Introduction
Lung cancer is the leading cause of
cancer-associated mortalities worldwide. Non-small cell lung cancer
(NSCLC), which constitutes 80% of lung cancer cases, and SCLC are
two major types (1,2). Lung cancer is a highly heterogeneous
disease and a large variety of factors are involved in its genesis
and progression (3). Bryant and
Cerfolio (4) have reported that
smoking is one of the most significant etiological factors that
contribute to lung cancer development and is associated with ~90%
of lung cancer cases. Furthermore, several genes have been
indicated to have a role in smoking-associated lung cancer
progression. For instance, Lv et al (5) reported that RBM5 inhibits the
proliferation of cigarette smoke-transformed BEAS-2B cells through
causing cell cycle arrest and apoptosis. Furthermore, polymorphisms
of CYPIA1 were indicated to be linked with the risk of
smoking-associated lung cancer risk in an Egyptian population
(6). However, the molecular
mechanisms underlying the smoking-associated genesis and
progression of lung cancer have remained largely elusive.
Long non-coding RNAs (lncRNAs) are a class of ncRNAs
of >200 nucleotides in length and no protein-coding function
(7). They have become a novel focus
of biological research, as they have been indicated to be important
regulators in various diseases by affecting a vast variety of
biological processes, including cell cycle, apoptosis and
differentiation (8). In lung cancer,
lncRNAs have been indicated to have an essential role in the
regulation of gene expression at the epigenetic, transcriptional
and post-transcriptional levels (9).
For instance, lncRNA HOXA distal transcript antisense RNA has been
reported to promote B-cell lymphoma-2 expression and induce
chemoresistance in SCLC by sponging microRNA (miR)-216a (10). In addition, lncRNA LINK-A interacts
with Phosphatidylinositol-3,4,5-trisphosphate [PtdIns(3,4,5)P3 or
PIP3] to hyperactivate AKT and confer resistance to AKT inhibitors
(11). However, apart from
metastasis associated lung adenocarcinoma (LUAD) transcript 1
(MALAT-1), colon cancer associated transcript 1 (CCAT-1) and long
intergenic non-coding RNA 94 (LINC00094), a limited number of
lncRNAs were identified to be associated with smoking-induced lung
cancer (12). Therefore,
identification of lncRNAs with a role in smoking-associated lung
cancer may provide novel insight to reveal mechanisms underlying
the smoking-induced genesis and progression of the malignancy.
In the present study, the public dataset GSE43458
was analyzed to identify differentially expressed lncRNAs and mRNAs
in smoking-associated lung cancer. Next, protein-protein
interaction (PPI) and co-expression networks were constructed to
identified hub mRNAs and lncRNAs in smoking-associated lung cancer.
Furthermore, gene ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway analyses were performed to explore the
potential roles of the differently expressed genes (DEGs). The
present study provides useful information to explore potential
candidate biomarkers for diagnosis, prognostication and drug
targets for smoking-associated lung cancer.
Materials and methods
Retrieval and pre-processing of
microarray data
The raw dataset GSE43458 (13) was downloaded from the gene expression
omnibus (GEO) website (https://www.ncbi.nlm.nih.gov/geo/) and pre-processed
by log2 transformation. A total of 110 samples were included in
GSE43458, which included 30 normal samples, 40 NSCLC tissues from
never-smoking patients and 40 NSCLC tissues from smoking patients.
Furthermore, The Cancer Genome Atlas (TCGA) (https://cancergenome.nih.gov/) LUAD dataset was
analyzed to identify smoking-associated miRNAs, including 46 normal
samples, 64 LUAD tissues from never-smoking patients and 372 LUAD
tissues from smoking patients. All sample data were normalized
using the linear models for microarray analysis (limma) package in
R version 3.3.0 (https://www.r-project.org/). The differentially
expressed mRNA and lncRNAs were identified by the limma method. The
DEGs were obtained with thresholds of |log fold change (FC)|>1.5
and P<0.001.
The hierarchical cluster analysis of differentially
expressed mRNAs and lncRNAs was performed using CLUSTER 3.0
(https://www.geo.vu.nl/~huik/cluster.htm), and the
hierarchical clustering heat map was visualized by Tree View
(14).
GO and KEGG pathway analysis
To identify functions of DEGs in smoking-associated
lung cancer, GO functional enrichment analysis was performed in the
categories biological process, cellular component and molecular
function. KEGG pathway enrichment analysis was also performed to
identify pathways enriched in smoking-associated lung cancer using
the Database for the Annotation, Visualization and Integrated
Discovery (DAVID; http://david.ncifcrf.gov/). P<0.05 was considered
to indicate a statistically significant difference.
lncRNA classification pipeline
In the present study, differently expressed lncRNAs
in lung cancer were identified by adopting the criteria reported by
Yang et al (15). In brief,
first, the GPL570 platform of the Affymetrix Human Genome U133 Plus
2.0 Array (Affymetrix Inc., Santa Clara, CA, USA) probe set ID was
mapped to the NetAffx Annotation Files (HG-U133 Plus 2.0
Annotations, CSV format, release 31, 08/23/10). The annotations
included the probe set ID, gene symbol and Refseq transcript ID.
Subsequently, the probe sets that were assigned a Refseq transcript
ID in the NetAffx annotations were extracted. In the present study,
only those labeled as ‘NR_’, indicating non-coding RNA in the
Refseq database, were retained. Finally, 2,448 annotated lncRNA
transcripts with corresponding Affymetrix probe IDs were generated.
lncRNAs with FC≥2 and P<0.05 were considered to be significantly
differentially expressed.
Construction of PPI network and module
analysis
In order to predict protein interactions, including
physical and functional associations, the present study used the
Search Tool for the Retrieval of Interacting Genes (STRING) to
construct the PPI network for DEGs (16). The interaction associations of the
proteins encoded by the DEGs were identified using STRING online
software, and a combined interaction score of >0.4 was used as
the cut-off criterion. In addition, Cytoscape software version
3.4.0 (http://www.cytoscape.org/) was used for
visualization of the PPI networks (17). Following the construction of the PPI
network, a module analysis of the network was performed using the
Mcode plugin (degree cut-off, ≥2; nodes with edges, ≥2-core)
(18). In addition, the Network
Analyzer was used to compute the basic properties of the PPI
network, including average clustering coefficient distribution,
closeness centrality, average neighborhood connectivity, node
degree distribution, shortest path length distribution and
topological coefficients (19).
Co-expression network construction and
analysis
In the present study, the Pearson correlation
coefficients of differently expressed mRNA-lncRNA pairs were
calculated according to their expression value. The co-expressed
DEG-lncRNA pairs with Pearson correlation coefficients with an
absolute value of ≥0.75 were selected and the co-expression network
was generated by using Cytoscape software. The Cytoscape Mcode
plug-in was applied for visualization of the co-expression
networks.
Survival analysis
Kaplan-Meier plots were generated to determine the
effect certain DEGs on patient survival (20). The patients were divided into two
groups according to the expression level of the gene of interest,
and differences in the survival rate were statistically analyzed.
The hazard ratio with 95% confidence intervals and log-rank P-value
were calculated and displayed.
Statistical analysis
The numerical data are expressed as the mean ±
standard deviation of at least three experiments. Statistical
comparisons between groups of normalized data were performed using
Student's t-test or Mann-Whitney U-test according to the test
conditions. P<0.05 was considered to indicate a statistically
significant difference with a 95% confidence level.
