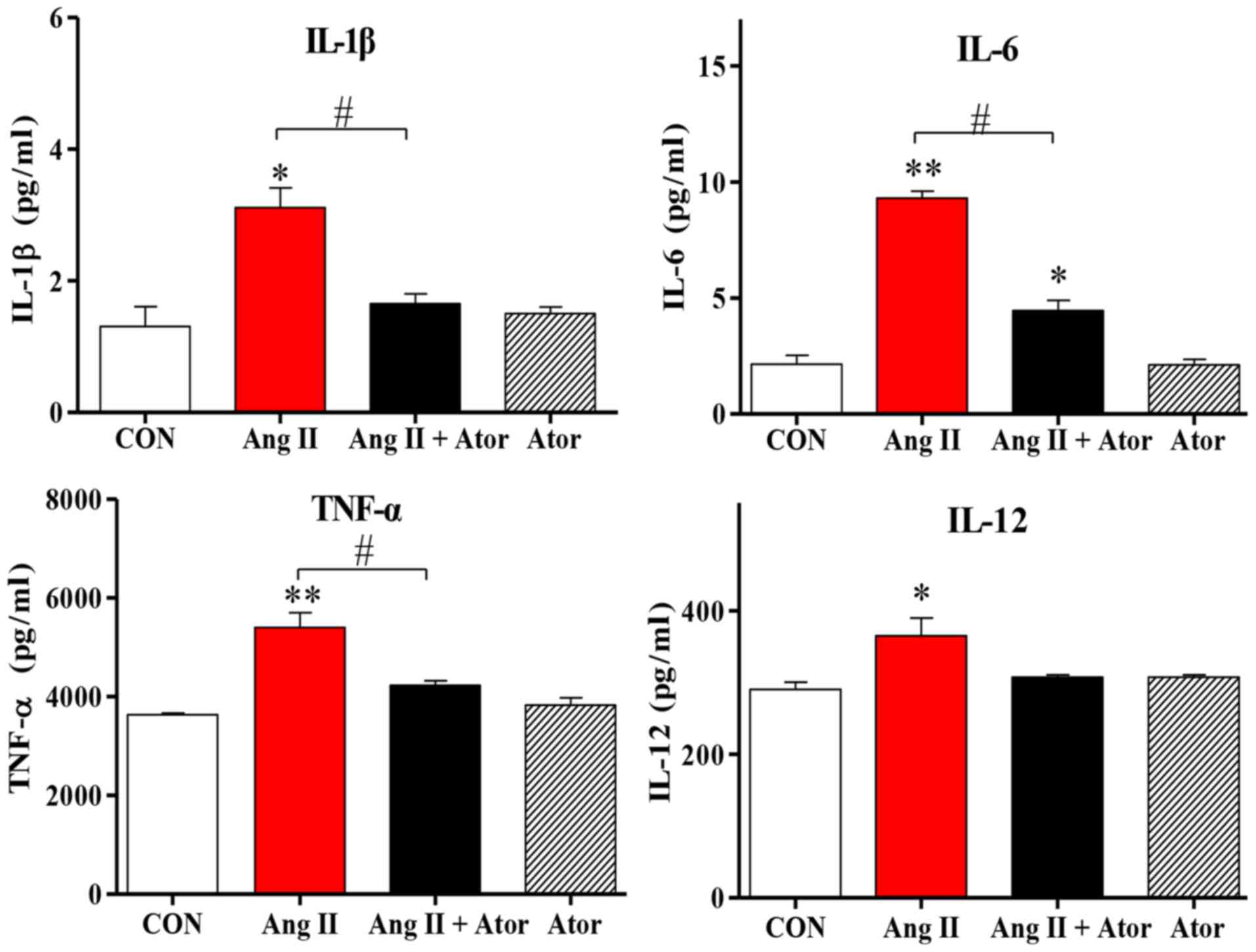
Cell injury may trigger a chain reaction of
inflammatory responses (22). The
present study examined the inflammatory response to treatment with
Ang II in HUVECs. Treatment with Ang II significantly increased
intracellular inflammation, as indicated by enhanced IL-1β, IL-12,
TNF-α and IL-6 cytokine expression levels, compared with the
untreated control group. However, the addition of Ator decreased
cytokine secretion compared with the Ang II treatment group.
Treatment with Ator alone did not cause any significant changes to
cytokine expression (Fig. 4). These
results suggested that Ator prevented cells from chronic
inflammation.
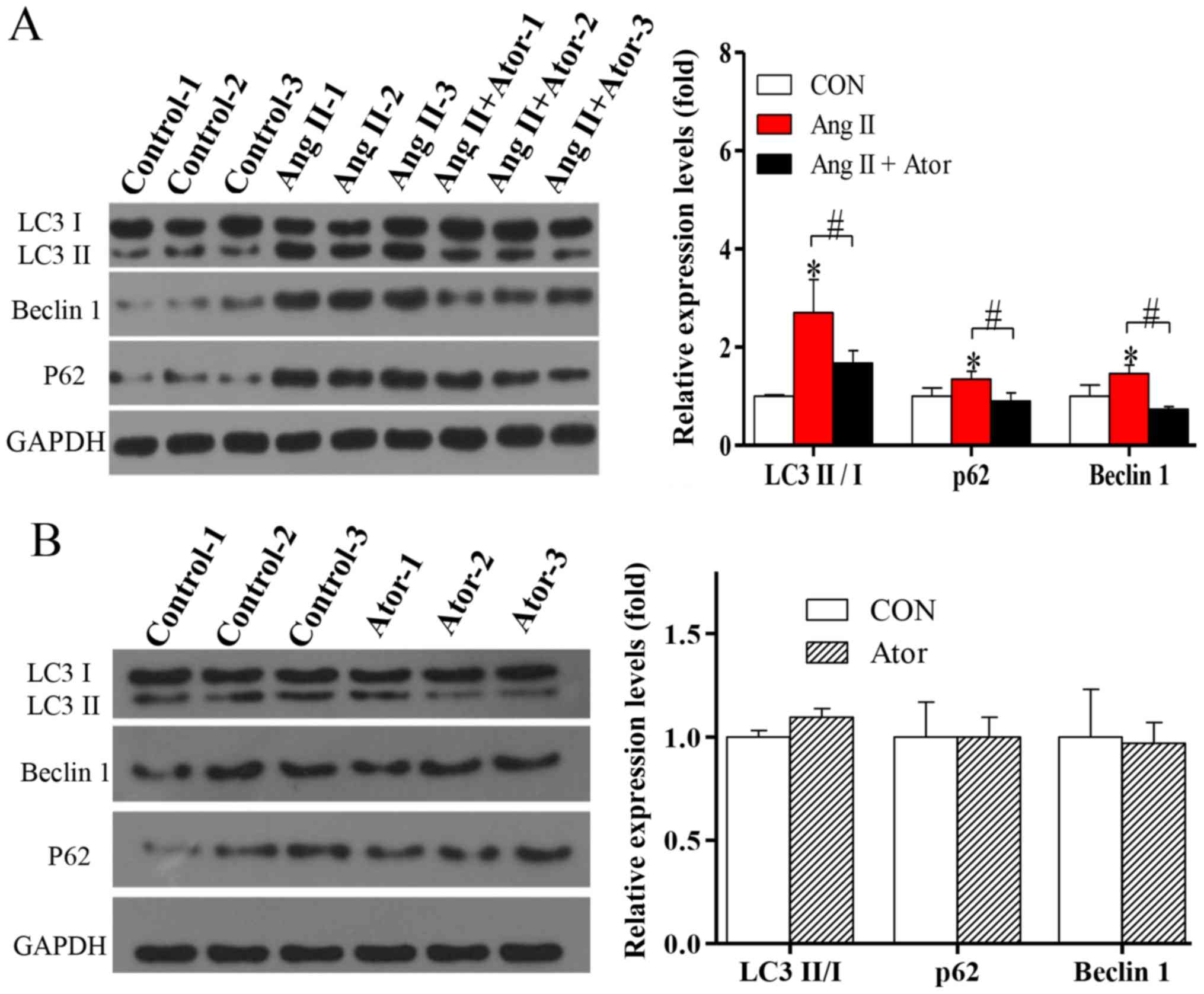
Autophagy is a fundamental cellular process in the
degradation of cellular organelles (23). Due to the Ang II-induced
mitochondrial damage observed, the effects of Ang II and Ator on
cell autophagy in HUVECs were analyzed. The exposure of HUVECs to
treatment with Ang II significantly enhanced protein expression
levels of LC3-II/-I (Fig. 5A). In
addition, the protein level of Beclin 1, which is known to regulate
autophagy via the Beclin 1/VPS34 complex was also significantly
increased following treatment with Ang II. To further confirm
autophagy, the protein level of p62, an important indicator of
autophagic flux, was analyzed. Expression levels of p62
significantly increased following treatment with Ang II (Fig. 5A), indicating inhibition of the
autophagic flux. The activation of autophagy and the suppression of
autophagic flux can lead to LC3-II and autophagosome accumulation
(24). The present study indicated
that in HUVECs, the activation of autophagy and suppression of
autophagic flux occur following treatment with Ang II. The
induction of autophagy and the blockade of autophagy flux can lead
to LC3II and autophagosome accumulation (24). The elevated levels of p62 suggested
that Ang II may not only induce autophagic activity but also block
