Introduction
P. aeruginosa is a ubiquitous microorganism
and one of the most frequent Gram-negative pathogens that cause
human opportunistic infections in burn and other immunocompromised
patients (1). These patients are the
most susceptible group to infection due to their compromised
natural defenses. Intensive care units, respiratory wards and burns
units are the main locations where P. aeruginosa infections
can spread (2). P. aeruginosa
also causes nosocomial pneumonia and cystic fibrosis with high
morbidity and mortality (3).
Antimicrobial therapy is recommended in the Health Technical
Memorandum 04–01 guidelines for the management of pneumonia
(4). However, the treatment of
pneumonia caused by P. aeruginosa has become increasingly
difficult owing to the elevated prevalence of drug resistance and
limited therapeutic options (5).
Thus, rapid and accurate detection of P. aeruginosa and
strict infection control is the most effective strategy in
preventing P. aeruginosa infection in burn wards (6).
The main conventional methods for detection of P.
aeruginosa include bacterial culture and immunological methods.
However, these methods exhibit specific shortcomings, such as being
time consuming or inaccurate (7).
Biochemical test kits, such as API 20 NE, are commonly used for
biochemical identification and exhibit a high oxidase-positive
misidentification rate in Gram-negative bacilli, including P.
aeruginosa (8). The polymerase
chain reaction (PCR) techniques established by Mullis et al
(9) specifically amplify the DNA
fragment with thermally stable DNA polymerase; these techniques
include single, multiplex, and quantitative PCR. PCR is a
high-quality, fast, and efficient method that is often utilized to
evaluate the specificity, sensitivity, and feasibility of other
detection methods (10). However,
this method is inconvenient for clinical testing given the
requirement of isothermal cyclic amplification (11). The present study aimed to develop an
efficient, time-saving, and inexpensive method for the detection of
P. aeruginosa.
A novel gene amplification method called
loop-mediated isothermal amplification (LAMP) was developed by
Notomi et al (12) in 2000,
and is possibly a feasible and cost-effective alternate scheme for
molecular diagnosis. This technique is based on a set of specific
primers with at least four primers to recognize six different
regions of the genome and produces a stem-loop DNA with several
inverted repeats and cauliflower-like structures with multiple
loops (13). The results can be
detected by fluorescence signal, turbidity, and the naked eye
(14). In the present study, a novel
specific gene of P. aeruginosa was obtained through
bioinformatics analysis. An efficient and accurate LAMP method was
built for the detection of P. aeruginosa and validated via
PCR assay.
Materials and methods
Bacterial strains and culturing
All 150 tested P. aeruginosa strains and 170
non-P. aeruginosa strains, including Escherichia coli
(ATCC25922), Klebsiella pneumoniae (ATCC700603),
Staphylococcus aureus (ATCC25923), Staphylococcus
epidermidis (ATCC12228), Enterococcus faecalis
(ATCC29212), and Shiga bacillus (ATCC25931) were kindly
provided by the First People's Hospital of Yunnan Province from
their burns ward (Yunnan, China). The strains were grown in
Luria-Bertani (LB) liquid medium (Biotopped Technology Co., Ltd.,
Taipei, Taiwan) in a shaking incubator (HZQ-F160 full temperature
double layer oscillator-cultivating box; Suzhou Pei Ying
Experimental Equipment Co., Ltd., Jiangsu, China) at 37°C and 180
rpm for 12 h. The bacterial genome was extracted using the TIANamp
genomic DNA kit (Tiangen Biotech Co., Ltd., Beijing, China)
following the manufacturer's protocol and then stored at −40°C for
further experiments (15).
Selection of specific genes of P.
aeruginosa
The formatted non-redundant nucleic acid database
was downloaded from National Center for Biotechnology Information
(NCBI; ftp://ftp.ncbi.nih.gov/blast/db) as the local
database. Local BLAST sequencing in BLAST software (version 2.7.1+;
http://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/)
was performed using the P. aeruginosa genome (KI518973.1) as
query sequence against this database. Potential specific genes were
further analyzed by online BLAST (version 2.7.1+; http://blast.ncbi.nlm.nih.gov/Blast.cgi)
with the database excluding or including the sequences of P.
aeruginosa. The specific gene of P. aeruginosa was
required to be highly conserved among the P. aeruginosa
strains and show no significant similarity to other species for
inclusion.