Results
Identification of differentially
expressed mRNAs in smoking-associated lung cancer
In the present study, the public gene expression
dataset GSE43458 from the GEO database was analyzed to identify
significantly differentially expressed RNAs between lung cancer and
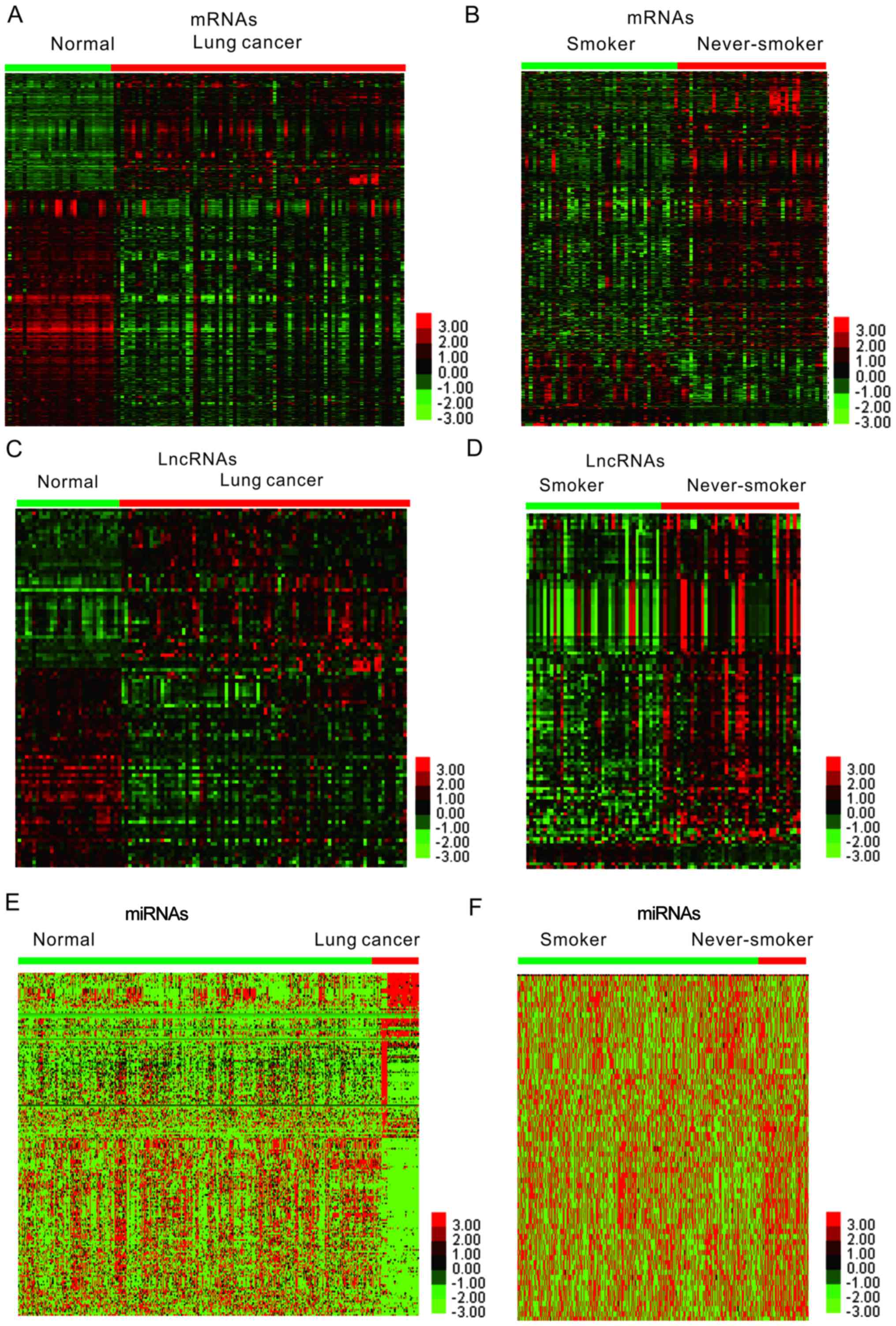
normal lung samples (Fig. 1).
Heatmaps generated by hierarchical clustering analysis of the DEGs
in lung cancer are presented in Fig. 1A
and B. A total of 729 up- and 1,485 downregulated mRNAs were
identified (Fig. 1A). Furthermore,
comparison of gene expression patterns between lung cancer samples
of smokers and non-smokers identified 610 mRNAs in the GSE43458
dataset (Fig. 1B).
smoking-associated
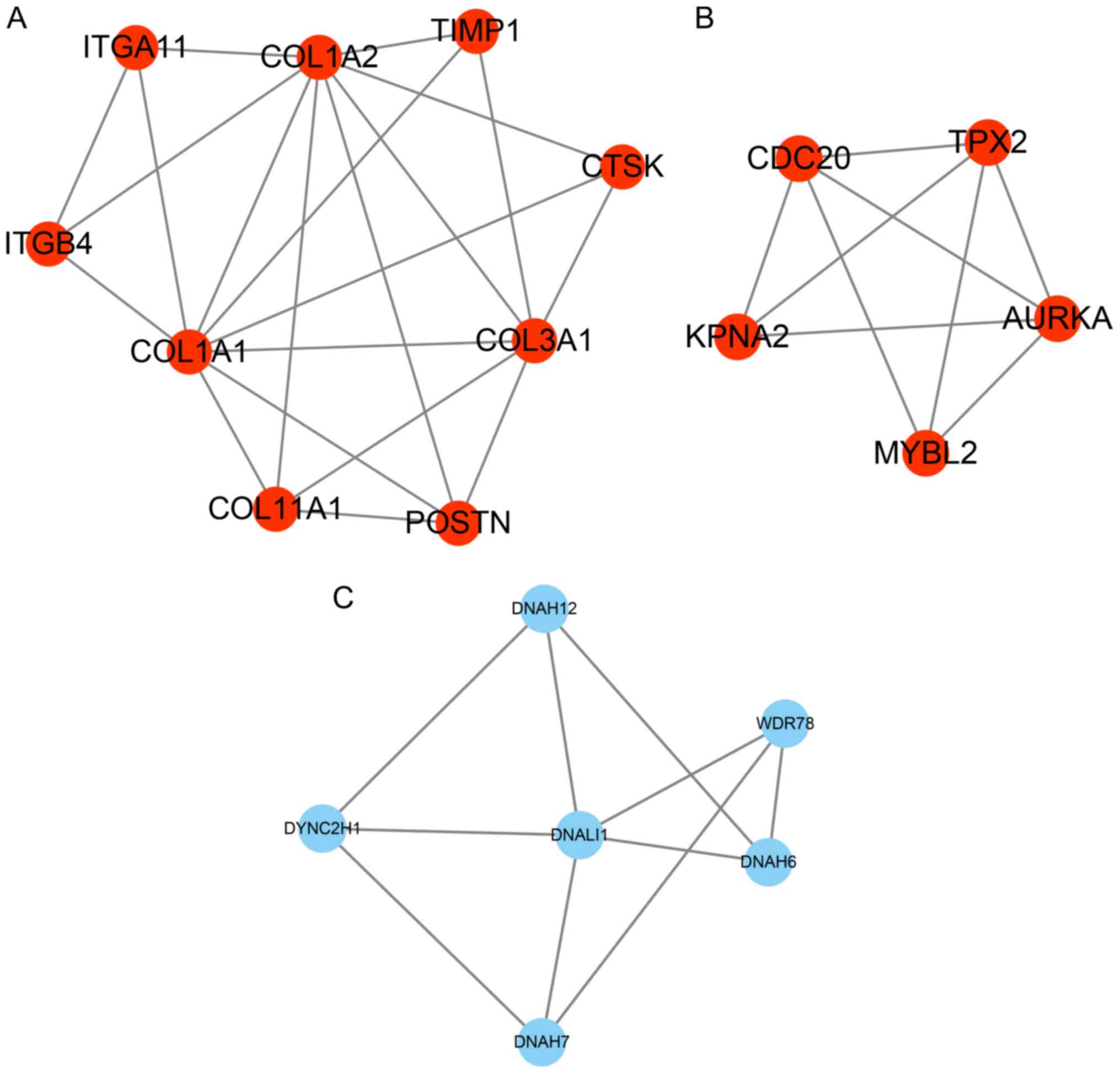
PPI network construction
PPI networks were constructed to predict the
interaction association among the 135 up- and 179 downregulated
proteins in smoking-associated lung cancer (combined score,
>0.4) by using the STRING database. A module analysis of the
network was then performed using the Mcode plugin (degree cut-off,
≥2; nodes with edges, ≥2-core). The PPI networks were constructed
by using Cytoscape and presented in Fig.