autophagy flux (24). The addition
of Ator significantly reversed Ang II-induced effects on the
expression of autophagy-associated proteins (Fig. 5A). Treatment with Ator alone did not
cause any significant alterations compared with the untreated
control (Fig. 5B). The present study
indicated that Ator attenuated Ang II-induced cellular autophagy,
and this may be attributed to its effect on intracellular ROS
generation.
To further investigate the effects of Ang II and
Ator on HUVECs, cellular apoptosis was analyzed by the TUNEL assay.
Cellular apoptosis was significantly increased following treatment
with Ang II, however the addition of Ator markedly suppressed Ang
II-induced apoptosis in HUVECs (Fig.
6A). Furthermore, treatment with Ang II significantly increased
expression levels of cellular apoptosis-associated proteins BAX and
caspase-3, and decreased the expression of BCL-2, compared with the
untreated control group (Fig. 6B).
The addition of Ator significantly reversed the effects of Ang II
on the expression of cellular apoptosis-associated proteins.
Treatment with Ator alone did not cause any significant alterations
compared with the untreated control (Fig. 6C). This study indicated that Ator
attenuated Ang II-induced cellular apoptosis.
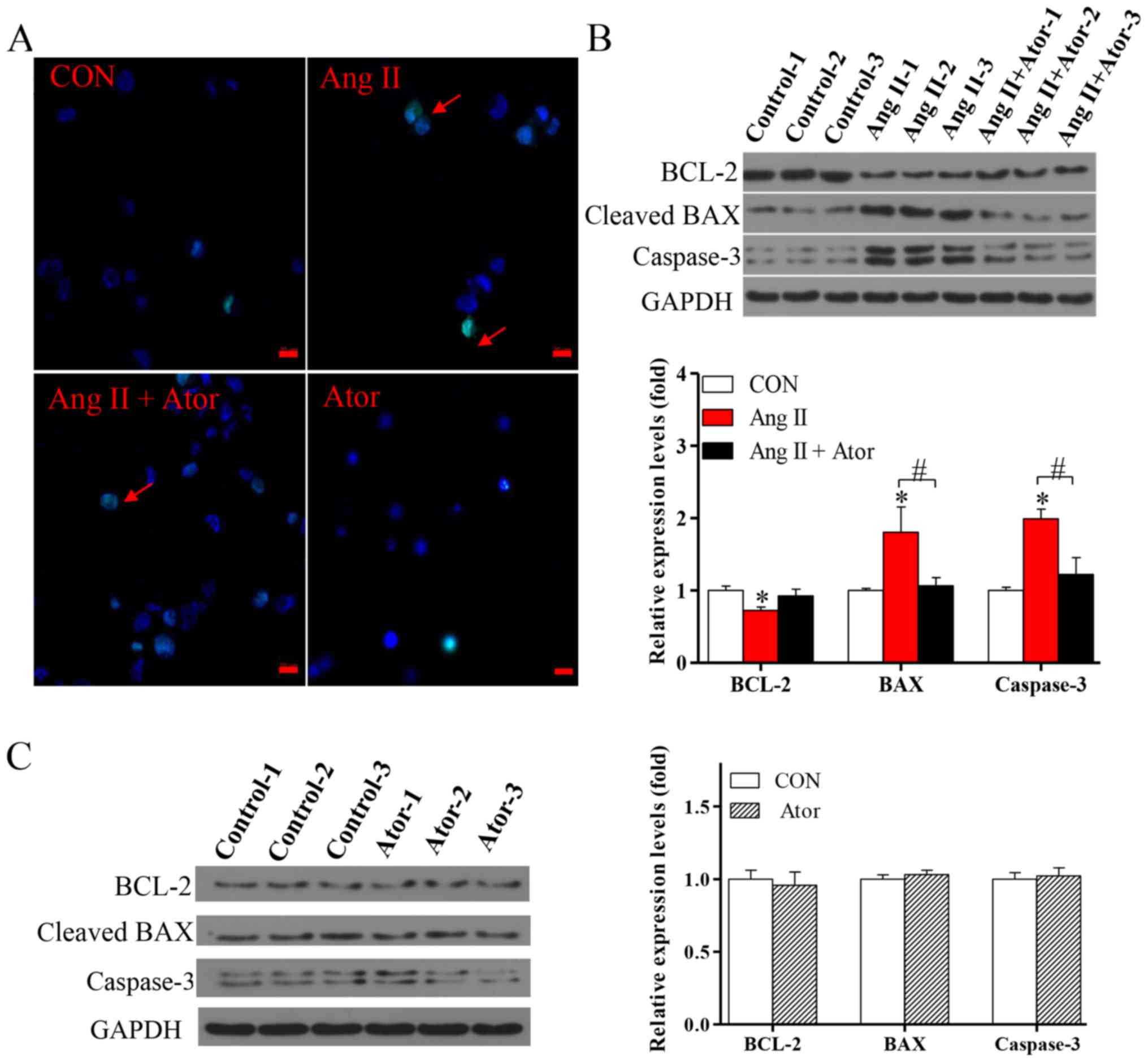 | Figure 6.Ator attenuates cell apoptosis in
HUVECs. (A) Terminal deoxynucleotidyl-transferase-mediated dUTP
nick-end labeling assay was used to examine cell apoptosis in
HUVECs following 24 h treatment with Ang II, Ator or both. Arrows
indicate apoptotic cells. The protein expression levels of BCL-2,
BAX and caspase-3 were determined using western blot analysis in
HUVECs following treatment with (B) Ang II and Ang II + Ator or (C)
Ator alone for 24 h. GAPDH was used as the loading control. Scale
bar, 20 µm. Data are presented as the mean ± standard error of the
mean, n=3. *P<0.05 vs. control group; #P<0.05 as indicated.