Initially, the genomic sequence of P.
aeruginosa and the non-redundant nucleic acid database of NCBI
were downloaded to the local server. Then, the potential specific
genes of P. aeruginosa were screened by sequence similarity
alignment. Online BLAST was used to further identify the screened
potential specific genes due to the slow update of the local
database. Two-step strategies using interspecies-specific and
intraspecies commonality were used to identify specific genes by
online BLAST. The first step excluded P. aeruginosa during
the alignment; the gene may be considered a possible target gene if
the alignment result is different. The second included P.
aeruginosa during alignment, with the highly conserved genes
considered as possible target genes. The retrieval range was
limited to the species with known sequences except for P.
aeruginosa. The specific gene of P. aeruginosa can only
be considered when the alignment differs from those of other
species, is similar to those of a few species or features a very
low similarity.
Primer design and reaction
Four oligonucleotide primers targeting the specific
gene identified using BLAST, hypothetical protein gene
(GenBank ID 882161), were designed using Primer Explorer version 5
software (http://primerexplorer.jp/lampv5/index.html) for the
LAMP assay. The outer primers F3 and B3 were used as the forward
and reverse primer in the PCR assays, respectively. Table I presents all the primers used in the
present study. PCR was performed using 2X Tsingke Master Mix, which
was purchased from Tsingke Biotech Co., Ltd (Kunming, China).
According to the manufacturer's protocol, the PCR reaction system
containing 12.5 µl 2X Tsingke Master Mix, 1.0 µl primers (10 µM)
and 1.0 µg DNA template from each strain was added with
nuclease-free water up to 25 µl volume. The reactions were
performed in a GeneAmp PCR System 9700 (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with the following amplification
conditions: Pre-denaturation at 95°C for 5 min, followed by 32
cycles of denaturation at 95°C for 30 sec, annealing at 57°C for 30
sec, extension at 72°C for 30 sec and a final extension at 72°C for
7 min. The PCR products were verified using gel electrophoresis on
a 2% agarose gel and stained with GelStain (Beijing Transgen
Biotech Co., Ltd., Beijing, China). LAMP assay reactions were
optimized at 65°C for 30 min and 80°C for 2 min. The system
contained 2.5 µl 10X Bio Labs buffer, 8.0 U Bst 2.0 DNA polymerase
[both New England Biolabs (Beijing) Ltd., Beijing, China], 5.2 mM
Mg2+, 1.4 mM deoxyribonucleotide, 0.2 µM of the outer
primer and 1.6 µM of the inner primer, with nuclease-free water up
to 25 µl volume. SYBR-Green I (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China), a nuclear dye, was added
into the LAMP reaction tubes for fluorescence visualization of the
LAMP products.
 | Table I.Loop-mediated isothermal
amplification primers for hypothetical protein gene. |
Table I.
Loop-mediated isothermal
amplification primers for hypothetical protein gene.
| Primer | Sequence
(5′-3′) | Size (bp) |
|---|
| F3 |
CAAGCGCAAGATAGTCGCC | 19 |
| B3 |
TCCGCTTGAACAGGCTGGTG | 20 |
| FIP |
GAAGATATCCGGCTGGTTGCTTTTCAAGAGGGAATGCCGCAGT | 43 |
| BIP |
AACGGATCATCGGCATCCTGGTTTTCATCGCCGTCCACAGGTAGA | 45 |
Construction and verification of
positive plasmids
The positive plasmids were constructed via the
following steps. The DNA fragment of target genes was obtained by
PCR and the genome DNA of P. aeruginosa as a template. The
target fragment was then inserted into the pMD 19-T simple vector
and transformed into E. coli JM109 cells using the E.
