Introduction
BMPs are a group of multi-functional growth factors
of the transforming growth factor β (TGF-β) superfamily. Binding of
BMP to BMP receptor type-2 (BMPR2) leads to phosphorylation of
BMPR1A, which activates its kinase activity and phosphorylates
regulatory Smads (R-Smads), including Smad1 at Ser463 and Ser465
(1). Phosphorylated R-Smads
(p-Smads) form complexes with common-partner Smads and translocate
into the nucleus to regulate transcription of target genes
(2).
Loss of function and mutations in the BMPR2 gene
have been identified in ~80% of patients with familial pulmonary
artery hypertension (PAH) and in 11–40% of patients with sporadic
PAH (3–5). Reduced expression levels of BMPR1A and
BMPR2 were also observed in the lungs of patients with idiopathic
and secondary PAH without detectable BMPR2 mutations (6,7) and in
experimental animal models of PAH (8,9).
Expression and phosphorylation levels of Smad1 significantly
decreased in lungs of rats with monocrotaline (MTC)-induced PAH
(10,11). The above evidence suggests that the
BMP receptor signaling is associated with the pathogenesis of PAH.
However, it remains to be elucidated how the BMP receptor signaling
causes PAH.
Smads have highly conserved N- and C-terminal
regions, termed mad homology (MH) 1 and MH2 domains, respectively
(12). The MH1 and MH2 domains are
bridged by a linker region with a variable length and consensus
amino acid sequences, which may be phosphorylated at Ser206 by
mitogen-activated protein kinase (MAPK) (13). The phosphorylation of Ser206 recruits
E3 ubiquitin-protein ligase SMURF1 to the linker region and leads
to degradation of Smad1 and interruption of nuclear translocation
(12,14). Activated MAPK signaling has been
observed to be positively associated with the occurrence and
development of PAH. However, it remains to be elucidated whether
MAPK contributes to PAH by promoting the degradation of Smad1
(15,16). Previous studies suggested the
involvement of the RhoA/Rho kinase pathway in the pathogenesis of
PAH induced by hypoxia, monocrotaline or high blood flow (17–19).
MAPK is one of the downstream signaling molecules of the RhoA/Rho
kinase pathway (20,21). The authors of the present study
hypothesized that the RhoA/Rho kinase signaling pathway may promote
the process of PAH by attenuating the BMP signaling pathway.
In the present study, using an experimental rat
model of PAH, a primary culture of PASMCs was established and used
to analyze cell proliferation under differential drug treatment.
The present study aimed to establish whether the RhoA/Rho kinase
signaling pathway activated by PDGF-BB enhances the proliferation
of PASMCs by suppressing the nuclear translocation of Smad1 induced
by BMP-2.
Materials and methods
Primary PASMC isolation and
culture
A total of 30 Male Sprague-Dawley (SD) rats (age, 4
weeks; weight 150–200 g) were purchased from Shanghai SLAC
Laboratory Animal Co., Ltd., (Shanghai, China). Animals were housed
under standard environmental conditions with a regulated
temperature (20–24°C) and humidity (45–50%), with a 12 h light/dark
cycle and free access to food and water. Animals were then divided
into two groups (n=15). One group were subcutaneously injected with
a single dose of MCT (40 mg/kg body weight; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) to induce PAH as previously reported
(22). The second group were
subcutaneously injected with an equal volume of normal saline as a
control and then fed normally for two weeks. The control SD rats
and PAH rats were subsequently anesthetized and pulmonary artery
systolic pressure was measured, as previously described, to
determine the establishment of the PAH model (23). All rats were sacrificed and the lungs
were harvested. Animal welfare and experimental procedures were
carried out in accordance with the Guide for the Care and Use of
Laboratory Animals, published by the Ministry of Science and
Technology of China (24), and were
approved by the Animal Ethics Committee of Shandong University.
