Introduction
Hearing loss is one of the most common birth defects
in China, and the incidence rate in newborns is 1/1,000–3/1,000
(1). Survey data from the World
Health Organization (http://www.who.int) have indicated that among 360
million individuals with hearing loss, there are 32 million
children. There are many factors related with hearing loss, and
genetic factors, environmental factors or interactions between the
two factors can cause deafness (2).
It is reported that 50% of deafness is associated with genetic
factors, of which 70% are classified into non-syndromic hearing
loss (NSHL), and approximately 80% of NSHL are caused by autosomal
recessive inheritance (3,4). Previous molecular etiology studies have
shown that four NSHL genes including gap junction β-2 (GJB2,
OMIM: 121011), GJB3 (OMIM: 603324), solute carrier family
26, member 4 (SLC26A4, OMIM: 605646) and mitochondrial
deoxyribonucleic acid 12SrRNA (mtDNA 12SrRNA, OMIM:
561000) are most common (5–9). Therefore, mutation screening of the
four genes in people provides an effective method for the diagnosis
of hearing loss. At the same time, prenatal diagnosis to the
families with inherited hearing loss or the risk of deafness before
the birth of other babies can avoid the birth of baby with hearing
loss.
In this study, the above pathogenic genes in 116
patients with hearing loss were directly sequenced by polymerase
chain reaction (PCR) amplified products, the causes of hearing loss
in family members were identified, and the technical support of
prenatal diagnosis was conducted, so as to guide families with
hearing loss delivering descents with normal hearing.
Patients and methods
Subjects
Following the principle of informed consent, 116
NSHL patients who were aged from 3 months to 39 years and had
genetic counseling in the Prenatal Diagnosis Center of Women and
Children's Health Care Hospital of Linyi (Linyi, China) from
January 2015 to June 2017 were collected. Hearing loss was >70
dB. Detailed medical history of deaf patients was collected, and
patients with systemic diseases, dysnoesia, syndromic hearing loss,
history of meningitis, otitis media or ear trauma were excluded.
The study was approved by the Ethics Committee of Women and
Children's Health Care Hospital of Linyi (Linyi, China). Signed
written informed consents were obtained from the patients or the
guardians.
Methods
DNA extraction
A total of 2 ml venous blood from patients was
collected by the ethylenediaminetetraacetic acid-Na (EDTA-Na)
anti-coagulation tube. For pregnant women who would receive
prenatal diagnosis, 10 ml amniotic fluid was extracted via
amniocentesis during 17–20 weeks of gestation. According to the
manufacturer's instructions, DNA was extracted by the kit (Tiangen
Biotech Co., Ltd., Beijing, China), a part of which was subjected
to quantitative and purity tests by an ultraviolet
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and preserved at −20°C.
PCR amplification and sequencing
analysis
PCR amplification reaction, PCR products
purification and sequencing analysis of target DNA fragment were
performed. Sequencing reaction was conducted by Shanghai Genome
Pilot Institutes of Genomics and Human Health (Shanghai, China)
with the use of an American ABI 373XL automated DNA sequencer
(Applied Biosystems; Thermo Fisher Scientific., Inc., Waltham, MA,
USA). Software including Chromas, BioEdit and SeqMan were used to
make comparison and analysis of sequencing data with National
Center for Biotechnology Information (NCBI) standard sequence and
Cambridge Reference Sequence (CRS).