coli JM109 Competent Cell kit [cat. no. 9052; both Takara
Biomedical Technology (Beijing) Co., Ltd., Beijing, China] at 0°C
for 30 min, 42°C for 45 sec and 0°C for 2 min, according to the
manufacturer's protocol. The 37°C preheated blank LB medium was
added into the transform system culture at 37°C and 180 rpm for 1
h. Positive clones were selected from the cultured bacterial
solution in the LB solid medium (Biotopped Technology Co., Ltd.,
Taipei, Taiwan) by spreading the bacteria around the plate, which
was cultured at 37°C for overnight. Then, following an overnight
culture at 37°C, the plasmid were extracted from the cells using
TIANprep Mini Plasmid kit (Tiangen Biotech Co., Ltd.), according to
the manufacturer's protocol and verified by sequencing. Finally,
the number of copies of recombinant plasmids was calculated by the
deduced polynomial model as follows:
C=X×10-9(2692+Y)×660×NA
where C denotes the copy of the recombinant plasmid,
X and Y represent the concentration of the recombinant plasmid and
the number of base pairs in the target fragment, respectively,
NA is Avogadro's constant, 2692 is the number of base
pairs in the vector, and 660 is the mean molecular weight of 1 base
pair.
As described above, we constructed positive plasmids
with the hypothetical protein gene. The plasmids were
transformed into JM109 competent cells. The positive clones were
used for bacterial liquid PCR. The sequence alignment of the result
was analyzed in NCBI following sequencing and the sequence
correctness was verified. The positive clones were then cultured in
LB liquid medium at 37°C and 180 rpm for 12 h prior to the
extraction of positive plasmids by TIANprep Mini Plasmid kit
(Tiangen Biotech Co., Ltd.), according to the manufacturer's
protocol. The plasmids were verified by gel electrophoresis on 2%
agarose gels and stained with GelStain, and then stored at −20°C
for further use.
Specificity of PCR and LAMP
reactions
A total of 150 clinical P. aeruginosa strains
and 170 non-P. aeruginosa strains, including 24 E.
coli, 32 K. pneumonia, 37 S. aureus, 15 S.
epidermidis, 17 E. faecalis, and 45 S. bacillus,
were tested in this study to evaluate the specificity of PCR and
LAMP reactions. The PCR test was used as the gold standard in
preliminary experiments for the specificity test prior to the LAMP
test. All experiments were repeated twice.
Sensitivity of PCR and LAMP
reactions
The sensitivity of PCR and LAMP reactions were
evaluated using two different templates, the serially diluted
10-fold positive plasmids (108−10° copies) and blood
sample mimicking infection. The counted P. aeruginosa strain
was serially diluted by 10-fold in PBS and then mixed with blood
from 3 healthy specific pathogen free female Kunming mice
(Laboratory Animal Center of Kunming Medical University, Kunming,
Yunnan; weight, 22–25 g; age, 5 weeks) at 1:1 proportion to mimic
infection. The mice were housed in the animal experiment center of
Kunming University of Science and Technology (Kunming, China) at
25°C with a 12/12 h light/dark cycle and access to food and water
ad libitum. The blood sample was lysed using a solution
containing 125 mM NaOH, 1 mM EDTA and 0.1% Tween-20, and then a
solution containing 125 mM HCl and 10 mM Tris-HCl, and the
suspension was used as the template for PCR and LAMP assays. All
experiments were repeated six times.
Results
Screening the specific gene and primer
design
Following the preliminary screening of possible
specific genes by local BLAST, 1,599 potential specific genes were
obtained. These genes were then used in the second screening by
online BLAST. Conclusively, 5 potential specific genes were
attained, and they met the criterion regarding interspecific
specificity and intraspecies universality. In the primer design
step, four primers were designed for the six regions of the target
gene in LAMP, and the primers were confirmed by primer BLAST and
PCR assay. Considering the above selection criteria, the
hypothetical protein gene was finally identified as the only
one that can be used in the detection of P. aeruginosa.