Intralobar pulmonary arteries (2nd branches; 100–400
µm diameter) were dissected, rinsed in cold PBS two times, and the
adventitia and endothelium were removed using scissors and a cotton
swab in PBS. Arteries were cut into small pieces and dissociated in
0.25% pancreatin solution (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C for 30 min. The dissociation solution was
removed following centrifugation at a speed of 200 × g for 5 min at
4°C, and 5 ml Dulbecco's Modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc.) was added to re-suspend the
pellets.
Collected cells were cultured in 25 ml culture
flasks for 3–5 days in 10% fetal bovine serum (FBS)-DMEM in a
humidified atmosphere of 5% CO2 at 37°C. Purity
of the cultures was assessed by morphological appearance using
phase-contrast microscopy (magnification, ×200) and
immunofluorescent staining using selective anti-α-smooth muscle
actin (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) antibody
observed under a fluorescence microscope at a magnification of
×200. Cells were used for experiments at passage 3–5.
PASMC treatment
PASMCs were cultured in 10% FBS-DMEM at 37°C in a
humidified atmosphere of 5% CO2 to 95% confluence. The
medium was changed to 0.1% FBS-DMEM for 24 h and, subsequently,
fresh 10% FBS-DMEM was added to synchronize the cell cycle to the
G0 phase. Rho, in combination with GTP, activates
downstream Rho-associated protein kinase (ROCK), phosphorylating
its downstream substrates (25).
PASMCs from the control rats were pretreated with the specific drug
Y-27632 (10 µM; Sigma-Aldrich; Merck KGaA) of ROCK inhibitor, or
U0126 (20 µM; Sigma-Aldrich; Merck KGaA) of MEK1/2 inhibitor, were
added 12 h prior to PDGF-BB (10 ng/ml; PeproTech, Inc., Rocky Hill,
NJ, USA) treatment to activate the Rho kinase. PASMCs from PAH rats
were pretreated 12 h prior to PDGF-BB or BMP-2 (2 ng/ml; PeproTech,
Inc., Rocky Hill, NJ, USA) treatment with the specific drugs
Y-27632 or U0126 to activate Smad1.
Immunofluorescence staining
PASMCs from PAH rats at different time-points
following treatment (0–210 min) were fixed in 4% paraformaldehyde
at room temperature for 20 min and permeabilized in 0.1% Triton
X-100 in PBS at room temperature for 20 min. Samples were then
blocked with 5% BSA at room temperature for 30 min. The fixed cells
were incubated with rabbit-phospho-Smad1 (phosphorylation site,
Ser463/465; 1:1,000; cat. no. 13820; Cell Signaling Technology,
Inc., Danvers, MA, USA) primary antibodies at 4°C overnight and a
Rhodamine Red-labeled goat anti-rabbit immunoglobulin G (1:2,000;
cat. no. 305-295-003; Jackson ImmunoResearch Laboratories, Inc.,
West Grove, PA, USA) secondary antibody at room temperature for 60
min to assess nuclear translocation of p-Smad1 using confocal
fluorescence microscopy (magnification, ×200). The nuclei were
counterstained with DAPI (Roche Diagnostics, Basel, Switzerland) at
room temperature for 3 min. A total of 10 images per group at each
time point were randomly selected to observe nuclear translocation
and calculated the percentage of cells with p-Smad1 (Ser463/465)
nuclear staining using Image J software (National Institute of
Health, Bethesda, MD, USA).
Western blotting
Nuclear proteins were isolated using nuclear protein
extraction kit (Bestbio Corporation, Nanjing, China) and the total
cellular proteins were extracted using total protein extraction kit
(Bestbio Corporation, Nanjing, JS, CN). Protein concentration was
detected using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). A total of 30 µg protein was loaded per lane and subjected
to 12% SDS-PAGE. Samples were then electrophoretically transferred
to nitrocellulose membranes. Following blocking with 5% BSA at room
temperature for 30 min, the membranes were incubated with the
following primary antibodies at 4°C overnight: Rabbit-ROCK1 (cat.