HUVECs, human umbilical vein endothelial cells; CON, untreated
control; Ator, atorvastatin; Ang II, angiotensin II; LC3,
microtubule-associated protein 1A/1B-light chain 3; BCL-2, B-cell
lymphoma 2; BAX, Bcl-2-associated X. |
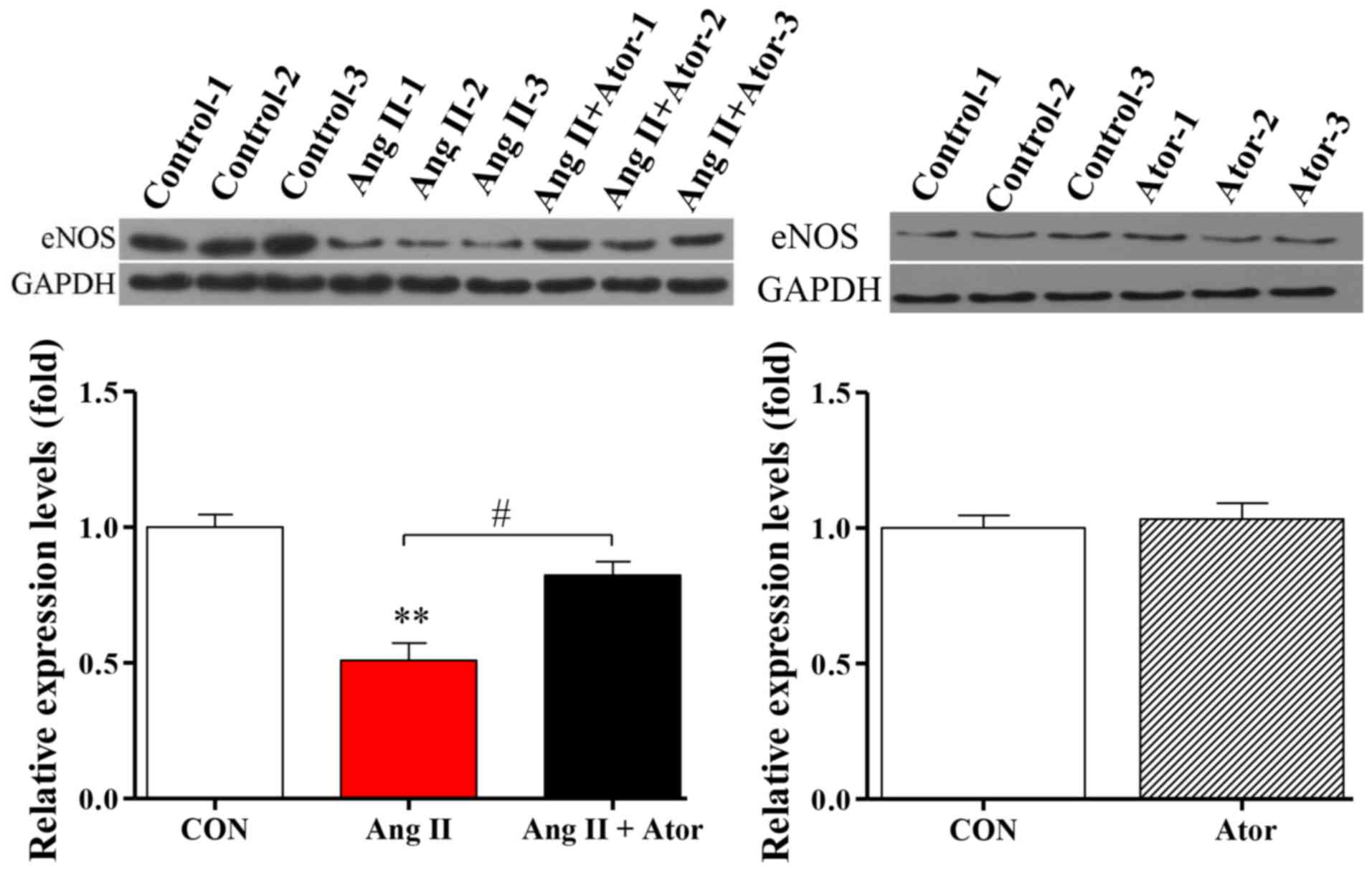
Ator modulates eNOS expression in
HUVECs
Injury can cause cellular dysfunction (25). To further investigate the biological
functions of Ang II and Ator in HUVECs, protein expression level of
eNOS, a key enzyme responsible for the proper function of the
vascular endothelium (26), was
analyzed by western blotting. Treatment with Ang II significantly
decreased the expression level of eNOS, compared with the untreated
control group (Fig. 7). The addition
of Ator significantly reversed the effects of Ang II on the
expression of eNOS. Expression levels in HUVECs treated with Ator
alone did not exhibit any significant changes compared with the
untreated control (Fig. 7).