Specificity of PCR and LAMP
reactions
The genomic DNA of P. aeruginosa and
non-P. aeruginosa were extracted as templates for the
specificity test of PCR and LAMP, as detailed above. Prior to LAMP
reaction, the PCR test was used for the specificity test, and all
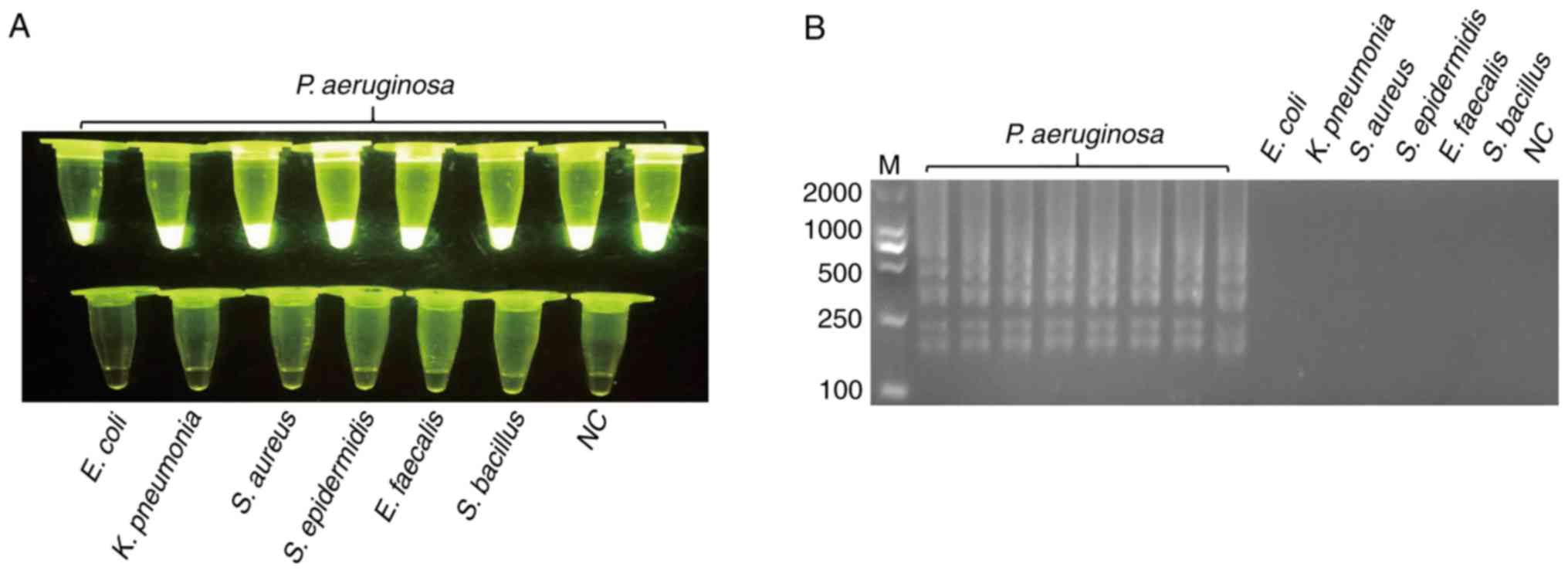
170 non-P. aeruginosa strains were negative. Fig. 1 presents the representative results.
Following validation by PCR, LAMP was used to test specificity.
SYBR-Green I was added to the LAMP reaction tubes for fluorescence
visualization. Fig. 2 presented the
results of LAMP; the findings of which were consistent with
PCR.
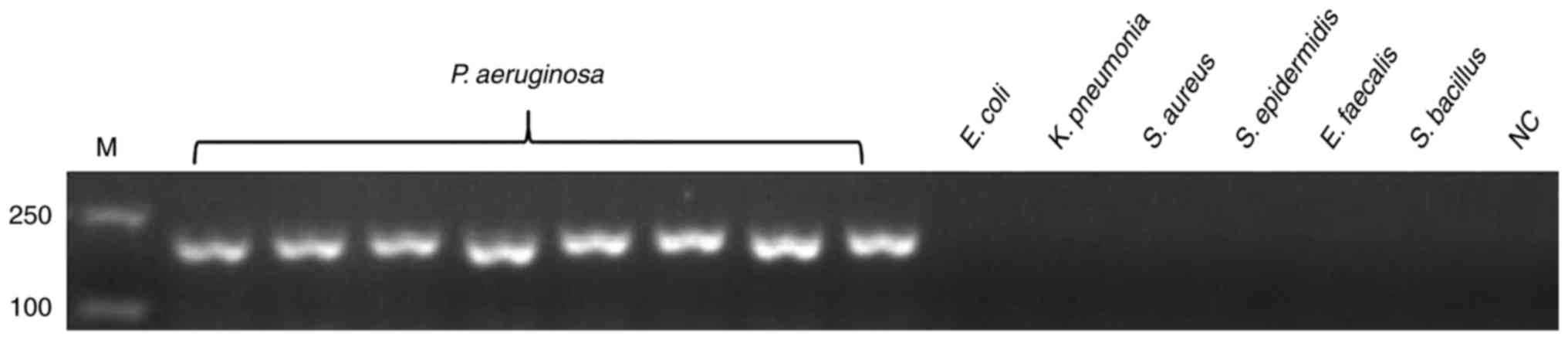 | Figure 1.Specificity of the PCR assay for
detecting the target gene of hypothetical protein gene.