2. For the upregulated genes in smoking-associated lung cancer,
2 distinct hub networks were identified, while 1 hub network was
identified for the downregulated genes in smoking-associated lung
cancer. Periostin (POSTN), collagen type III α 1 chain (COL3A1),
COL1A1, COL1A2, cathepsin K (CTSK), integrin subunit β 4 (ITGB4),
tissue inhibitor of metalloproteinases 1 (TIMP1), ITGA11, COL11A1,
MYB proto-oncogene like 2 (MYBL2), karyopherin subunit α 2 (KPNA2),
aurora kinase A (AURKA), TPX2, microtubule nucleation factor (TPX2)
and cell division cycle 20 (CDC20) were identified as key
upregulated genes in smoking-associated lung cancer (Fig. 2A and B). Furthermore, a total of 6
genes [dynein axonemal heavy chain 7 (DNAH7), dynein cytoplasmic 2
heavy chain 1 (DYNC2H1), WD repeat domain 78 (WDR78), DNAH6, DNAH12
and dynein axonemal light intermediate chain 1 (DNALI1)] were the
key downregulated genes in smoking-associated lung cancer (Fig. 2C).
Functional analysis of deregulated
genes in smoking-associated lung cancer
In order to explore the functional roles of the hub
genes in smoking-associated lung cancer, the DepMap dataset
(https://depmap.org/portal/depmap/)
was analyzed. DepMap aims to identify novel diagnostic and
therapeutic targets for human cancers by integrating large-scale
datasets, including CRISPR-associated protein 9 nuclease screening
and small hairpin RNA screening. As presented in Fig. 3, by analyzing CRISPR (Broad Avana) in
the DepMap dataset, it was observed that knockout of CTSK, ITGA11,
MYBL2, KPNA2, DNAH7, AURKA and TPX2 significantly suppressed
(CERES<0) and knockout of DNAH7 significantly promoted
(CERES>0) the proliferation of human cancer cells, including
lung cancer.
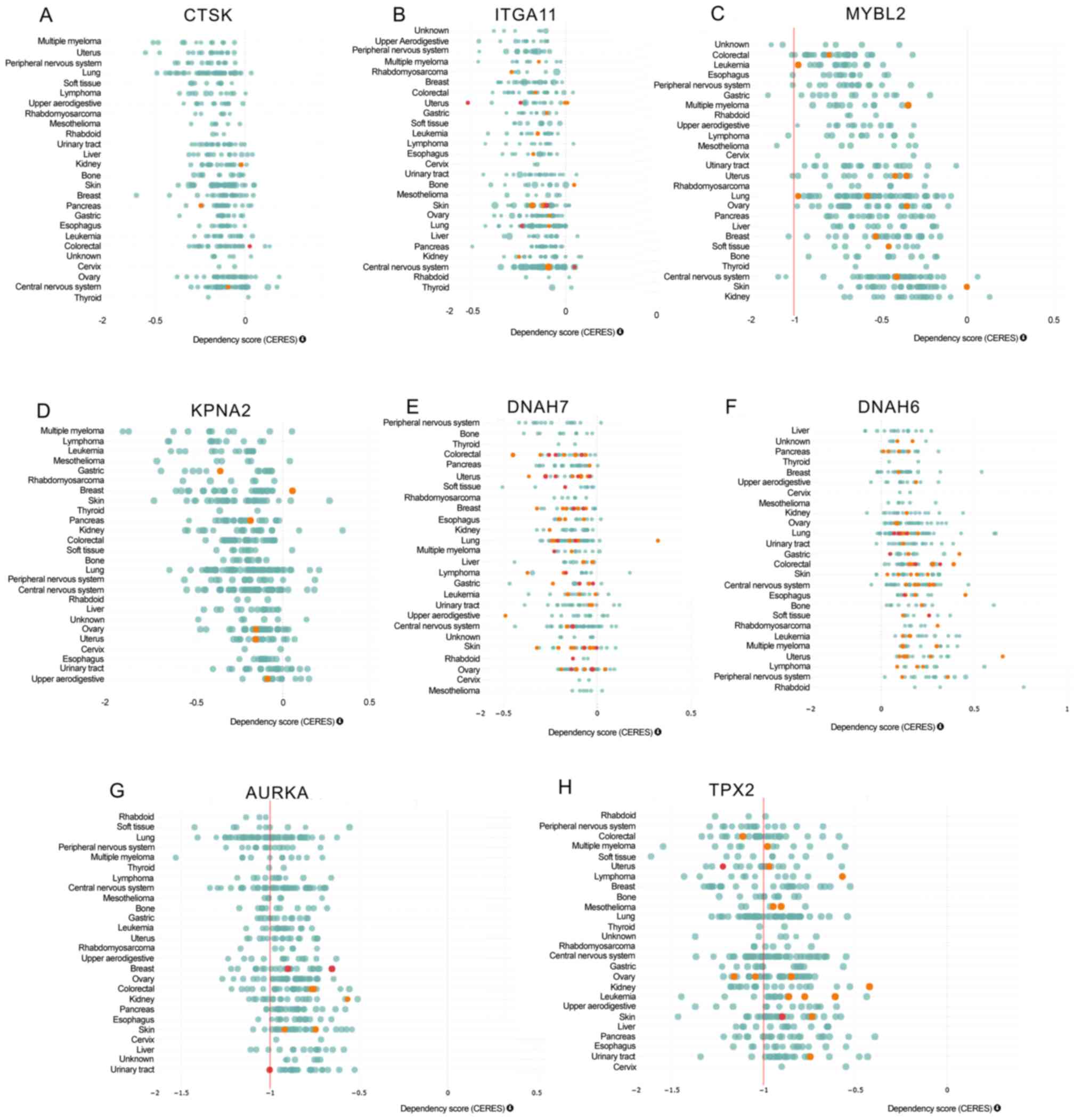 | Figure 3.Functional analysis of deregulated
genes in smoking-associated lung cancer. The DepMap analysis
indicates that knockout of (A) CTSK, (B) ITGA11, (C) MYBL2, (D)
KPNA2, (E) DNAH7, (F) AURKA and (G) TPX2 significantly suppressed
(CERES<0) and (H) knockout of DNAH7 significantly promoted
(CERES>0) the proliferation of human cancer cells, including
lung cancer. CTSK, cathepsin K; ITGA11, integrin subunit α 11;
MYBL2, MYB proto-oncogene like 2; KPNA2, karyopherin subunit α 2;
DNAH7, dynein axonemal heavy chain 7; AURKA, aurora kinase A; TPX2,
TPX2, microtubule nucleation factor. |
Influence of deregulated genes in
smoking-associated lung cancer on patient survival
To evaluate the prognostic value of 20 hub genes
selected by Mcode, Kaplan-Meier-analysis was employed. The median
expression level of each target gene among all lung cancer samples
was used as the cut-off point to divide all cases into high and low
expression groups. The overall survival rates were observed to be
lower in AURKA-high compared to AURKA-low expression groups of lung
cancer patients, and the same trends were identified for ITGB4,
COL1A1, CDC20, MYBL2, POSTN, COL11A1, TPX2, COL3A1 and TIMP1
(P<0.05; Fig. 4A-J). Furthermore,
the results indicated that a high mRNA expression of DNALI1, DNAH6,
CTSK, DYNC2H1, DNAH12 and WDR78 was associated with better overall
survival of lung cancer patients (Fig.
4K-P).