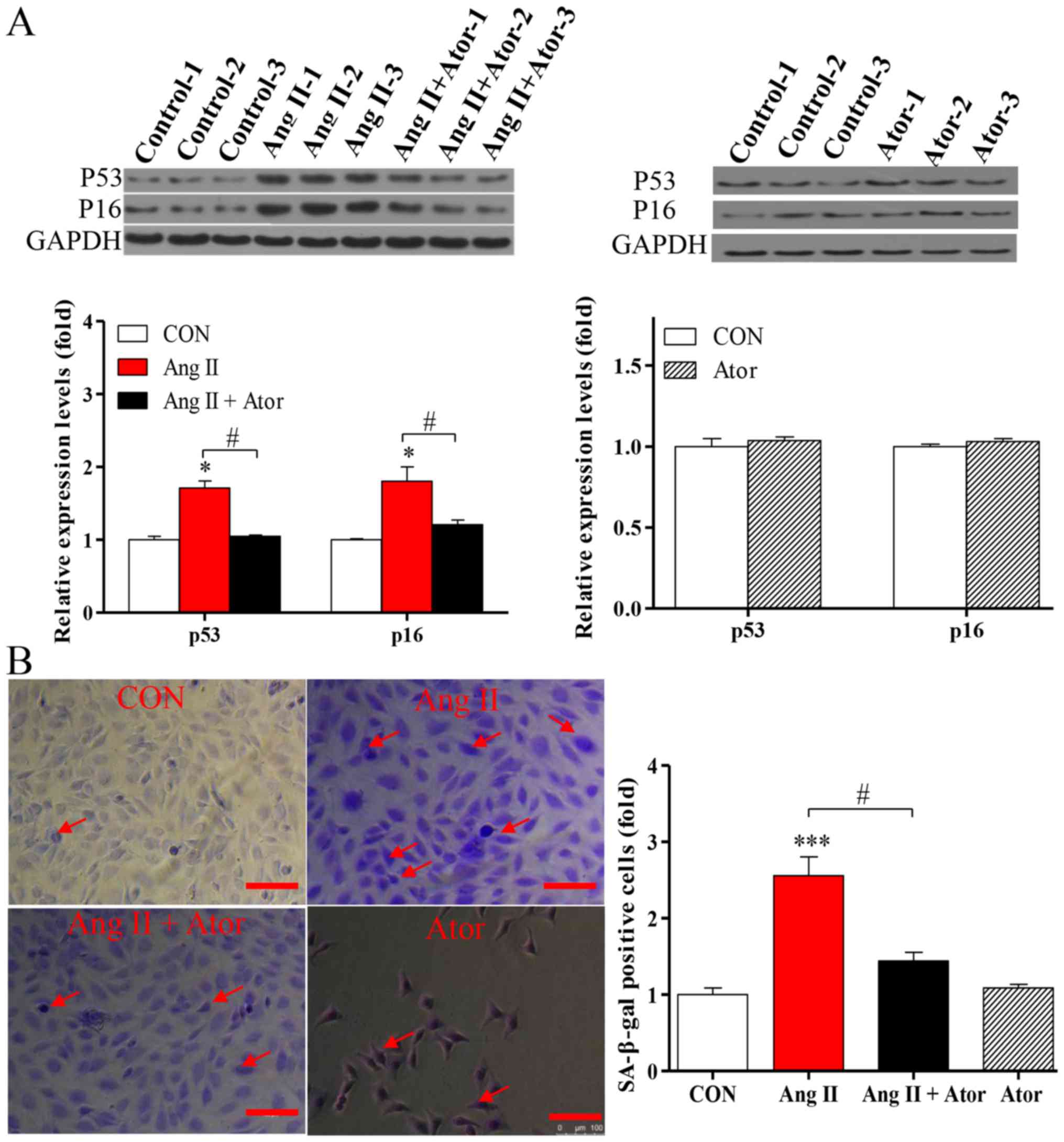
Ator delays aging in HUVECs
Apoptosis may enhance aging (19). To determine the effects of Ator on
the process of aging in HUVECs, the expression levels of
senescence-associated markers p53 and p16 were analyzed by western
blotting. Treatment with Ang II significantly increased the
expression levels of p53 and p16, compared with the untreated
control. The addition of Ator significantly reversed the effect
observed (Fig. 8A). Cells treated
with Ator alone did not exhibit any significant changes, compared
with the untreated control (Fig.
8A). These results indicated that Ang II-induced apoptosis may
be associated with the process of cellular senescence, while Ator
may be involved in delaying the aging process. To further
investigate the process of aging in HUVECs, the level of SA-β-gal
expression was analyzed by utilizing a commercial SA-β-gal staining
kit. The proportion of senescent cells was significantly increased
in HUVECs treated with Ang II (Fig.
8B). Following treatment with Ator, the proportion of senescent
cells decreased significantly compared with the Ang II-only
treatment group (Fig. 8B). These
results are consistent with those for previously described
senescence markers (27). In
addition, HUVECs treated with Ator alone did not exhibit any
significant changes in expression levels of the
senescence-associated markers compared with the untreated control
(Fig. 8B). Therefore, Ator reversed
the aging induced by Ang II, exhibiting anti-aging activity.