Genomic DNA of P. aeruginosa was used as the template for
PCR in lanes 1–8, the template in lanes 9–14 were ordinal of
control strains, E. coli, K. pneumonia, S. aureus, S.
epidermidis, E. faecalis and S. bacillus, whereas lane
15 is NC. All experiments were repeated twice. PCR, polymerase
chain reaction; NC, negative control; M, marker. |
Sensitivity of PCR and LAMP
reactions
The positive plasmids and bacterial solution were
serially diluted 10-fold based on the calculated copy number and
plate counting, respectively. The positive plasmids were directly
used as templates for PCR and LAMP assay. Figs. 3 and 4
demonstrate the extreme sensitivities, which can reach up to 10°
copies per reaction, of PCR and LAMP assay. The bacterial solutions
were mixed with the blood of mice with a volume ratio of 1:1. These
mixtures were lysed by direct extraction reagent and then used for
the LAMP assay. Fig. 4 indicates
that the LAMP assay exhibits a high sensitivity that may reach up
to 10° copies per reaction. SYBR-Green I into was also added to the
LAMP reaction tubes for fluorescence visualization detection
(Fig. 5).
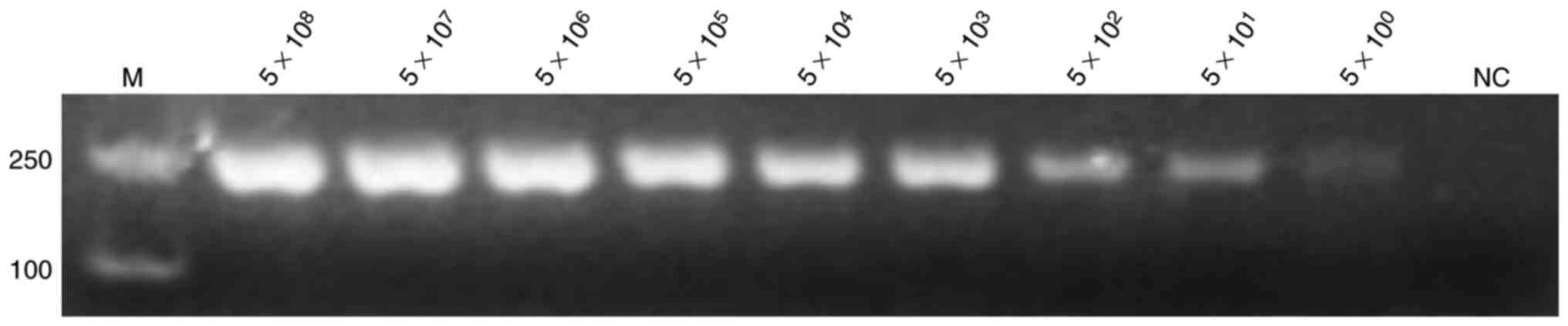 | Figure 3.Sensitivity of the polymerase chain
reaction assay for detection of the hypothetical protein
gene. The positive plasmid was used as a template at
concentrations of 5×108, 5×107,
5×106, 5×105, 5×104,
5×103, 5×102, 5×101 and 5×10° per
reaction, with nuclease-free water used as template for NC. All
experiments were repeated six times. NC, negative control; M,
marker. |
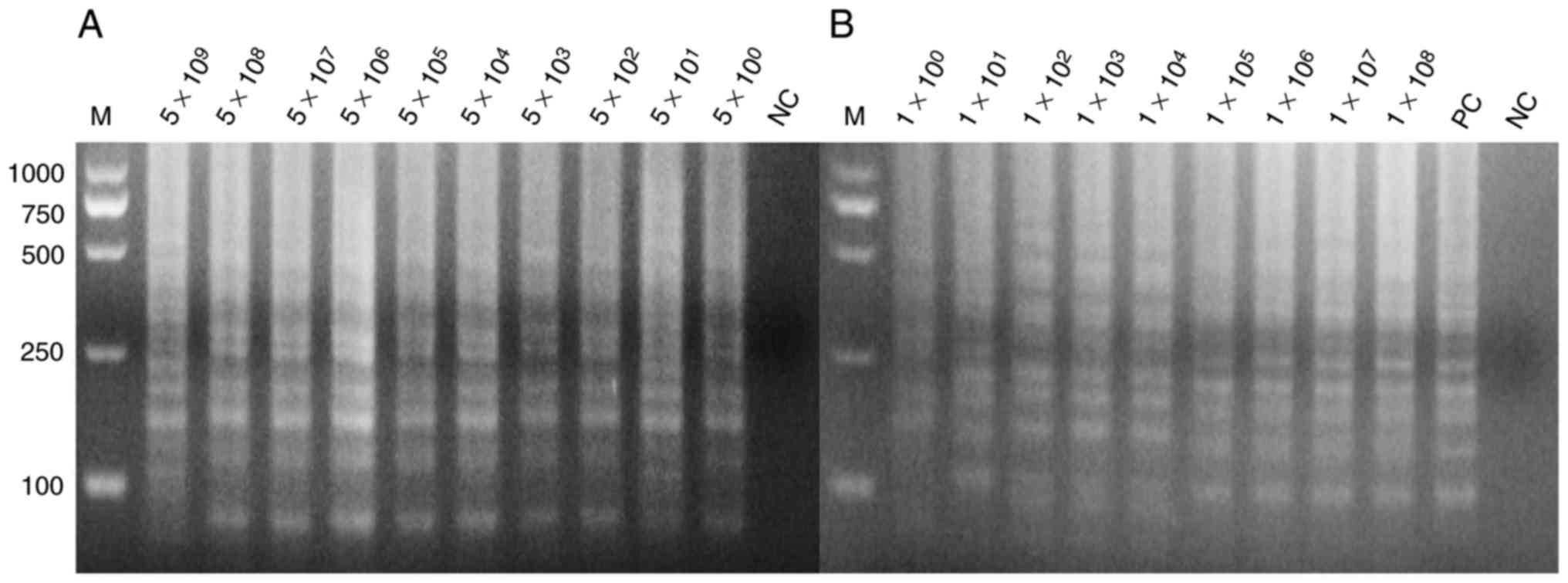 | Figure 4.Sensitivity of the loop-mediated