Results
In this study, 31 mutation sites in the four common
genes in Chinese were detected in 116 patients with hearing loss,
and 51 patients were diagnosed with definite pathogenic mutations
with a positive rate of 43.96%. The remaining 65 patients had no
definite pathogenic mutations. The sequencing results are shown in
Table I. A total of 35 patients were
diagnosed with definite GJB2 mutation-induced deafness,
accounting for 30.17% (35/116) of all subjects. Among them,
homozygous mutations were identified in 21 patients, and compound
heterozygous mutations were found in 14 patients. Definite
GJB2 gene mutation-induced hearing loss was identified in 14
patients, of which the prevalence rate was 12.07% among all
subjects. Among them, homozygous mutations were identified in 7
patients, and compound heterozygous mutations were found in 7
patients. A total of 2 (2/116, 1.72%) patients were found carrying
mtDNA 12SrRNA 1555A>G mutations, and they had
administration history of aminoglycoside. No GJB3 gene
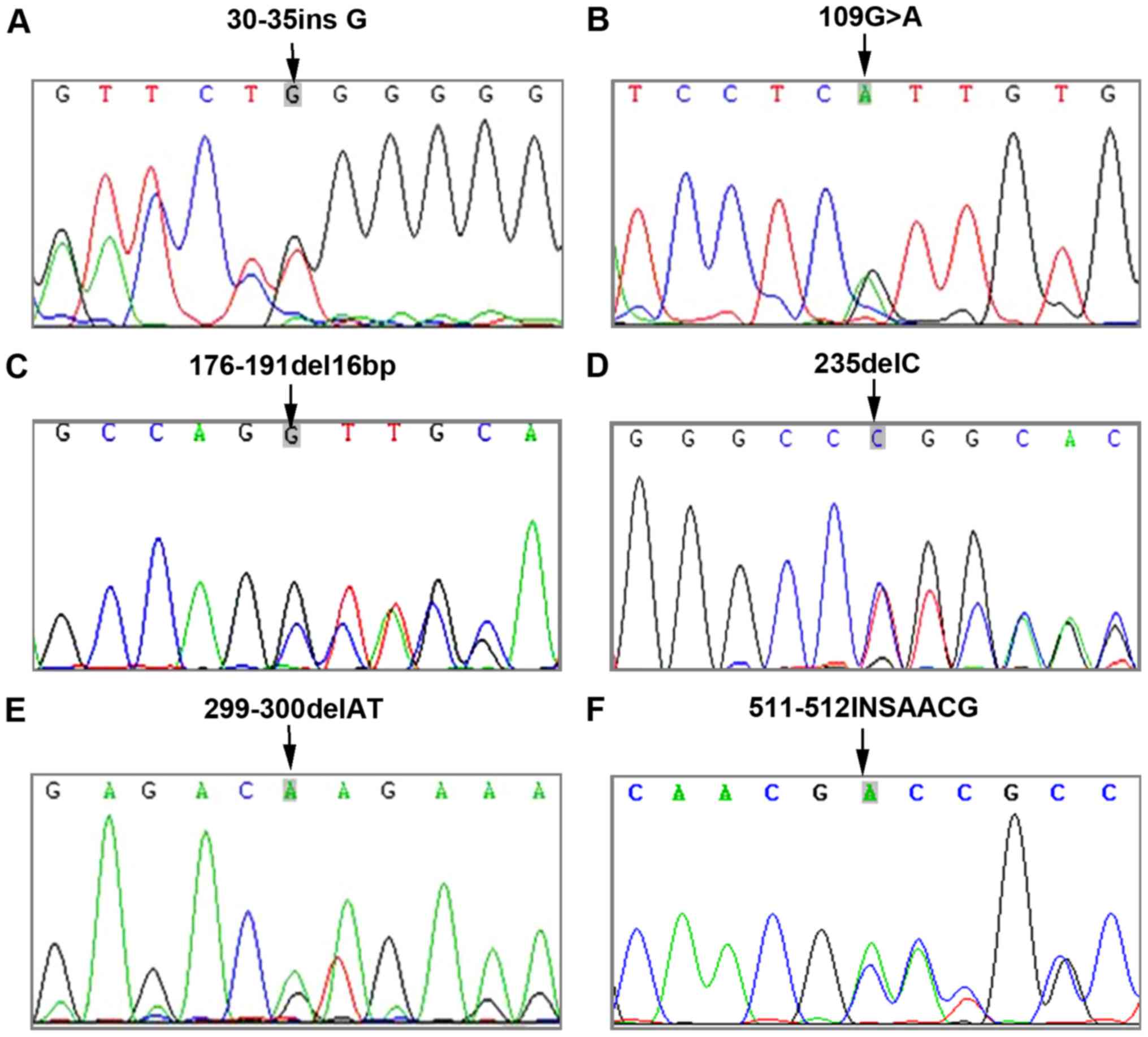
mutation site was detected. All in all, a total of six GJB2
mutation sites (Fig. 1) and five
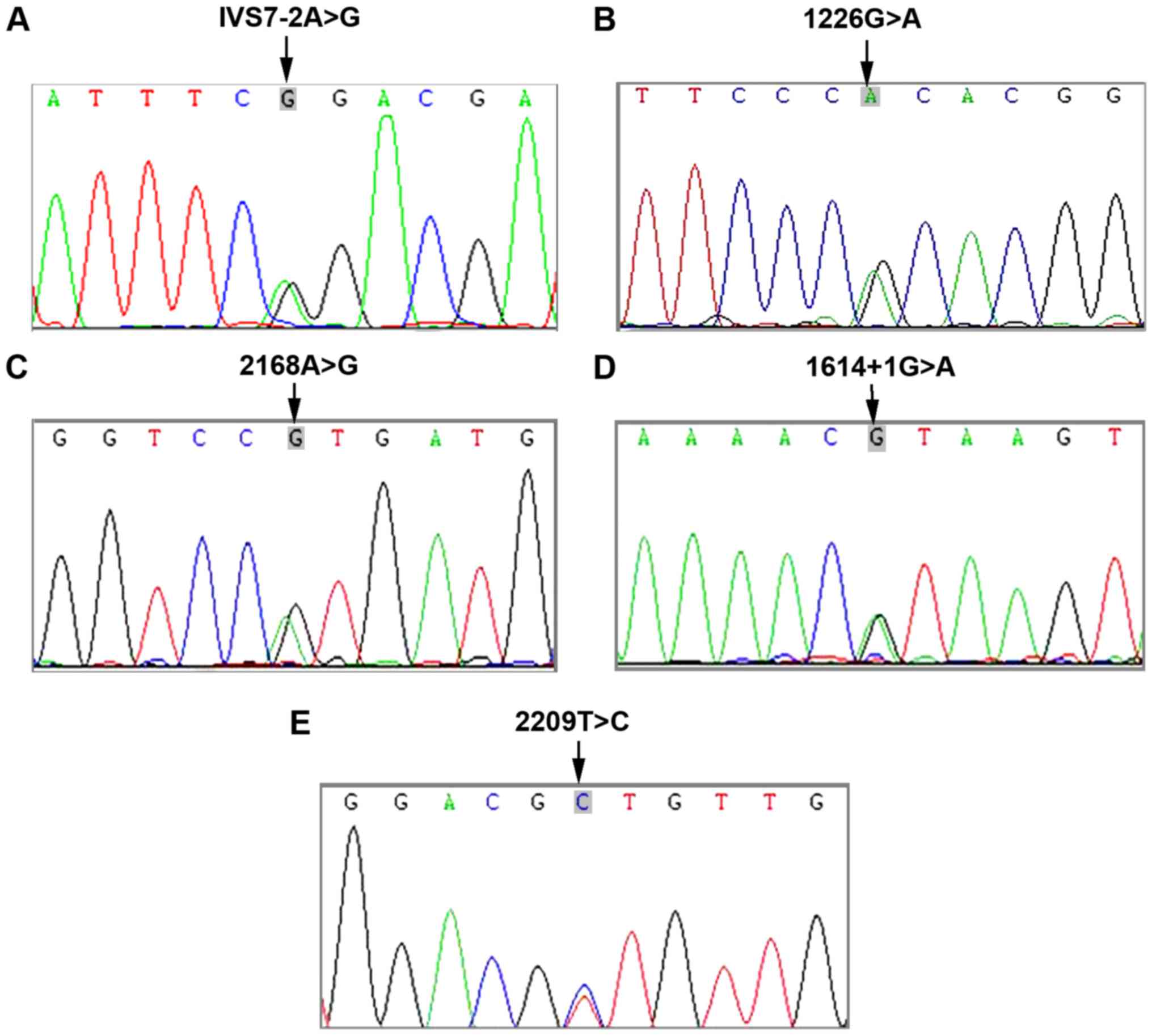
SLC26A4 mutation sites were identified (Fig. 2).
 | Table I.Results of genetic test of 51 patients
with hearing loss. |
Table I.
Results of genetic test of 51 patients
with hearing loss.