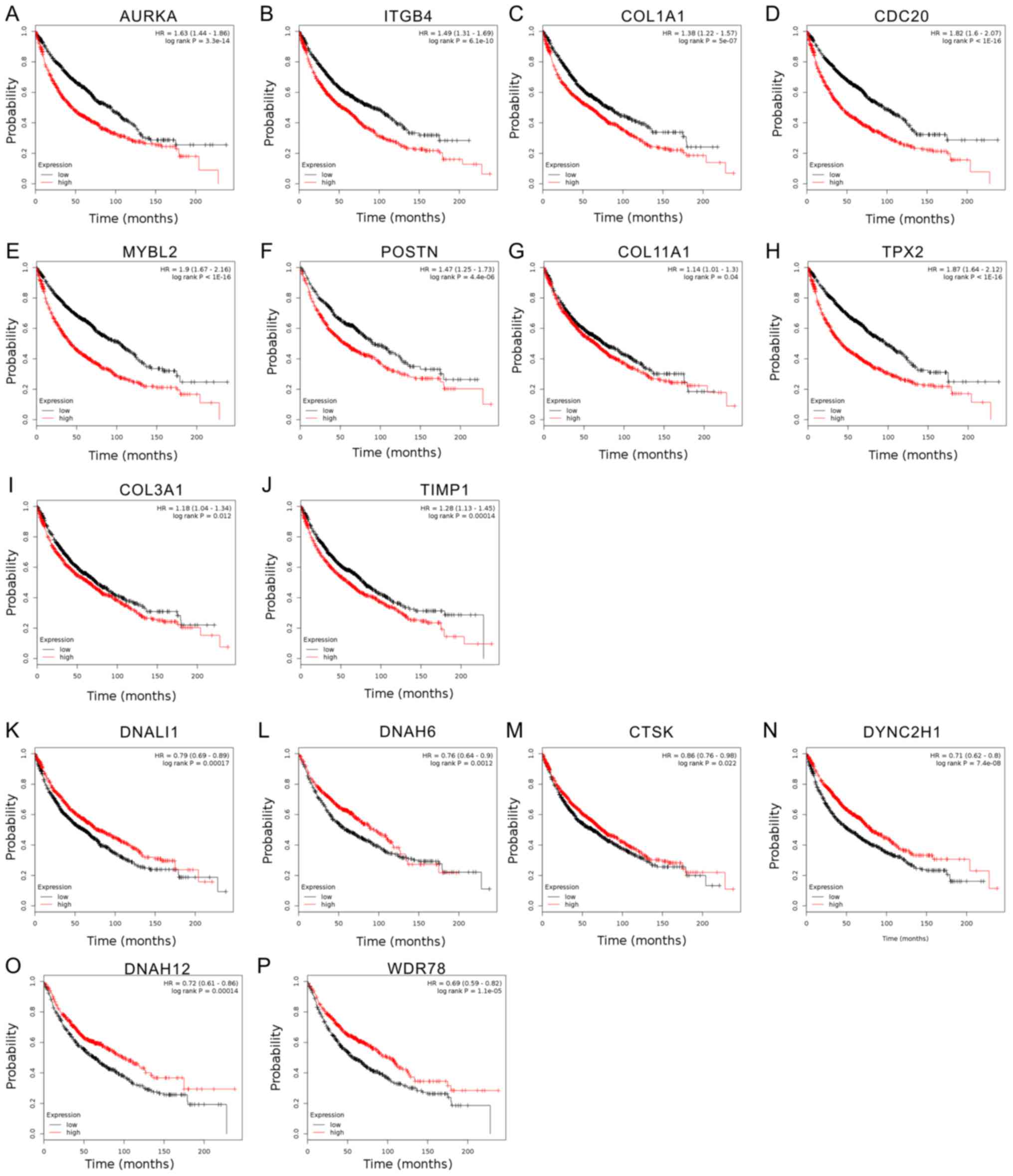 | Figure 4.Analysis of the influence of
deregulated genes in smoking-associated lung cancer on survival of
lung cancer patients. Kaplan-Meier analysis of the effect of 16 hub
genes, including (A) AURKA, (B) ITGB4, (C) COL1A1, (D) CDC20, (E)
MYBL2, (F) POSTN, (G) COL11A1, (H) TPX2, (I) COL3A1, (J) TIMP1, (K)
DNALI1, (L) DNAH6, (M) CTSK, (N) DYNC2H1, (O) DNAH12 and (P) WDR78,
on the survival of lung cancer patients. AURKA, aurora kinase A;
ITGB4, integrin subunit β 4; COL3A1, collagen type III α 1 chain;
CDC20, cell division cycle 20; MYBL2, MYB proto-oncogene like 2;
POSTN, periostin; TPX2, TPX2, microtubule nucleation factor; TIMP1,
tissue inhibitor of metalloproteinases 1; DNAH6, dynein axonemal
heavy chain 6; DNALI1, dynein axonemal light intermediate 1; CTSK,
cathepsin K; DYNC2H1, dynein cytoplasmic 2 heavy chain 1; WDR78, WD
repeat domain 78. |
Identification of differently
expressed lncRNAs in smoking-associated lung cancer
Various lncRNAs have been revealed to be associated
with the progression of numerous different types of human cancer,
including lung cancer. However, few studies have focused on
smoking-associated lncRNAs in lung cancer. In the present study, an
lncRNA classification pipeline was used to identify differently
expressed lncRNAs in smoking-associated lung cancer. Heatmaps
generated by hierarchical clustering analysis of the differentially
expressed lncRNAs in lung cancer are presented in Fig. 1C and D.
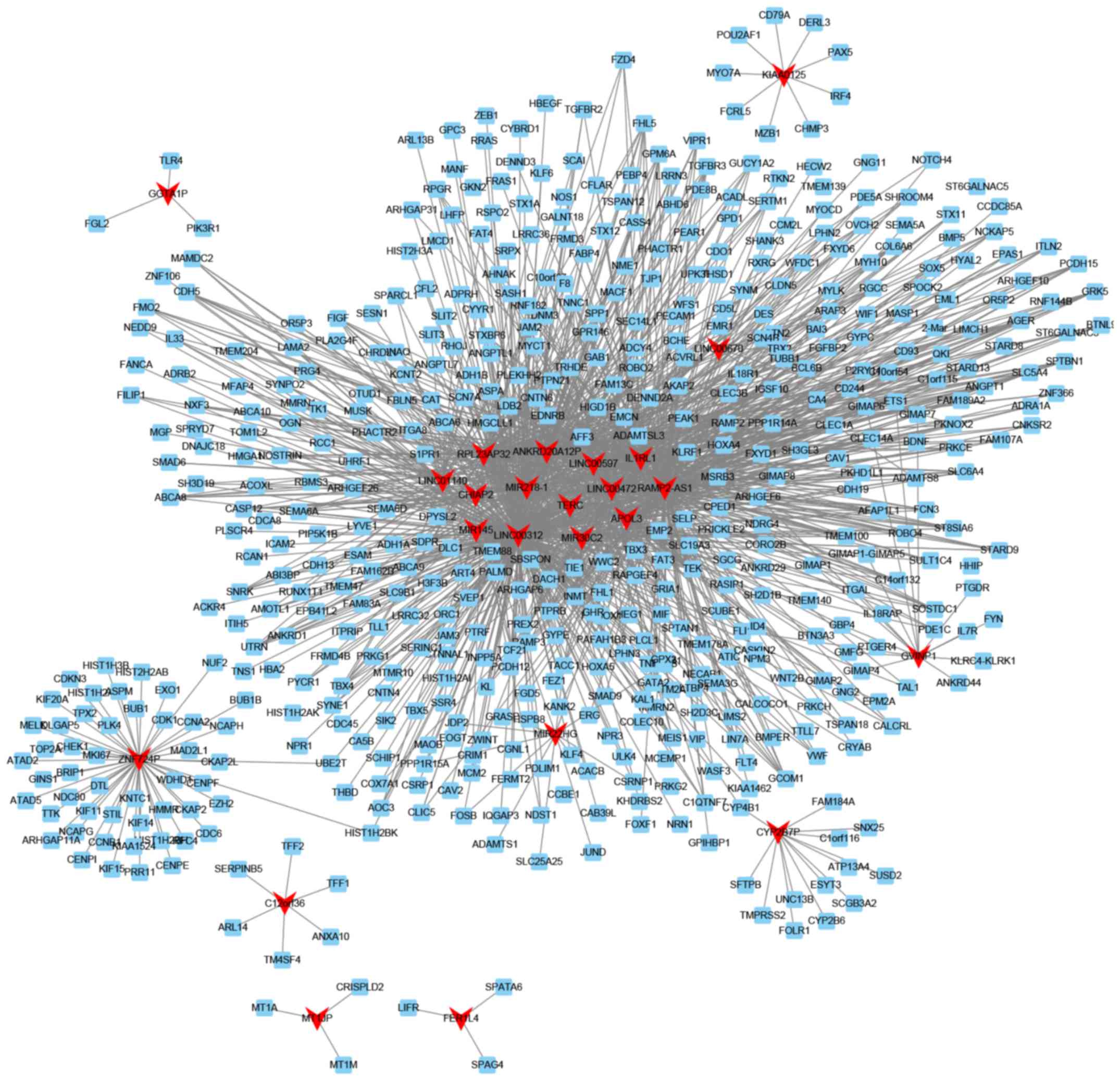
Co-expression network analysis of
lncRNAs in smoking-associated lung cancer
To predict the potential functional roles of these
lncRNAs, the Pearson correlation coefficient of lncRNA-mRNA pairs
was first calculated. The co-expressed mRNA-lncRNA pairs with an
absolute value of the Pearson correlation coefficient of ≥0.75 were
selected to construct co-expression networks with Cytoscape. The
network presented in Fig. 5 includes
24 lncRNAs and 580 mRNAs. Certain lncRNAs, including relaxin family
peptide receptor 1 (RXFP1), receptor activity modifying protein
2-antisense RNA 1 (RAMP2-AS1), LINC00312 and LINC00472, were
co-expressed with >100 mRNAs with an absolute value of the
Pearson correlation coefficient of ≥0.75. These lncRNAs were
identified as key lncRNAs in smoking-associated lung cancer.