no. 4035), mouse-MEK1 (cat. no. 2352), rabbit-phospho-MEK1/2
(Ser217/221; cat. no. 9154), mouse-ERK1/2 (cat. no. 4696),
rabbit-phospho-ERK1/2 (Thr202/Tyr204; cat. no. 4376),
rabbit-phospho-Smad1 (Ser206; cat. no. 9553), rabbit-phospho-Smad1
(Ser463/465; cat. no. 13820), rabbit-Smad1 (cat. no. 9743; 1:1,000;
all Cell Signaling Technology, Inc.), mouse-β-actin (1:1,000
dilution; cat. no. 21800; Signalway Antibody LLC, College Park, MD,
USA) or rabbit-histone H3.1 (1:500; cat. no. 32667; Signalway
Antibody LLC). Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G
(cat. no. 115-005-166) or goat anti-rabbit immunoglobulin G (cat.
no. 111-005-144; both 1:2,000; Jackson ImmunoResearch Laboratories,
Inc.) at room temperature for 60 min. Bands were identified through
enhanced chemiluminescence (ECL) using the Immobilon™ Western
Chemiluminescent HRP Substrate (EMD Millipore, Billerica, MA, USA).
Scanned images were inverted and quantified by densitometric
analysis with Quantity One 1-D software (version 4.6.9; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Histone H3.1 and β-actin
were used as loading controls for nuclear and total cellular
proteins, respectively.
Cell proliferation analyses
Cell proliferation was analyzed using Cell Counting
kit (Dojindo Molecular Technologies, Inc., Kumamuto, Japan)
according to the manufacturer's protocol. Briefly, PAH-PASMCs were
cultured in a 96-well plate overnight and were pre-treated with the
specific drug (Y-27632 or U0126) 12 h prior to the PDGF-BB or BMP-2
treatment for 24 h. A total of 10 µl of the WST-8 solution from the
cell counting kit was added to each well and incubated for another
4 h. Cell viability was analyzed by measuring the absorbance at a
wavelength of 570 nm using a microplate reader. The optical density
value was considered proportional to the number of living
cells.
Statistical analysis
Data are presented as the mean ± standard deviation,
one-way analysis of variance followed by a post-hoc Bonferroni's
test was performed using SPSS software (version 16.0; SPSS, Inc.,
Chicago, IL, USA) to compare the differences between groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RhoA/Rho signaling pathway is involved
in PAH through regulating the phosphorylation and degradation of
Smad1
To study the alterations in the RhoA/Rho and BMP
signaling in PAH, the protein expression levels of ROCK1, MEK1 and
ERK, as well as total Smad1 and p-Smad1 in PASMCs from the control
and PAH rats were determined. The expression levels of ROCK1
increased in PASMCs from the PAH group compared with the control
rats (Fig. 1A). The same effect was
observed for the phosphorylated forms of MEK1, ERK and Smad1
(Ser206), but not the total proteins (Fig. 1A). The increased phosphorylation of
Smad1 at Ser206 and decreased expression levels of Smad1 were
observed in PAH-PASMC compared with the control PASMCs (Fig. 1B). By contrast, the nuclear
expression levels of p-Smad1 (Ser463/465), which stimulates
transcription of target genes, decreased in PASMCs from the rat
models of PAH compared with the control rats (Fig. 1B). These results indicate that the
Rho signaling pathway was activated in PAH compared with the
control group, leading to the degradation and decreased
translocation of Smad1 into the nucleus.