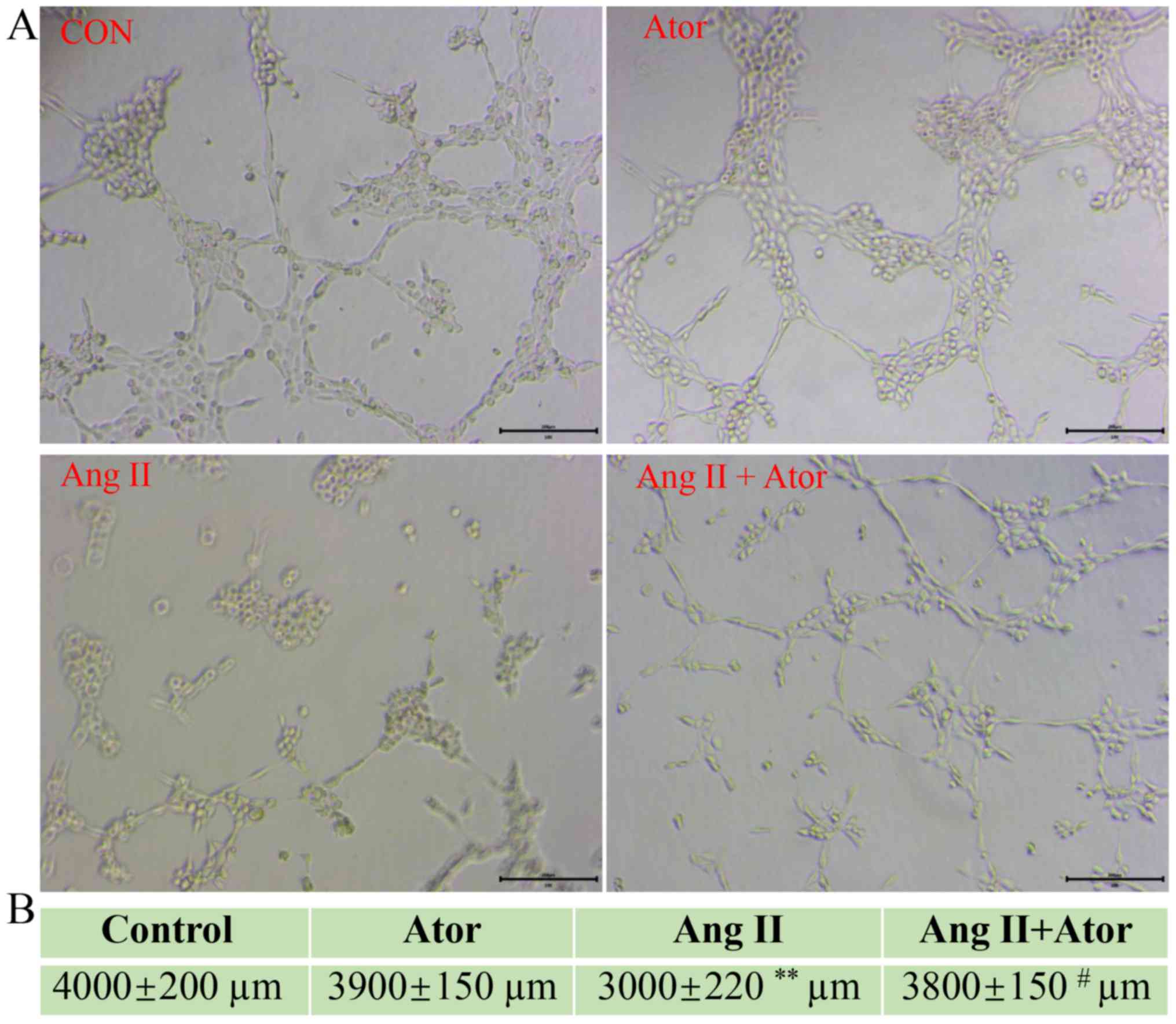
Ator reverses Ang II-induced tube
formation damage
HUVECs are the most widely used cell type used for
studying vasculature and angiogenesis (28–30).
Excessive damage to HUVECs may influence their biological function
(31). The effect of Ang II and Ator
on tube formation in HUVECs was analyzed by tube formation assay
(Fig. 9A). The length of the
tube-like vasculature of HUVECs was significantly shortened in
response to treatment with Ang II, compared with the control group
(Fig. 9B). However, the addition of
Ator significantly reversed the effect observed with Ang II alone.
Cells treated with Ator alone did not exhibit any obvious changes
in length, compared with the untreated control (Fig. 9B). The present study demonstrated the
ability of Ator to reverse Ang II-induced cellular dysfunction. A
series of cellular responses, including oxidative stress, cellular
apoptosis, inflammatory response, autophagy, expression of eNOS and
the angiogenic function of HUVECs all returned to normal following
treatment with Ator.
Discussion
As Ang II may cause damage to human umbilical vein
endothelial cells (HUVECs), an MTT assay was performed to examine
cell viability. Ang II significantly decreased HUVEC viability in a
dose-dependent manner. However, Ator reversed the Ang II-induced
cell death in a dose-dependent manner. These results demonstrated a
critical role for Ator in Ang II-induced cell death in HUVECs.
Several cellular responses were further examined in the present
study, including oxidative stress, inflammatory response, MMP and
autophagy.
ROS are well-known indicators of oxidative stress
and their expression is associated with a number of cellular
responses (7). Treatment with Ang II
induced high levels of ROS, however not when combined with Ator
treatment. Increased levels of ROS may target the cell membrane,
mitochondrial membrane or intracellular biological molecules,
causing cell damage and/or cell death (32). Mitochondria are one of the main
sources of ROS under conditions of oxidative stress (21). An increasing number of reports have
demonstrated that elevated ROS can attack mitochondria, causing
mitochondrial damage (21). This
study investigated the effects of Ang II and Ator on MMP, an
important marker of mitochondrial function (33). Following treatment with Ang II, a
significant decrease in MMP was observed and this decrease in MMP
was attenuated by the addition of Ator. The current study revealed
that exposure to Ang II caused mitochondrial stress, leading to a
loss of mitochondrial function, which was partially reversed by the
addition of Ator.
Ang II can induce chronic inflammation in cells
(34). Although the inflammatory
response may be important for preventing cell damage, chronic
inflammation is harmful for living cells (35). The present study demonstrated that
Ator attenuated Ang II-induced inflammation.
Homeostatic imbalance can lead to the activation of
several signaling pathways for the prevention of cell damage or
death. Autophagy is a well-known, regulated mechanism promoting the
survival of cells under metabolic stress (36). During autophagy, LC3 protein is
converted from its cytosolic form (LC3-I) into its enzymatic
counterpart, LC3-II, which is recruited to autophagosomal membranes
(24). In the present study, changes
in LC3-II/I expression levels revealed that Ator reversed Ang
II-induced autophagy. Autophagy can be activated during the
degradation of abnormal proteins and following damage to cellular
organelles (37). Additionally,
autophagy was generally considered a survival mechanism; however,
when damage to cells is excessive, autophagic flux can be
inhibited, initiating autophagic cell death (38). In the current study, the
pro-inflammatory response and autophagic activity results have a
similar trend, suggesting that a potential association exists
between inflammation and autophagy. Previous studies have
demonstrated that autophagy was associated with innate immunity;
autophagy can regulate inflammation by regulating the secretion of
inflammatory mediators (39–41). Autophagy may promote inflammation in
the presence of ROS-damaged mitochondria (42), which is consistent with the results
of the present study. Several reports have demonstrated that
abnormal autophagy in cells can lead to death (24,43,44). In
the present study, the cellular apoptosis assay identified Ang
II-induced cell death, which was attenuated by treatment with Ator.
By releasing cytochromes and proapoptotic-associated proteins into
the cytosol, mitochondria serve a key role in apoptosis (45). Mitochondrial dysfunction may cause an
increase in ROS, leading to cellular apoptosis (46,47). In
the present study, cellular apoptosis may be ascribed to a disorder
of the intracellular microenvironment, including an increase in
ROS, mitochondrial dysfunction and/or autophagic activity.
Furthermore, Ator may attenuate these cellular responses preventing
Ang II-induced cytotoxicity in HUVECs.