| Gene | Mutation site | No. of patients
(case) | Total no. (case) | Proportion (%) |
|---|
| GJB2 | 235delC/235delC | 18 | 35 | 30.17 |
|
|
235delC/299-300delAT | 7 |
|
|
|
|
235delC/176-191del16bp | 1 |
|
|
|
|
235delC/511-512insAACG | 1 |
|
|
|
|
235delC/30-35insG | 1 |
|
|
|
|
299-300delAT/299-300delAT | 2 |
|
|
|
|
109G>A/109G>A | 1 |
|
|
|
|
109G>A/176-191del16bp | 1 |
|
|
|
|
109G>A/299-300delAT | 2 |
|
|
|
| 176-191del16bp/ |
|
|
|
|
| 511-512insAACG | 1 |
|
|
| SLC26A4 |
IVS7-2A>G/IVS7-2A>G | 7 | 14 | 12.07 |
|
|
IVS7-2A>G/1226G>A | 2 |
|
|
|
|
IVS7-2A>G/2168A>G | 3 |
|
|
|
|
IVS7-2A>G/1614+1G>A | 1 |
|
|
|
|
IVS7-2A>G/2009T>C | 1 |
|
|
| 12SrRNA | 1555A>G | 2 | 2 | 1.72 |
| Total |
|
| 51 | 43.96 |
Among 116 patients with hearing loss, 17 were
identified with mutations in the GJB2 or SLC26A4
gene. Subsequently, their parents received tests of genes
associated with hearing loss, which indicated that they were
carriers. Sequencing results are shown in Table II. All of the 17 families had the
need for further fertility. During their next pregnancy, prenatal
diagnosis was performed. The results suggested that the genotypes
of 6 fetuses were consistent with those of the probands, so the 6
families decided to terminate the pregnancy. No mutation was found
in 3 fetuses. The genotypes of the other 5 children were consistent
with those of their parents. Follow-up results showed that their
fetuses had normal hearing after birth.
| Table II.Sequencing results of the family
members of the 17 hearing loss patients with prenatal
diagnosis. |
Table II.
Sequencing results of the family
members of the 17 hearing loss patients with prenatal
diagnosis.
| No. of case | Proband | Father | Mother | Fetus | Follow-up
results |
|---|
|
1b |
IVS7-2A>G/2009T>C | 2009T>C/WT | IVS7-2A>G/WT | WT/WT | Normal hearing |
|
2a |
299-300delAT/299-300delAT |
299-300delAT/WT |
299-300delAT/WT |
299-300delAT/299-300delAT | Induced labor |
|
3a |
235delC/235delC | 235delC/WT | 235delC/WT | 235delC/WT | Normal hearing |
|
4a |
235delC/235delC | 235delC/WT | 235delC/WT |
235delC/235delC | Induced labor |
|
5a |
235delC/235delC | 235delC/WT | 235delC/WT |
235delC/235delC | Induced labor |
|
6a |
235delC/235delC | 235delC/WT | 235delC/WT | WT/WT | Normal hearing |
|
7a | 35insG/235delC | 235delC/WT | 35insG/WT | 235delC/WT | Normal hearing |
|
8a |
235delC/235delC | 235delC/WT | 235delC/WT | 235delC/WT | Normal hearing |
|
9a |
235delC/235delC | 235delC/WT | 235delC/WT | 235delC/WT | Normal hearing |
| 10b |
IVS7-2A>G/IVS7-2A>G |
IVS7-2A>G/WT |
IVS7-2A>G/WT |
IVS7-2A>G/IVS7-2A>G | Induced labor |
| 11b |
IVS7-2A>G/1226G>A |
IVS7-2A>G/WT | 1226G>A/WT | 1226G>A/WT | Normal hearing |
| 12a |
235delC/511-512insAACG |
511-512insAACG/WT | 235delC/WT | 235delC/WT | Induced labor |
| 13a |
235delC/235delC | 235delC/WT | 235delC/WT | 235delC/WT | Induced labor |
| 14a |
235delC/235delC | 235delC/WT | 235delC/WT | WT/WT | Normal hearing |
| 15b |
IVS7-2A>G/IVS7-2A>G |
IVS7-2A>G/WT |
IVS7-2A>G/WT |
IVS7-2A>G/IVS7-2A>G | Induced labor |
| 16a |
235delC/235delC | 235delC/WT | 235delC/WT |
235delC/235delC | Induced labor |
| 17b |
IVS7-2A>G/IVS7-2A>G |
IVS7-2A>G/WT |
IVS7-2A>G/WT |
IVS7-2A>G/WT | Normal hearing |
Discussion
Due to genetic heterogeneity and phenotypic
diversity shown in patients with hearing loss, providing proper
genetic counseling for patients is still a great challenge
clinically. Up to now, hundreds of genes causing hereditary hearing
loss have been identified. Most of NSHL are induced by mutations in
the four genes including GJB2, SLC26A4, mtDNA 12SrRNA
and GJB3 (5–9). The findings provide theoretic basis for
the implementing of prenatal diagnosis of deafness genes. In this
study, Sanger sequencing was employed to analyze the 31 mutation
sites in these four genes in 116 NSHL patients.