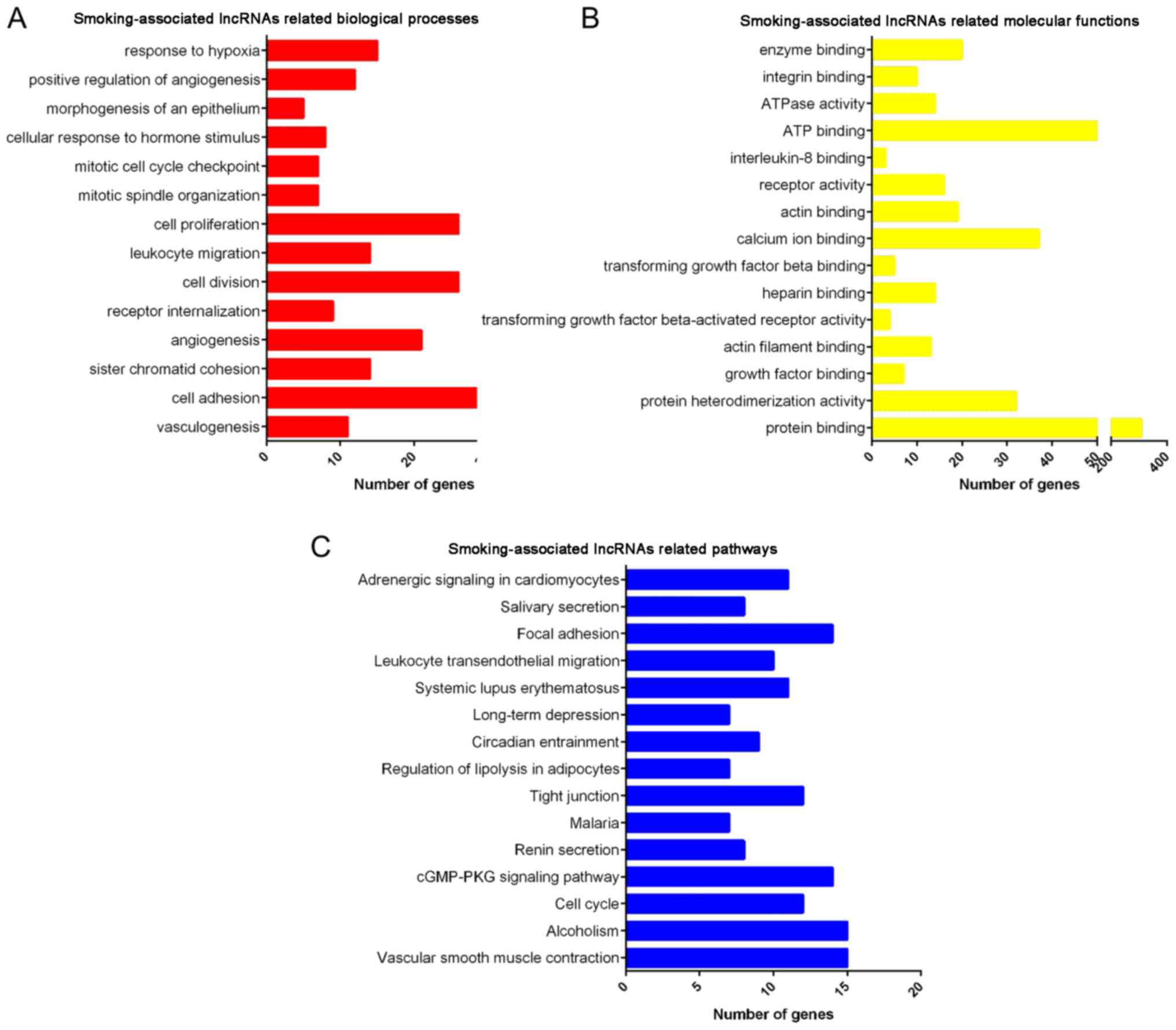
GO and KEGG analysis of deregulated
lncRNAs in smoking-associated lung cancer
Furthermore, GO and KEGG enrichment analyses were
performed for the differentially expressed lncRNAs (Fig. 5). GO analysis revealed that lncRNAs
that were deregulated in smoking-associated lung cancer were mainly
involved in regulating vasculogenesis, cell adhesion, sister
chromatid cohesion, angiogenesis, receptor internalization, cell
division, leukocyte migration, cell proliferation, mitotic spindle
organization and mitotic cell cycle checkpoint (Fig. 6A). Furthermore, it was identified
that the deregulated lncRNAs were enriched in GO terms in the
category molecular function associated with protein binding,
protein heterodimerization activity, growth factor binding, actin
filament binding and transforming growth factor β-activated
receptor activity (Fig. 6B).
KEGG pathway analysis revealed that the deregulated
lncRNAs were primarily enriched in pathways associated with
vascular smooth muscle contraction, alcoholism, cell cycle, cyclic
guanosine monophosphate cGMP)/protein kinase cGMP-dependent 1
signaling pathway, renin secretion, malaria, tight junction,
regulation of lipolysis in adipocytes, circadian entrainment and
long-term depression (Fig. 6C).