In the current study, the expression level of eNOS
in HUVECs was evaluated. The results indicated that HUVECs
expressed an abnormal level of eNOS following treatment with Ang
II. Ator may be beneficial for the normal function of HUVECs as it
was able to reverse the Ang II-induced effects on eNOS expression.
These results were consistent with previous studies in which
statins were shown to modulate the expression of endoglin, a
glycoprotein strongly associated with the expression and activity
of eNOS (48–50). The expression of eNOS may be involved
in endothelial dysfunction (51). It
may be hypothesized that Ator modulated eNOS expression in HUVECs
by regulating cellular homeostasis.
Numerous studies have demonstrated that cellular
responses, including oxidative stress, apoptosis and mitochondrial
dysfunction, may lead to accelerated aging (19,52,53). In
the current study, Ator delayed Ang II-induced cellular senescence,
efficiently reversing the aging process in HUVECs and indicating a
potential anti-aging activity.
The present study examined the angiogenic function
of HUVECs using a tube formation assay. Ang II significantly
decreased the length of the tube-like vasculature, consistent with
a previous study (30). However,
following treatment with Ator, the length of the tube-like
vasculature increased and angiogenesis function in HUVECs was
recovered. The Ang II-induced angiogenic dysfunction in HUVECs was
reversed by treatment with Ator.
There were several limitations to this study. Given
the limited study duration, there is a possibility that treatment
with Ator may be more beneficial to HUVECs at a larger dose and/or
for a longer period of time. In addition, the involvement of
several signaling pathways including, the mitogen-activated protein
kinase pathway, nuclear factor κ light chain enhancer of activated
B cells pathway, Rho kinase pathway and the protein kinase B
pathway, require further investigation. The potential anti-aging
activity of Ator should also be further evaluated.
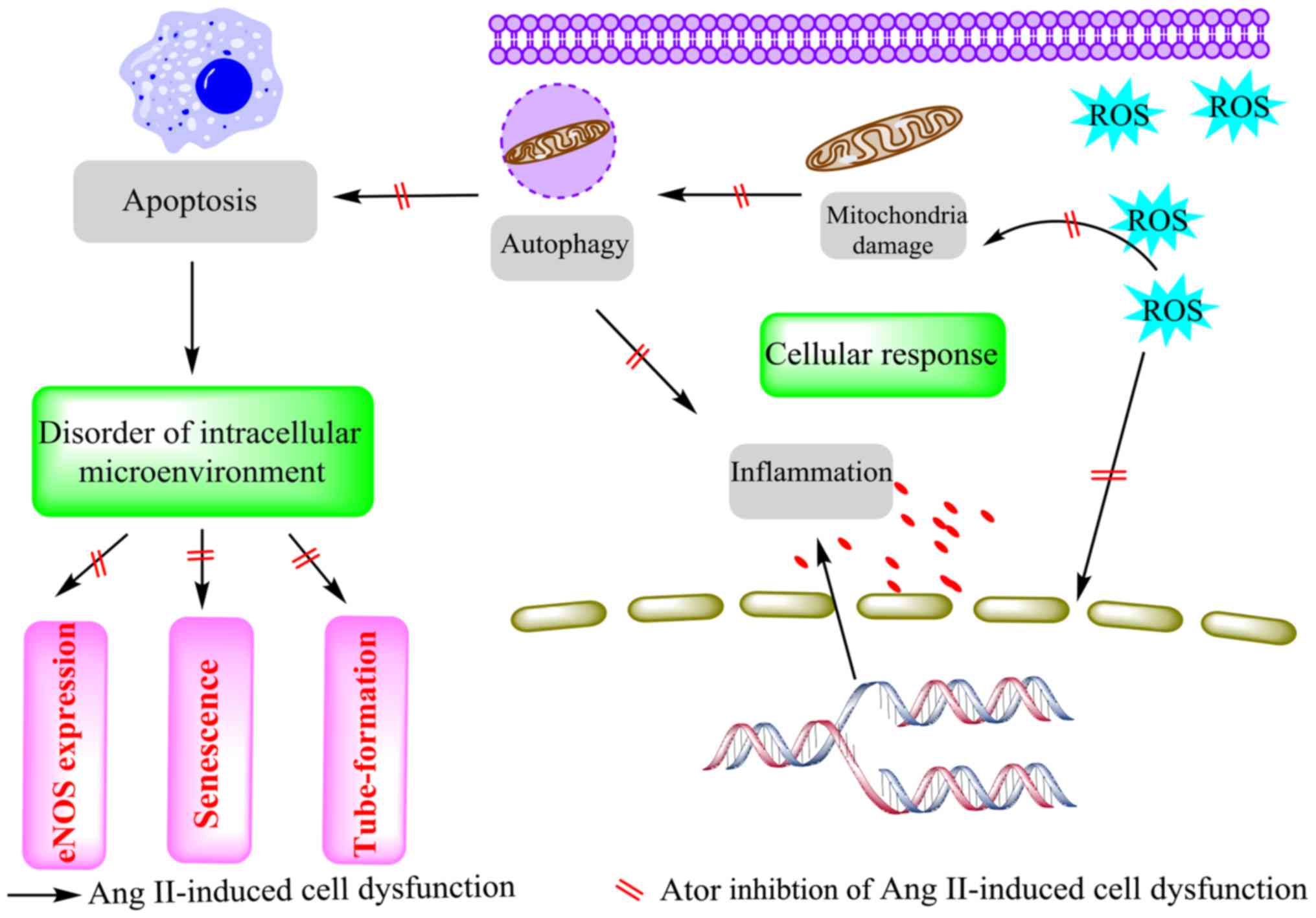
In conclusion, the results of the present study
demonstrated that Ator inhibited Ang II-induced cytotoxicity in
HUVECs by modulating a series of cellular responses (Fig. 10). In general, treatment of HUVECs
with Ang II caused cytotoxic effects. These cytotoxic effects were
associated with an increase in ROS production and significant
deficits in mitochondrial activity. These changes caused activation
of the inflammatory response, autophagy and apoptosis. Treatment
with Ator significantly reversed these adverse cellular responses
and maintained cellular homeostasis. Furthermore, eNOS expression,
which is associated with the function of HUVECs (51), was reinstated by treatment with Ator.
Alteration in the cellular microenvironment ultimately leads to
cell aging (54). In the current
study, Ator delayed Ang II-induced aging by maintaining cellular
homeostasis. Additionally, Ang II-induced dysfunction of
angiogenesis in HUVECs was reversed by the addition of Ator
treatment. As a lipid-lowering drug, atorvastatin can lower
cholesterol in the blood by inhibiting HMG-CoA reductase; however,
it may also modulate many non-lipid effects, including anti-tumor
effects, and the suppression of atherosclerosis and thrombosis.