In this study, 35 (30.17%) patients carrying
GJB2 mutations were detected, among which, 21 patients had
GJB2 genetic homozygous mutations, and 14 had GJB2
compound heterozygous mutations. GJB2 235delC had the
highest percentage, followed by 299_300delAT. Epidemiological
studies of GJB2 mutations in deaf patients in different
countries have revealed that 46% of Hungarian patients with NSHL
were caused by GJB2 mutations, 24.3% of patients with NSHL
among Americans were induced by GJB2 mutations, and 12.2–33%
of patients with NSHL among Chinese were due to GJB2
mutations (10). In this study, it
was found that 30.17% of patients with NSHL were caused by
GJB2 mutations, which suggested that the GJB2 gene
mutation plays an important role in hereditary NSHL. The types of
GJB2 mutation are different in different regions or races.
Studies of Yu et al (11) and
Dai et al (12) have
suggested that the mutation types of the most common GJB2 in
the Chinese people are 235delC, 299_300delAT and 176_191del16bp. In
this study, 235delC accounted for the highest percentage, followed
by 299_300delAT and 109G>A. A study by Zhang et al
(13) found that the mutation types
of GJB2 in patients with HSHL are consistent in Linyi.
SLC26A4 gene mutations will cause large
vestibular aqueduct syndrome, of which the mutation frequency ranks
second in patients with NSHL, next to GJB2 gene (14). Chai et al (15) reported that the incidence rate of
SLC26A4 gene mutations in Chinese patients with NSHL is
11.2%, and in this study it was 12.07%. A study on patients with
NSHL in China by Yuan et al (16) showed that the most common
SLC26A4 mutations in China are IVS7-2A>G, 2168A>G and
1174A>T. In this study, it was found that the most common
mutation in SLC26A4 was IVS7-2A>G, which is consistent
with the findings of Yuan et al (16).
In this study, 2 patients were found carrying the
mtDNA 12SrRNA 1555A>G mutations, which accounted for
1.72%. Carrying mtDNA 12SrRNA mutations can lead to the
occurrence of aminoglycoside drug-induced deafness (17). However, the proportion of patients
with drug-induced deafness is small due to the strict application
requirements of aminoglycosides in clinical practice. In this
study, no carrier of the definite GJB3 mutation site was
discovered, which might be resulted from the small sample size.
Therefore, further verification is needed.
In this study, prenatal diagnosis to 17 families
with hearing loss induced by definite pathogenic mutation suggested
that all their parents were carriers. Moreover, genetic analysis
and genetic counseling showed that genotypes of 6 fetuses were in
line with those of probands, so the 6 families decided to terminate
the pregnancy. No mutation was found in 3 fetuses, and the
genotypes of the other 8 fetuses were consistent with their
parents. Follow-up results showed that the 11 fetuses had normal
hearing after birth.
The deafness genes including GJB2, GJB3 and
SLC26A4 are mainly autosomal recessively inherited, while
12SrRNA is maternally inherited (18,19).
Detection of deafness couples and parents of patients with
mutations in GJB2, SLC26A4 and 12SrRNA genes were
performed to determine the genotypes and estimate recurrence risk
on the basis of genotypes. If the pathogenic genes carried by
parents are located on different genes, their descendants are at a
lower risk of illness and do not need prenatal diagnosis. If the
parents are carriers of the same gene mutation (GJB2 or
SLC26A4), their offspring have a 25% risk of deafness.
Prenatal diagnosis is recommended for a further birth. If mothers
carry 12SrRNA mutations, for their siblings, mother as well
as offspring, the administration of aminoglycosides should be
banned for life, which can effectively prevent the occurrence of
deafness.
To sum up, application of gene sequencing is very
useful to the genetic testing for patients with NSHL. In addition,
this method is simple in operation and can be applied in clinical
practice, providing accurate genetic counseling clinically.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL drafted the manuscript. HL and JQ were mainly
devoted to DNA extraction. JZ and YH were responsible for PCR. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Women and Children's Health Care Hospital of Linyi (Linyi, China).
Signed written informed consents were obtained from the patients or
the guardians.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Morton CC and Nance WE: Newborn hearing
screening - a silent revolution. N Engl J Med. 354:2151–2164. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shearer AE and Smith RJ: Genetics:
Advances in genetic testing for deafness. Curr Opin Pediatr.
24:679–686. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bozdoğan ST, Kuran G, Yüregir OO, Aslan H,
Haytoğlu S, Ayaz A and Arıkan OK: The prevalence of gap junction
protein beta 2 (GJB2) mutations in non syndromic sensorineural
hearing loss in Çukurova region. J Int Adv Otol. 11:118–121. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoefsloot LH, Feenstra I, Kunst HP and
Kremer H: Genotype phenotype correlations for hearing impairment:
Approaches to management. Clin Genet. 85:514–523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fang Y, Gu M, Wang C, Suo F, Wang G and
Xia Y: GJB2 as well as SLC26A4 gene mutations are
prominent causes for congenital deafness. Cell Biochem Biophys.