isothermal amplification assay for detection of the hypothetical
protein gene. (A) The positive plasmids as template at
concentrations of 5×109, 5×108,
5×107, 5×106, 5×105,
5×104, 5×103, 5×102,
5×101 and 5×10° per reaction, with nuclease-free water
as template in lane 1 for NC. (B) The blood sample being direct
lysed as template at concentrations of 1×108,
1×107, 1×106, 1×105,
1×104, 1×103, 1×102,
1×101 and 1×10° per reaction the sample order from
1×10°−1×108, respectively, with positive plasmids as the
template in lane 11 as PC, and nuclease-free water as the template
in lane 12 as NC. All the experiments were repeated six times. PC,
positive control; NC, negative control; M, marker. |
 | Figure 5.Visualization of LAMP. (A)
Sensitivity of the LAMP assay for detecting the target gene of
hypothetical protein gene; the templates were the positive
plasmids, serially 10-fold diluted as 5×109,
5×108, 5×107, 5×106,
5×105, 5×104, 5×103,
5×102, 5×101 and 5×10° per reaction. (B)
Sensitivity of the LAMP assay for detection of the hypothetical
protein gene; the templates were mixed with mouse blood,
serially 10-fold diluted as 1×108, 1×107,
1×106, 1×105, 1×104,
1×103, 1×102, 1×101 and 1×10° per
reaction, respectively, with positive plasmids as the template as
PC. LAMP, loop-mediated isothermal amplification; PC, positive
control; NC, negative control. |
Discussion
P. aeruginosa must be controlled in related
infections, and an effective method for the detection must be
evaluated and used. Various methods, including culturing and
PCR-based amplification method, have been utilized for the
detection of this pathogen (7).