These findings highlight the non-lipid effects of Ator and its
potential for clinical application.
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
The analyzed datasets generated during this study
are available from the corresponding author on reasonable
request.
Authors' contributions
HD and BS designed and performed the study. RD
helped analyze the data. HZ directed this paper and is the
corresponding author.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Whaley-Connell A, Johnson MS and Sowers
JR: Aldosterone: Role in the cardiometabolic syndrome and resistant
hypertension. Prog Cardiovasc Dis. 52:401–409. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Touyz RM: The role of angiotensin II in
regulating vascular structural and functional changes in
hypertension. Curr Hypertens Rep. 5:155–164. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Olkowicz M, Chlopicki S and Smolenski RT:
Perspectives for angiotensin profiling with liquid
chromatography/mass spectrometry to evaluate ACE/ACE2 balance in
endothelial dysfunction and vascular pathologies. Pharmacol Rep.
67:778–785. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gallo S, Sala V, Gatti S and Crepaldi T:
Cellular and molecular mechanisms of HGF/Met in the cardiovascular
system. Clin Sci (Lond). 129:1173–1193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kawai T, Forrester SJ, O'Brien S, Baggett
A, Rizzo V and Eguchi S: AT1 receptor signaling pathways in the
cardiovascular system. Pharmacol Res. 125:4–13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lucas AM, Caldas FR, da Silva AP, Ventura
MM, Leite IM, Filgueiras AB, Silva CG, Kowaltowski AJ and Facundo
HT: Diazoxide prevents reactive oxygen species and mitochondrial
damage, leading to anti-hypertrophic effects. Chem Biol Interact.
261:50–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim W, Yang C, Jeong M, Bazer FW and Song
G: Coumestrol induces mitochondrial dysfunction by stimulating ROS
production and calcium ion influx into mitochondria in human
placental choriocarcinoma cells. Mol Hum Reprod. 23:786–802. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meng XM, Ren GL, Gao L, Yang Q, Li HD, Wu
WF, Huang C, Zhang L, Lv XW and Li J: NADPH oxidase 4 promotes
cisplatin-induced acute kidney injury via ROS-mediated programmed
cell death and inflammation. Lab Invest. 98:63–78. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsai CY, Chen CY, Chiou YH, Shyu HW, Lin
KH, Chou MC, Huang MH and Wang YF: Epigallocatechin-3-gallate
suppresses human herpesvirus 8 replication and induces ROS leading
to apoptosis and autophagy in primary effusion lymphoma cells. Int
J Mol Sci. 19(pii): E162017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tseng CY, Wang JS and Chao MW: Causation
by diesel exhaust particles of endothelial dysfunctions in
cytotoxicity, pro-inflammation, permeability, and apoptosis induced
by ROS generation. Cardiovasc Toxicol. 17:384–392. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hong S, Kwon J, Kim DW, Lee HJ, Lee D and
Mar W: Mulberrofuran G protects ischemic injury-induced cell death
via inhibition of NOX4-mediated ROS generation and ER stress.
Phytother Res. 31:321–329. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marchi S, Giorgi C, Suski JM, Agnoletto C,
Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti
F, et al: Mitochondria-ros crosstalk in the control of cell death
and aging. J Signal Transduct. 2012:3296352012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng J, Li G, Chen S, Bihl J, Buck J, Zhu
Y, Xia H, Lazartigues E, Chen Y and Olson JE: Activation of the
ACE2/Ang-(1–7)/Mas pathway reduces oxygen-glucose
deprivation-induced tissue swelling, ROS production, and cell death
in mouse brain with angiotensin II overproduction. Neuroscience.
273:39–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schachter M: Chemical, pharmacokinetic and
pharmacodynamic properties of statins: An update. Fundam Clin
Pharmacol. 19:117–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang ZX, Wang YZ, Jia BB, Mao GX, Lv YD,
Wang GF and Yu H: Downregulation of miR-146a, cyclooxygenase-2 and
advanced glycation end-products in simvastatin-treated older
patients with hyperlipidemia. Geriatr Gerontol Int. 16:322–328.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Čarnická S, Adameová A, Nemčeková M,
Matejíková J, Pancza D and Ravingerová T: Distinct effects of acute
pretreatment with lipophilic and hydrophilic statins on myocardial
stunning, arrhythmias and lethal injury in the rat heart subjected
to ischemia/reperfusion. Physiol Res. 60:825–830. 2011.PubMed/NCBI
|
|
17
|
Davignon J: Beneficial cardiovascular
pleiotropic effects of statins. Circulation. 109(23): Suppl
1:III39–III43. 2004.PubMed/NCBI
|
|
18
|
Cui W, Matsuno K, Iwata K, Ibi M,
Katsuyama M, Kakehi T, Sasaki M, Ikami K, Zhu K and Yabe-Nishimura
C: NADPH oxidase isoforms and anti-hypertensive effects of
atorvastatin demonstrated in two animal models. J Pharmacol Sci.
111:260–268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ren C, Hu X, Li X and Zhou Q: Ultra-trace
graphene oxide in a water environment triggers Parkinson's
disease-like symptoms and metabolic disturbance in zebrafish
larvae. Biomaterials. 93:83–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Assmus B, Urbich C, Aicher A, Hofmann WK,
Haendeler J, Rössig L, Spyridopoulos I, Zeiher AM and Dimmeler S:
HMG-CoA reductase inhibitors reduce senescence and increase
proliferation of endothelial progenitor cells via regulation of
cell cycle regulatory genes. Circ Res. 92:1049–1055. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brand MD and Nicholls DG: Assessing
mitochondrial dysfunction in cells. Biochem J. 435:297–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang X, Zhang J, Guo F, Wei L and Zhou Q:
Oxidative stress and inflammatory responses in the liver of swamp
eel (Monopterus albus) exposed to carbon tetrachloride.
Aquaculture. 496:232–238. 2018. View Article : Google Scholar
|
|
23
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Wang X, Miao Y, Chen Z, Qiang P,
Cui L, Jing H and Guo Y: Magnetic ferroferric oxide nanoparticles
induce vascular endothelial cell dysfunction and inflammation by
disturbing autophagy. J Hazard Mater. 304:186–195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Q, Lai S, Hou X, Cao W, Zhang Y and
Zhang Z: Protective effects of PI3K/Akt signal pathway induced cell
autophagy in rat knee joint cartilage injury. Am J Transl Res.
10:762–770. 2018.PubMed/NCBI
|
|
26
|
Chatterjee A, Black SM and Catravas JD:
Endothelial nitric oxide (NO) and its pathophysiologic regulation.