Key lncRNAs downregulated in
smoking-associated lung cancer
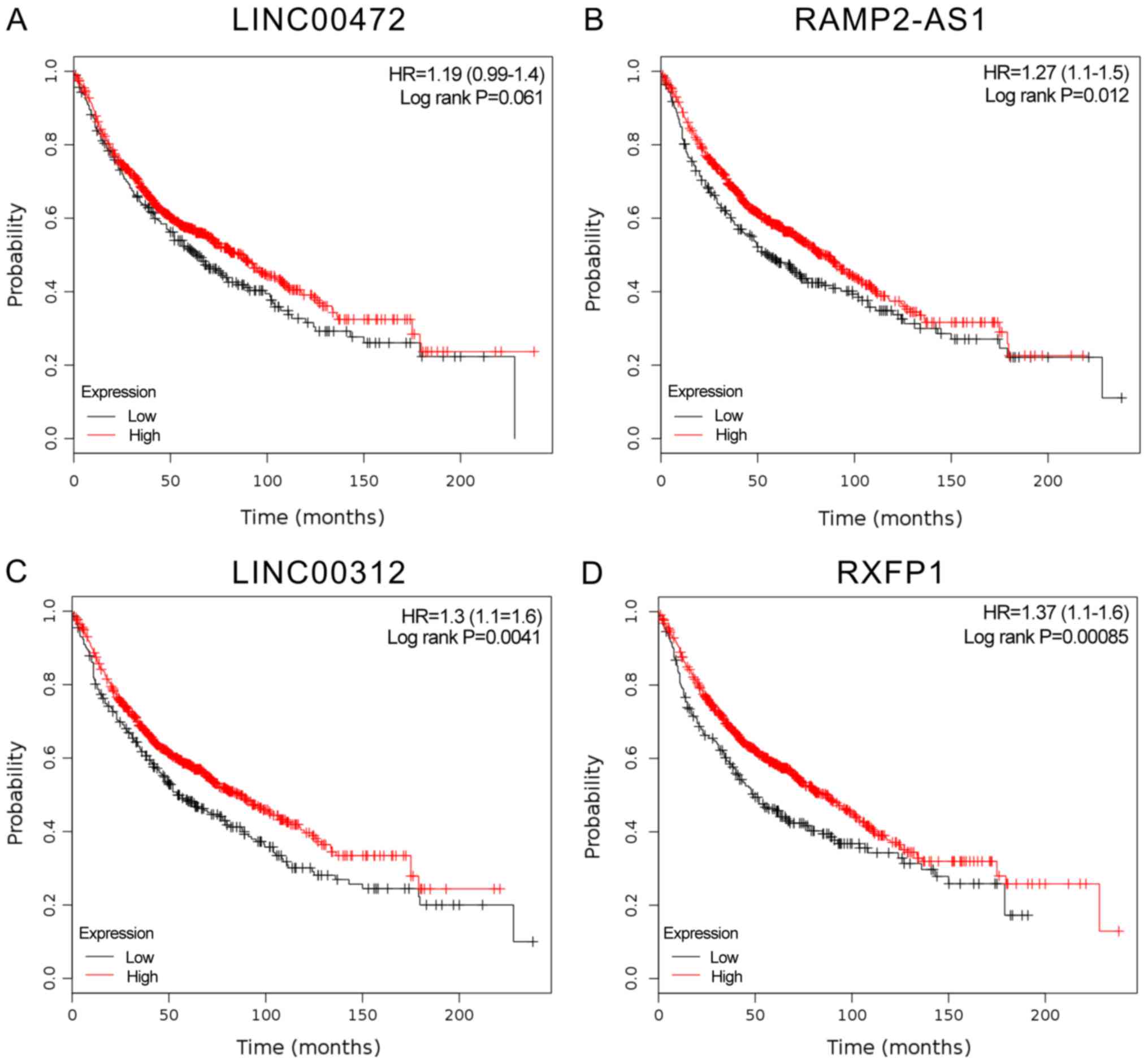
In the present study, RXFP1, RAMP2-AS1, LINC00312
and LINC00472 were identified as key lncRNAs in smoking-associated
lung cancer. However, their prognostic value and functional roles
in lung cancer have remained elusive. Therefore, their specific
expression patterns were analyzed in other public datasets from
TCGA. The results indicated that RXFP1, RAMP2-AS1, LINC00312 and
LINC00472 may act as tumor suppressors and were significantly
downregulated in LUAD (Fig. 7).
Furthermore, Kaplan-Meier-analysis was used to reveal their
potential prognostic value. As presented in Fig. 7, higher expression levels of RXFP1,
RAMP2-AS1, LINC00312 and LINC00472 were significantly associated
with a longer overall survival time (Fig. 8).
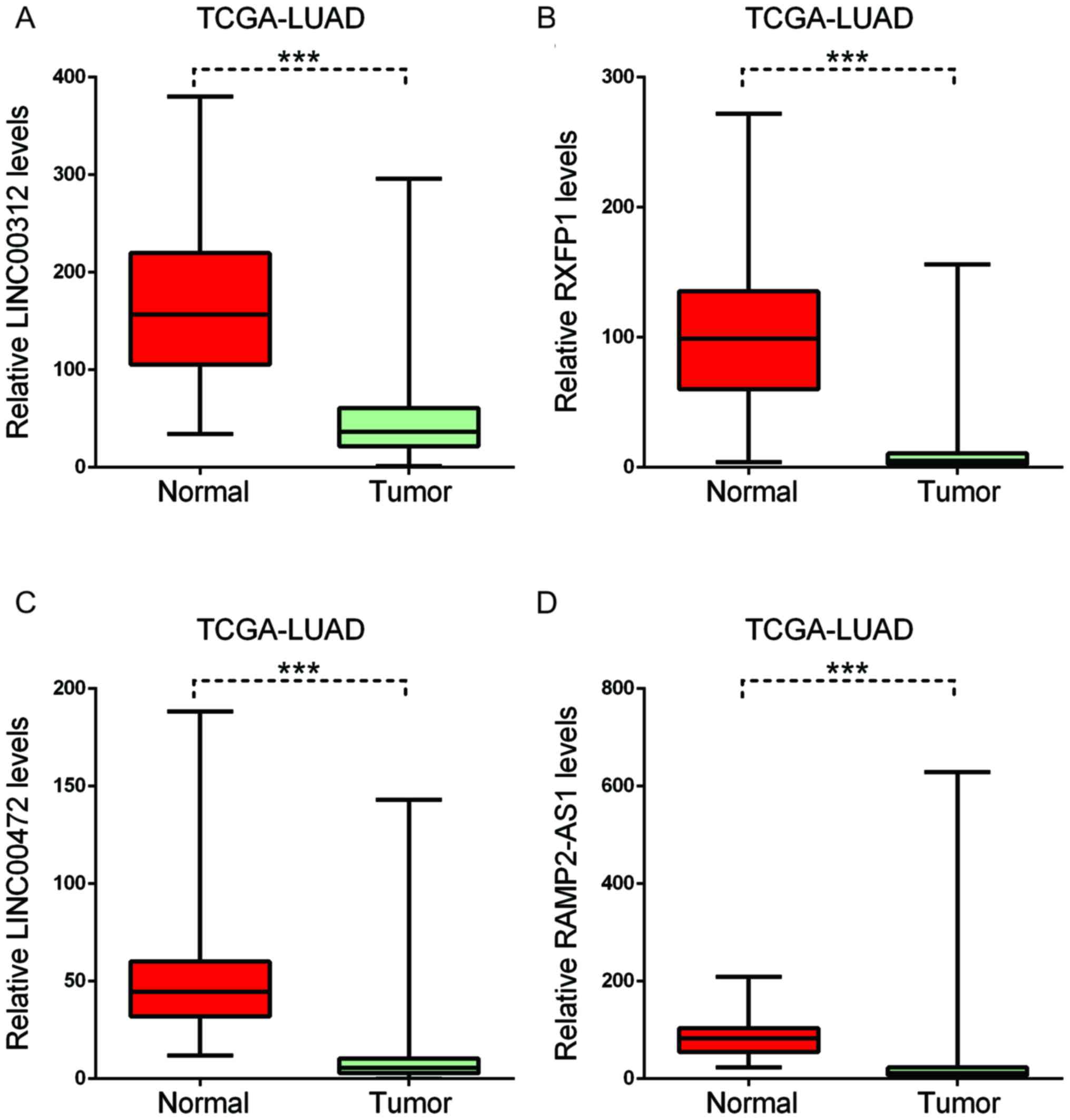 | Figure 7.Key long non-coding RNAs in
smoking-associated lung cancer were downregulated. In LUAD, (A)
LINC00312, (B) RXFP1, (C) LINC00472 and (D) RAMP2-AS1 were
downregulated. Boxplots indicate the minimum, maximum, median and
upper and lower quartiles of gene expression levels in each group.
The red boxplot indicates gene expression in normal groups, the
blue boxplot indicates gene expression in tumor groups.