Conventional methods, such as culturing, are experience-dependent
and time-consuming (7). In addition
to diagnostic or detection application, PCR is useful for cloning
and other molecular biology areas, but has not been broadly
established in clinical set-up given the requirement for a
high-precision thermal cycler, which cannot be afforded by
low-level medical institutions (16).
LAMP is a novel DNA amplification technique
(17). This method is widely used in
various fields of biological science, including bacterial, viral
and parasitic infections, and has the potential to be used as a
simple detection assay due to its simplicity, ruggedness and low
cost (18). In contrast to PCR
technology, wherein a series of alternating temperature steps or
cycles is carried out, the LAMP assay requires no thermal cycler
and is carried out at a constant temperature of 60–65°C (19). Additionally, compared with the
traditional method of isolation and identification and PCR assay
(24–48 and 2–3 h, respectively), LAMP is rapid, simple, and
convenient (8).
In the present study, a novel specific gene was
identified, named hypothetical protein gene (GenBank ID:
882161), which can be used for identification of P.
aeruginosa. Hypothetical protein gene was initially used
as a target gene for the detection of P. aeruginosa.
Hypothetical protein gene was highly conserved in P.
aeruginosa, however functional studies have not yet been
conducted. In the present study, four oligonucleotide primers were
designed for LAMP assay, and the outer primers F3 and B3 were used
as the forward and reverse primers, respectively, in PCR assays for
the hypothetical protein gene detection; the reaction
conditions were optimal (optimizing data not shown). The results
demonstrated that the hypothetical protein gene is specific
to P. aeruginosa. Therefore, the LAMP system was utilized to
detect P. aeruginosa due to its simplicity, and rapid, easy
detection, which may appropriately meet the demands in developing
countries with insufficient facilities and health resources.
Furthermore, it was attempted to use the LAMP assay to detect the
antibiotics resistance gene of P. aeruginosa. However, due
to the rapid mutation and multiple subtypes of the drug resistance
gene, the primers for LAMP reaction cannot correctly match with the
target fragment and thus lead to false negative amplification.
P. aeruginosa infection has become one of the
most serious nosocomial infections (20). Thus, establishment of a fast and
effective detection method is required urgently. The LAMP
technology offers the opportunity to resolve such burden for its
simplicity, rapidity, and high sensitivity. With the emergence of
multidrug-resistant bacteria, the LAMP technology can also be
applied for detecting resistant genes (21).
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from
Yunnan Science and Technology Commission (grant nos. 2015BC001 and
2015DH010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL wrote the manuscript and performed experiments.
YSh constructed the positive plasmids. GY performed the specificity
of PCR assay experiments. X-SX performed the BLAST experiments and
proofread the manuscript. XM collected the strains and performed
the sensitivity of LAMP experiments. YF performed data analysis.
YSo and A-MZ designed the study.
Ethics approval and consent to
participate
The current study was approved by the ethics
committee of Kunming University of Science and Technology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moradali MF, Ghods S and Rehm BH:
Pseudomonas aeruginosa lifestyle: A paradigm for adaptation,
survival, and persistence. Front Cell Infect Microbiol. 7:392017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nathwani D, Raman G, Sulham K, Gavaghan M
and Menon V: Clinical and economic consequences of
hospital-acquired resistant and multidrug-resistant Pseudomonas
aeruginosa infections: A systematic review and meta-analysis.
Antimicrob Resist Infect Control. 3:322014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sana TG, Berni B and Bleves S: The T6SSs
of Pseudomonas aeruginosa strain PAO1 and their effectors:
Beyond bacterial-cell targeting. Front Cell Infect Microbiol.