Vascul Pharmacol. 49:134–140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen L, Xia W and Hou M: Mesenchymal stem
cells attenuate doxorubicin-induced cellular senescence through the
VEGF/Notch/TGF-β signaling pathway in H9c2 cardiomyocytes. Int J
Mol Med. 42:674–684. 2018.PubMed/NCBI
|
|
28
|
Skrzypek K, Nibbelink MG, Karbaat LP,
Karperien M, van Apeldoorn A and Stamatialis D: An important step
towards a prevascularized islet macroencapsulation device-effect of
micropatterned membranes on development of endothelial cell
network. J Mater Sci Mater Med. 29:912018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nashimoto Y, Hayashi T, Kunita I, Nakamasu
A, Torisawa Y, Nakayama M, Takigawa-Imamura H, Kotera H, Nishiyama
K, Miura T and Yokokawa R: Integrating perfusable vascular networks
with a three-dimensional tissue in a microfluidic device. Integr
Biol (Camb). 9:506–518. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kou B, Vatish M and Singer DR: Effects of
angiotensin II on human endothelial cells survival signalling
pathways and its angiogenic response. Vascul Pharmacol. 47:199–208.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Buharalioglu CK, Song CY, Yaghini FA,
Ghafoor HU, Motiwala M, Adris T, Estes AM and Malik KU: Angiotensin
II-induced process of angiogenesis is mediated by spleen tyrosine
kinase via VEGF receptor-1 phosphorylation. Am J Physiol Heart Circ
Physiol. 301:H1043–H1055. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jagadeeswaran R, Vazquez BA, Thiruppathi
M, Ganesh BB, Ibanez V, Cui S, Engel JD, Diamond AM, Molokie RE,
DeSimone J, et al: Pharmacological inhibition of LSD1 and mTOR
reduces mitochondrial retention and associated ROS levels in the
red blood cells of sickle cell disease. Exp Hematol. 50:46–52.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Márquez-Ramírez SG, Delgado-Buenrostro NL,
Chirino YI, Iglesias GG and López-Marure R: Titanium dioxide
nanoparticles inhibit proliferation and induce morphological
changes and apoptosis in glial cells. Toxicology. 302:146–156.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen L, Chen DQ, Wang M, Liu D, Chen H,
Dou F, Vaziri ND and Zhao YY: Role of RAS/Wnt/β-catenin axis
activation in the pathogenesis of podocyte injury and
tubulo-interstitial nephropathy. Chem Biol Interact. 273:56–72.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alvarenga EM, Souza LK, Araújo TS,
Nogueira KM, Sousa FB, Araújo AR, Martins CS, Pacífico DM, de C
Brito GA, Souza EP, et al: Carvacrol reduces irinotecan-induced
intestinal mucositis through inhibition of inflammation and
oxidative damage via TRPA1 receptor activation. Chem Biol Interact.
260:129–140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yorimitsu T and Klionsky DJ: Autophagy:
Molecular machinery for self-eating. Cell Death Differ. 12 (Suppl
2):S1542–S1552. 2005. View Article : Google Scholar
|
|
38
|
Li C, Liu H, Sun Y, Wang H, Guo F, Rao S,
Deng J, Zhang Y, Miao Y, Guo C, et al: PAMAM nanoparticles promote
acute lung injury by inducing autophagic cell death through the
Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol. 1:37–45. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Deretic V, Saitoh T and Akira S: Autophagy
in infection, inflammation and immunity. Nat Rev Immunol.
13:722–737. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saitoh T and Akira S: Regulation of innate
immune responses by autophagy-related proteins. J Cell Biol.
189:925–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vesa J, Su H, Watts GD, Krause S, Walter
MC, Martin B, Smith C, Wallace DC and Kimonis VE: Valosin
containing protein associated inclusion body myopathy: abnormal
vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul
Disord. 19:766–772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mishra AR, Zheng J, Tang X and Goering PL:
Silver nanoparticle-induced autophagic-lysosomal disruption and
NLRP3-inflammasome activation in HepG2 cells is size-dependent.
Toxicol Sci. 150:473–487. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang G, Wang S, Zhong L, Dong X, Zhang W,
Jiang L, Geng C, Sun X, Liu X, Chen M and Ma Y: 6-Gingerol induces
apoptosis through lysosomal-mitochondrial axis in human hepatoma G2
cells. Phytother Res. 26:1667–1673. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lane N: Mitochondrial disease: Powerhouse
of disease. Nature. 440:600–602. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bouchier-Hayes L, Lartigue L and Newmeyer
DD: Mitochondria: Pharmacological manipulation of cell death. J
Clin Invest. 115:2640–2647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Toporsian M, Gros R, Kabir MG, Vera S,
Govindaraju K, Eidelman DH, Husain M and Letarte M: A role for
endoglin in coupling eNOS activity and regulating vascular tone
revealed in hereditary hemorrhagic telangiectasia. Circ Res.
96:684–692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shyu KG, Wang BW, Chen WJ, Kuan P and Hung
CR: Mechanism of the inhibitory effect of atorvastatin on endoglin
expression induced by transforming growth factor-beta1 in cultured
cardiac fibroblasts. Eur J Heart Fail. 12:219–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Giordano A, Romano S, Monaco M, Sorrentino
A, Corcione N, Di Pace AL, Ferraro P, Nappo G, Polimeno M and
Romano MF: Differential effect of atorvastatin and tacrolimus on
proliferation of vascular smooth muscle and endothelial cells. Am J
Physiol Heart Circ Physiol. 302:H135–H142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zemankova L, Varejckova M, Dolezelova E,
Fikrova P, Jezkova K, Rathouska J, Cerveny L, Botella LM, Bernabeu
C, Nemeckova I and Nachtigal P: Atorvastatin-induced endothelial
nitric oxide synthase expression in endothelial cells is mediated
by endoglin. J Physiol Pharmacol. 66:403–413. 2015.PubMed/NCBI
|
|
52
|
von Muhlinen N, Horikawa I, Alam F,
Isogaya K, Lissa D, Vojtesek B, Lane DP and Harris CC: p53 isoforms
regulate premature aging in human cells. Oncogene. 37:2379–2393.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Isaev NK, Genrikhs EE, Oborina MV and
Stelmashook EV: Accelerated aging and aging process in the brain.
Rev Neurosci. 29:233–240. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Moruno-Manchon JF, Uzor NE, Kesler SR,
Wefel JS, Townley DM, Nagaraja AS, Pradeep S, Mangala LS, Sood AK
and Tsvetkov AS: Peroxisomes contribute to oxidative stress in
neurons during doxorubicin-based chemotherapy. Mol Cell Neurosci.
86:65–71. 2018. View Article : Google Scholar : PubMed/NCBI
|