***P<0.001. LUAD, lung adenocarcinoma; LUSC, lung squamous cell
carcinoma; TCGA, The Cancer Genome Atlas; RXFP1, relaxin family
peptide receptor 1; RAMP2-AS1, receptor activity modifying protein
2-antisense RNA 1; LINC00312, long intergenic non-protein coding
RNA 312. |
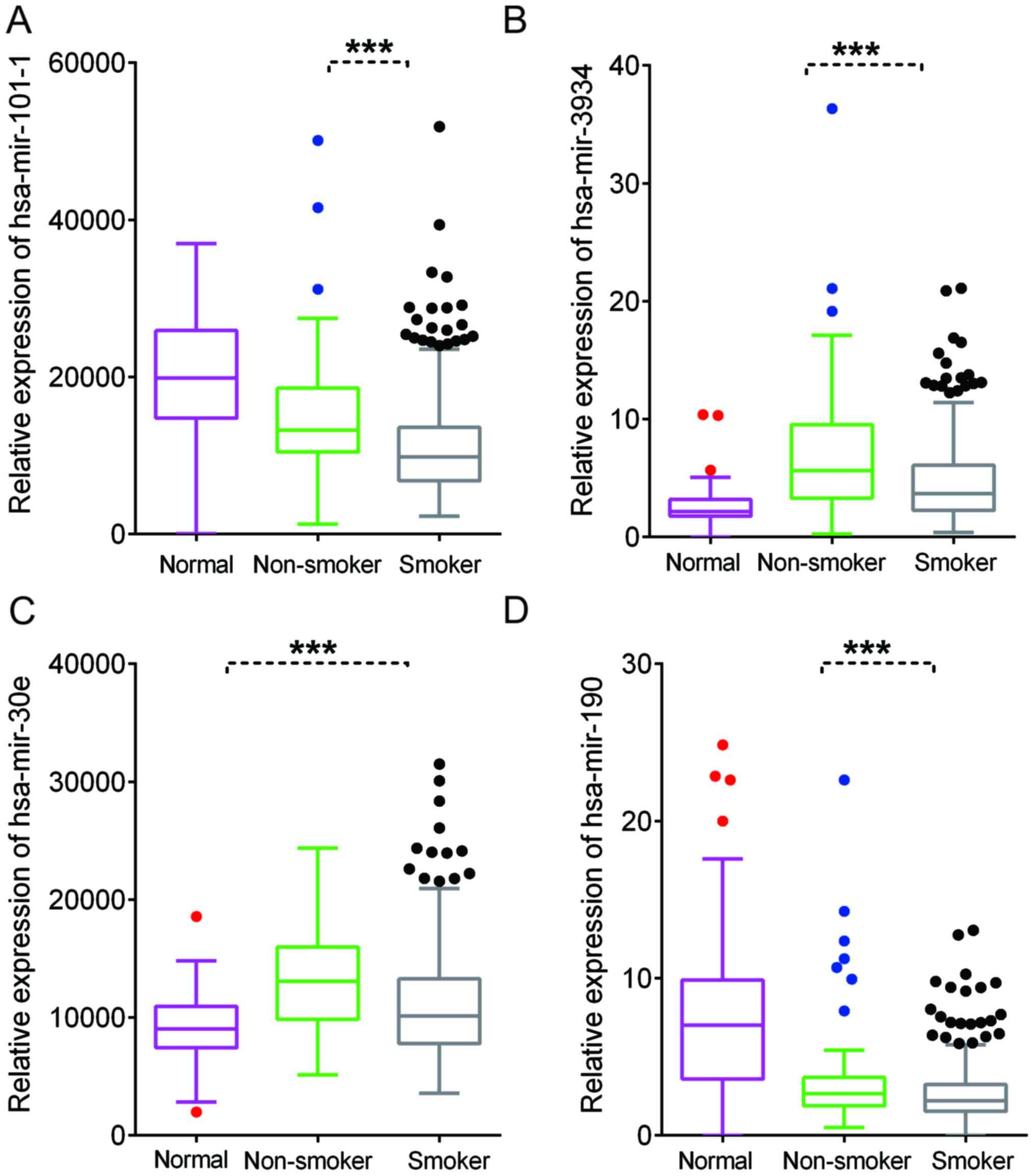
Identification of differently
expressed miRNAs in smoking-associated lung cancer
Emerging studies have demonstrated that miRNAs have
crucial roles in lung cancer. In the present study, the TCGA LUAD
dataset was analyzed to identify differently expressed miRNAs in
smoking-associated lung cancer. A total of 53 miRNAs were
identified to be dysregulated in lung cancer samples of smokers.
Furthermore, 107 miRNAs were identified to be upregulated and 101
miRNAs were indicated to be downregulated in lung cancer compared
with normal samples (Fig. 1E-F).
Finally, Homo sapiens (hsa)-miR-3934, hsa-miR-101-1,
hsa-miR-30e and hsa-miR-190 were identified as differently
expressed miRNAs in smoking-associated lung cancer (Fig. 9).
Discussion
Lung cancer is the leading cause of
cancer-associated mortalities worldwide. Smoking is one of the most
significant etiological contributors to lung cancer development
(21). However, the molecular
mechanisms underlying smoking-induced initiation and progression of
lung cancer have remained largely elusive. In present study, the
public dataset GSE43458 was analyzed, and 135 up- and 179
downregulated mRNAs in smoking-associated lung cancer were
identified. Furthermore, PPI networks were constructed to identify
hub genes. A total of 6 downregulated genes (DNAH7, DYNC2H1, WDR78,
DNAH6, DNAH12 and DNALI1) and 14 upregulated genes (POSTN, COL3A1,
COL1A1, COL1A2, CTSK, ITGB4, TIMP1, ITGA11, COL11A1, MYBL2, KPNA2,
AURKA, TPX2 and CDC20) were identified as hub genes in
smoking-associated lung cancer. Of note, AURKA has been previously
reported to be upregulated in the tumor tissues of smoking patients
(22). Furthermore, the DepMap
dataset was analyzed to evaluate the potential functions of these
hub genes, revealing that CTSK, ITGA11, MYBL2, KPNA2, DNAH7, AURKA,
TPX2 and DNAH7 are involved in regulating the proliferation of
human cancer cells, including lung cancer. Most of these hub genes
were identified to be associated with smoking-associated lung
cancer for the first time in the present study, to the best of our
knowledge.
miRNAs and lncRNAs have been indicated to serve as
important regulators in various diseases, including lung cancer, by
affecting various biological processes, including cell cycle,
apoptosis and invasion. For instance, lncRNA small nucleolar RNA
host gene 20 was reported to promote NSCLC cell proliferation and
migration by epigenetically silencing P21 expression (23). lncRNA MetaLnc9 was also reported to
facilitate lung cancer metastasis via the phosphoglycerate kinase
1-activated AKT/mammalian target of rapamycin pathway (24). Furthermore, several lncRNAs,
including MALAT1, CCAT1 and LINC00094, were identified to be
associated with smoking-induced lung cancer (12). In present study, differently
expressed genes were screened in lung cancer tissues of smokers vs.
non-smokers. A total of 11 up- and 13 downregulated lncRNAs in
smoking-associated lung cancer were identified in the present
study. TCGA LUAD dataset was screened to identify hsa-miR-3934,
hsa-miR-101-1, hsa-miR-30e and hsa-miR-190 as differentially
expressed miRNAs in smoking-associated lung cancer. Furthermore, a
co-expression network analysis revealed that certain key lncRNAs,
including RXFP1, RAMP2-AS1, LINC00312 and LINC00472. A GO and KEGG
enrichment analysis indicated that these lncRNAs were associated
with vasculogenesis, cell adhesion, sister chromatid cohesion,
angiogenesis, receptor internalization, cell division, cell
proliferation, cell cycle and cGMP/PKG signaling pathway.
In previous studies, LINC00312 and LINC00472 have
been indicated to be associated with lung cancer progression. For
instance, Zhu et al (25)
reported that LINC00312 was downregulated in NSCLC tissues and
correlated with a poor clinical outcome. Functional experiments
indicated that LINC00312 may inhibit cell proliferation and promote
apoptosis in vitro and in vivo. Furthermore, Tian
et al (26) observed that
LINC00312 is downregulated in lung cancer. The functional roles of
LINC00472 in lung cancer have been revealed by bioinformatics
analyses. For instance, Sui et al (27) and Zhu et al (28) reported that LINC00472 was
downregulated in lung cancer by constructing an lncRNA-mediated
competitive endogenous RNA network. To the best of our knowledge,
the present study was the first to identify that LINC00312 and
LINC00472 are associated with smoking-induced lung cancer.
In the present study, the prognostic value of the
hub genes and lncRNAs in lung cancer was determined. Kaplan-Meier
analysis revealed that high mRNA expression of DNAH7, DYNC2H1,
WDR78, DNAH6, DNAH12 and DNALI1 was associated with a longer
overall survival time in lung cancer patients. However, the overall
survival time of lung cancer patients with high POSTN expression
were shorter compared with those with low POSTN expression, and the
same trends were identified for COL3A1, COL1A1, COL1A2, CTSK,
ITGB4, TIMP1, ITGA11, COL11A1, MYBL2, KPNA2, AURKA, TPX2 and CDC20.