6:612016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walker J and Moore G: Pseudomonas
aeruginosa in hospital water systems: Biofilms, guidelines, and
practicalities. J Hosp Infect. 89:324–327. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding C, Yang Z, Wang J, Liu X, Cao Y, Pan
Y, Han L and Zhan S: Prevalence of Pseudomonas aeruginosa
and antimicrobial-resistant Pseudomonas aeruginosa in
patients with pneumonia in mainland China: A systematic review and
meta-analysis. Int J Infect Dis. 49:119–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Potron A, Poirel L and Nordmann P:
Emerging broad-spectrum resistance in Pseudomonas aeruginosa
Acinetobacter baumannii Mechanisms and epidemiology. Int J
Antimicrob Agents. 45:568–585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jami Al-Ahmadi G: Zahmatkesh Roodsari R.
Fast and specific detection of Pseudomonas aeruginosa from
other pseudomonas species by PCR. Ann Burns Fire Disasters.
29:264–267. 2016.PubMed/NCBI
|
|
8
|
Ghosh R, Nagavardhini A, Sengupta A and
Sharma M: Development of Loop-Mediated Isothermal Amplification
(LAMP) assay for rapid detection of Fusarium oxysporum f. sp.
ciceris-wilt pathogen of chickpea. BMC Res Notes. 8:402015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mullis K, Faloona F, Scharf S, Saiki R,
Horn G and Erlich H: Specific enzymatic amplification of DNA in
vitro: The polymerase chain reaction. 1986. Biotechnology.
24:17–27. 1992.PubMed/NCBI
|
|
10
|
Duś I, Dobosz T, Manzin A, Loi G, Serra C
and Radwan-Oczko M: Role of PCR in Helicobacter pylori diagnostics
and research-new approaches for study of coccoid and spiral forms
of the bacteria. Postepy Hig Med Dosw (Online). 67:261–268. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee PL: DNA amplification in the field:
Move over PCR, here comes LAMP. Mol Ecol Resour. 17:138–141. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Notomi T, Okayama H, Masubuchi H, Yonekawa
T, Watanabe K, Amino N and Hase T: Loop-mediated isothermal
amplification of DNA. Nucleic Acids Res. 28:E632000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karthik K, Rathore R, Thomas P, Arun TR,
Viswas KN, Agarwal RK, Manjunathachar HV and Dhama K: Loop-mediated
isothermal amplification (LAMP) test for specific and rapid
detection of Brucella abortus in cattle. Vet Q. 34:174–179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mori Y, Nagamine K, Tomita N and Notomi T:
Detection of loop-mediated isothermal amplification reaction by
turbidity derived from magnesium pyrophosphate formation. Biochem
Biophys Res Commun. 289:150–154. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brewster JD and Paoli GC: DNA extraction
protocol for rapid PCR detection of pathogenic bacteria. Anal
Biochem. 442:107–109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seki M, Yamashita Y, Torigoe H, Tsuda H,
Sato S and Maeno M: Loop-mediated isothermal amplification method
targeting the lytA gene for detection of Streptococcus pneumoniae.
J Clin Microbiol. 43:1581–1586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murray L, Edwards L, Tuppurainen ES,
Bachanek-Bankowska K, Oura CA, Mioulet V and King DP: Detection of
capripoxvirus DNA using a novel loop-mediated isothermal
amplification assay. BMC Vet Res. 9:902013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohtsuka K, Yanagawa K, Takatori K and
Hara-Kudo Y: Detection of Salmonella enterica in naturally
contaminated liquid eggs by loop-mediated isothermal amplification,
and characterization of Salmonella isolates. Appl Environ
Microbiol. 71:6730–6735. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhai S, Liu C, Zhang Q, Tao C and Liu B:
Detection of two exogenous genes in transgenic cattle by
loop-mediated isothermal amplification. Transgenic Res.
21:1367–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Poole K: Pseudomonas aeruginosa
Resistance to the max. Front Microbiol. 2:652011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng J, Tang S, Liu L, Kuang X, Wang X, Hu
S and You S: Development of a loop-mediated isothermal
amplification (LAMP) assay for rapid and specific detection of
common genetically modified organisms (GMOs). Int J Food Sci Nutr.
66:186–196. 2015. View Article : Google Scholar : PubMed/NCBI
|