73:41–44. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai P, Stewart AK, Chebib F, Hsu A,
Rozenfeld J, Huang D, Kang D, Lip V, Fang H, Shao H, et al:
Distinct and novel SLC26A4/Pendrin mutations in
Chinese and U.S. patients with nonsyndromic hearing loss. Physiol
Genomics. 38:281–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Z, Li R, Chen J, Liao Z, Zhu Y, Qian Y,
Xiong S, Heman-Ackah S, Wu J, Choo DI, et al: Mutational analysis
of the mitochondrial 12S rRNA gene in Chinese pediatric
subjects with aminoglycoside-induced and non-syndromic hearing
loss. Hum Genet. 117:9–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia JH, Liu CY, Tang BS, Pan Q, Huang L,
Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, et al: Mutations in the
gene encoding gap junction protein beta-3 associated with autosomal
dominant hearing impairment. Nat Genet. 20:370–373. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma D, Zhang J, Luo C, Lin Y, Ji X, Hu P
and Xu Z: Genetic counseling for patients with nonsyndromic hearing
impairment directed by gene analysis. Mol Med Rep. 13:1967–1974.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai ZY, Sun BC, Huang SS, Yuan YY, Zhu YH,
Su Y and Dai P: Correlation analysis of phenotype and genotype of
GJB2 in patients with non-syndromic hearing loss in China.
Gene. 570:272–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu F, Han DY, Dai P, Kang DY, Zhang X, Liu
X, Zhu QW, Yuan YY, Sun Q, Xue DD, et al: Mutation of GJB2
gene in nonsyndromic hearing impairment patients: Analysis of 1190
cases. Zhonghua Yi Xue Za Zhi. 87:2814–2819. 2007.(In Chinese).
PubMed/NCBI
|
|
12
|
Dai P, Yu F, Han B, Liu X, Wang G, Li Q,
Yuan Y, Liu X, Huang D, Kang D, et al: GJB2 mutation
spectrum in 2,063 Chinese patients with nonsyndromic hearing
impairment. J Transl Med. 7:262009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang F, Xiao Y, Xu L, Zhang X, Zhang G,
Li J, Lv H, Bai X and Wang H: Mutation analysis of the common
deafness genes in patients with nonsyndromic hearing loss in Linyi
by SNP scan assay. Biomed Res Int. 2016:13029142016.PubMed/NCBI
|
|
14
|
Du W, Guo Y, Wang C, Wang Y and Liu X: A
systematic review and meta-analysis of common mutations of
SLC26A4 gene in Asian populations. Int J Pediatr
Otorhinolaryngol. 77:1670–1676. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chai Y, Huang Z, Tao Z, Li X, Li L, Li Y,
Wu H and Yang T: Molecular etiology of hearing impairment
associated with nonsyndromic enlarged vestibular aqueduct in East
China. Am J Med Genet A. 161A:2226–2233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan Y, Guo W, Tang J, Zhang G, Wang G,
Han M, Zhang X, Yang S, He DZZ and Dai P: Molecular epidemiology
and functional assessment of novel allelic variants of
SLC26A4 in non-syndromic hearing loss patients with enlarged
vestibular aqueduct in China. PLoS One. 7:e499842012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O'Sullivan M, Rutland P, Lucas D, Ashton
E, Hendricks S, Rahman S and Bitner-Glindzicz M: Mitochondrial
m.1584A 12S m62A rRNA methylation in families with m.1555A>G
associated hearing loss. Hum Mol Genet. 24:1036–1044. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao GD, Li SX, Chen DL, Feng HQ, Zhao SB,
Liu YJ, Guo LL, Yang ZM, Zhang XF, Sun CX, et al: Combination of
hearing screening and genetic screening for deafness-susceptibility
genes in newborns. Exp Ther Med. 7:218–222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim MA, Kim YR, Sagong B, Cho HJ, Bae JW,
Kim J, Lee J, Park HJ, Choi JY, Lee KY, et al: Genetic analysis of
genes related to tight junction function in the Korean population
with non-syndromic hearing loss. PLoS One. 9:e956462014. View Article : Google Scholar : PubMed/NCBI
|