Conversely, higher expression levels of lncRNA RXFP1, RAMP2-AS1,
LINC00312 and LINC00472 were significantly associated with a longer
overall survival time. These results suggested that these RNAs may
provide novel tools for the diagnosis and prognostication, as well
as drug targets for smoking-associated lung cancer.
Of note, the present study has several limitations.
First, the expression pattern of key regulatory RNAs in
smoking-associated lung cancer should be further validated. The
correlation between the expression of regulatory RNAs in
smoking-associated lung cancer and clinicopathological features of
the patients, including age, sex, Grade, T stage, N stage, smoking
status and survival status, should be further evaluated.
Furthermore, it was demonstrated that smoking-associated genes
predicted the outcome of NSCLC patients. However, it may be
appropriate to assess whether the dysregulation of
smoking-associated genes are associated with smoking and
non-smoking patients with NSCLC. This may be problematic as the
number of non-smoking patients with NSCLC is limited. In addition,
previous studies have demonstrated that the competing endogenous
RNA network (ceRNA) serves crucial roles in cancer progression. In
the current study, smoking associated lncRNAs, miRNAs and mRNAs
were identified. Therefore, the construction of smoking associated
ceRNA networks in NSCLC may provide useful information to
understand the potential mechanisms that underly cancer
progression. In addition, the functional roles of these regulatory
genes should be further validated by performing loss/gain of
function assays.
In conclusion, the present bioinformatics study
identified 314 mRNAs, 24 lncRNAs and 4 miRNAs that are deregulated
in smoking-associated NSCLC. PPI network analysis identified 20 hub
genes in smoking-associated lung cancer, including DNAH7, DYNC2H1,
WDR78, COL3A1, COL1A1 and COL1A2. Co-expression network analysis
indicated that RXFP1, RAMP2-AS1, LINC00312 and LINC00472 are key
lncRNAs. Furthermore, GO and KEGG analysis indicated that these
smoking-associated lncRNAs are enriched in a variety of functions
and pathways, including cell proliferation and the cGMP/PKG
signaling pathway. Of note, these hub genes and lncRNAs were
associated with the prognosis of lung cancer patients. Although
further validation of the present results is required, the present
study provides useful information to further explore potential
candidate biomarkers for the diagnosis, prognostication and
utilization as drug targets for smoking-associated lung cancer.
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XD and YC conceived and designed the present study.
YC, YP and YJ collected and assembled the data. YC, LS and XD
analyzed and interpreted the data. All authors contributed to
writing the manuscript and approved the final version.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bhattacharjee A, Richards WG, Staunton J,
Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et
al: Classification of human lung carcinomas by mRNA expression
profiling reveals distinct adenocarcinoma subclasses. Proc Natl
Acad Sci USA. 98:13790–13795. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molina JR, Yang PG, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Pro. 83:584–594.
2008. View Article : Google Scholar
|
|
3
|
Roggli VL, Vollmer RT, Greenberg SD,
Mcgavran MH, Spjut HJ and Yesner R: Lung-Cancer heterogeneity: A
blinded and randomized study of 100 consecutive cases. Hum Pathol.
16:569–579. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bryant A and Cerfolio RJ: Differences in
epidemiology, histology, and survival between cigarette smokers and
never-smokers who develop non-small cell lung cancer. Chest.
132:185–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lv XJ, Du YW, Hao YQ, Su ZZ, Zhang L, Zhao
LJ and Zhang J: RNA-binding motif protein 5 inhibits the
proliferation of cigarette smoke-transformed BEAS-2B cells through
cell cycle arrest and apoptosis. Oncol Rep. 35:2315–2327. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hamdy SI, Hiratsuka M, Narahara K, Endo N,
El-Enany M, Moursi N, Ahmed MSE and Mizugaki M: Genotyping of four
genetic polymorphisms in the CYP1A2 gene in the Egyptian
population. Br J Clin Pharmacol. 55:321–324. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mattick JS and Makunin IV: Non-coding RNA.
Hum Mol Genet. 15:R17–R29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wan XC, Huang WH, Yang S, Zhang YL, Pu HL,
Fu FQ, Huang Y, Wu H, Li T and Li Y: Identification of
androgen-responsive lncRNAs as diagnostic and prognostic markers
for prostate cancer. Oncotarget. 7:60503–60518. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kornienko AE, Guenzl PM, Barlow DP and
Pauler FM: Gene regulation by the act of long non-coding RNA
transcription. BMC Biol. 11:2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun Y, Hu B, Wang Q, Ye M, Qiu Q, Zhou Y,
Zeng F, Zhang X, Guo Y and Guo L: Long non-coding RNA HOTTIP
promotes BCL-2 expression and induces chemoresistance in small cell
lung cancer by sponging miR-216a. Cell Death Dis. 9:852018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin A, Hu Q, Li C, Xing Z, Ma G, Wang C,
Li J, Ye Y, Yao J, Liang K, et al: The LINK-A lncRNA interacts with
PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT
inhibitors. Nat Cell Biol. 19:238–251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li SC, Sun X, Miao SC, Liu J and Jiao WJ:
Differential protein-coding gene and long noncoding RNA expression
in smoking-related lung squamous cell carcinoma. Thorac Cancer.
8:672–681. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kabbout M, Garcia MM, Fujimoto J, Liu DD,
Woods D, Chow CW, Mendoza G, Momin AA, James BP, Solis L, et al:
ETS2 mediated tumor suppressive function and MET oncogene
inhibition in human non-small cell lung cancer. Clin Cancer Res.
19:3383–3395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Page RD: TreeView. Glasgow University;
Glasgow, UK: 2001
|
|
15
|
Yang J, Lin J, Liu T, Chen T, Pan S, Huang
W and Li S: Analysis of lncRNA expression profiles in non-small
cell lung cancers (NSCLC) and their clinical subtypes. Lung Cancer.
85:110–115. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39:D561–D568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T. Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Assenov Y, Ramirez F, Schelhorn SE,
Lengauer T and Albrecht M: Computing topological parameters of
biological networks. Bioinformatics. 24:282–284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hecht SS: Cigarette smoking and lung
cancer: Chemical mechanisms and approaches to prevention. Lancet
Oncol. 3:461–469. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Li BG, Yang Q, Zhang PY and Wang
HT: Prognostic value of Aurora kinase A (AURKA) expression among
solid tumor patients: A systematic review and meta-analysis. Jpn J
Clin Oncol. 45:629–636. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen ZY, Chen X, Chen P, Yu SX, Nie FQ, Lu
BB, Zhang T, Zhou Y, Chen QN, Wei CC, et al: Long non-coding RNA
SNHG20 promotes non-small cell lung cancer cell proliferation and
migration by epigenetically silencing of P21 expression. Cell Death
Dis. 8:e30922017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu T, Zhao YJ, Hu ZX, Li J, Chu DD, Zhang
JW, Li Z, Chen B, Zhang X, Pan HY, et al: MetaLnc9 facilitates lung
cancer metastasis via a PGK1-activated AKT/mTOR pathway. Cancer
Res. 77:5782–5794. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu Q, Lv T, Wu Y, Shi X, Liu H and Song
Y: Long non-coding RNA 00312 regulated by HOXA5 inhibits tumour
proliferation and promotes apoptosis in Non-small cell lung cancer.
J Cell Mol Med. 21:2184–2198. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian Z, Wen S, Zhang Y, Shi X, Zhu Y, Xu
Y, Lv H and Wang G: Identification of dysregulated long non-coding
RNAs/microRNAs/mRNAs in TNM I stage lung adenocarcinoma.
Oncotarget. 8:51703–51718. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sui J, Li YH, Zhang YQ, Li CY, Shen X, Yao
WZ, Peng H, Hong WW, Yin L, Pu YP and Liang GY: Integrated analysis
of long non-coding RNA-associated ceRNA network reveals potential
lncRNA biomarkers in human lung adenocarcinoma. Int J Oncol.
49:2023–2036. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu TG, Xiao X, Wei Q, Yue M and Zhang LX:
Revealing potential long non-coding RNA biomarkers in lung
adenocarcinoma using long non-coding RNA-mediated competitive
endogenous RNA network. Braz J Med Biol Res. 50:e62972017.
View Article : Google Scholar : PubMed/NCBI
|