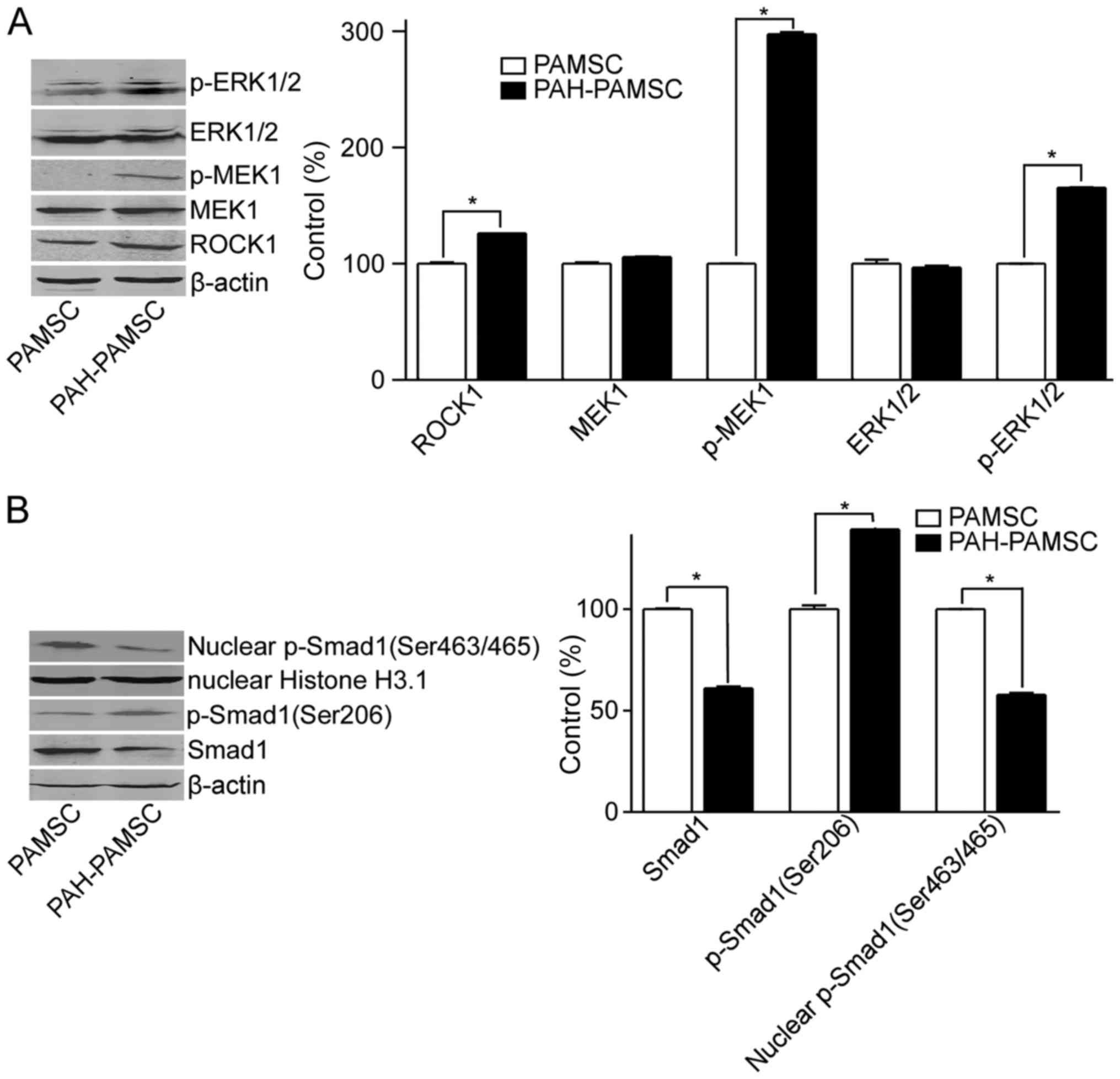 | Figure 1.Expression levels of Smad1 and
proteins associated with the Rho signaling pathway. PASMCs were
isolated from pulmonary arteries of control rats or rat models of
PAH and cultured. Total and nuclear proteins were extracted. (A)
Protein levels of p-ERK1/2, ERK1/2, p-MEK1, MEK1 and ROCK1 in
PASMCs and PAH-PASMCs were measured by western blotting. (B) Total
Smad1, p-Smad1 (Ser206) and nuclear Smad1 (Ser463/465) protein
expression levels were measured by western blotting in PASMCs and
PAH- PASMCs. *P<0.05. Data are presented as the mean ± standard
deviation, n=3. PAH, pulmonary artery hypertension; ROCK1,
Rho-associated protein kinase 1; p, phosphorylated; PASMC,
pulmonary artery smooth muscle cell; ERK1/2,
mouse-mitogen-activated protein kinase; MEK1, dual specificity
mitogen-activated protein kinase kinase; Smad1, mothers against
decapentaplegic homolog 1. |
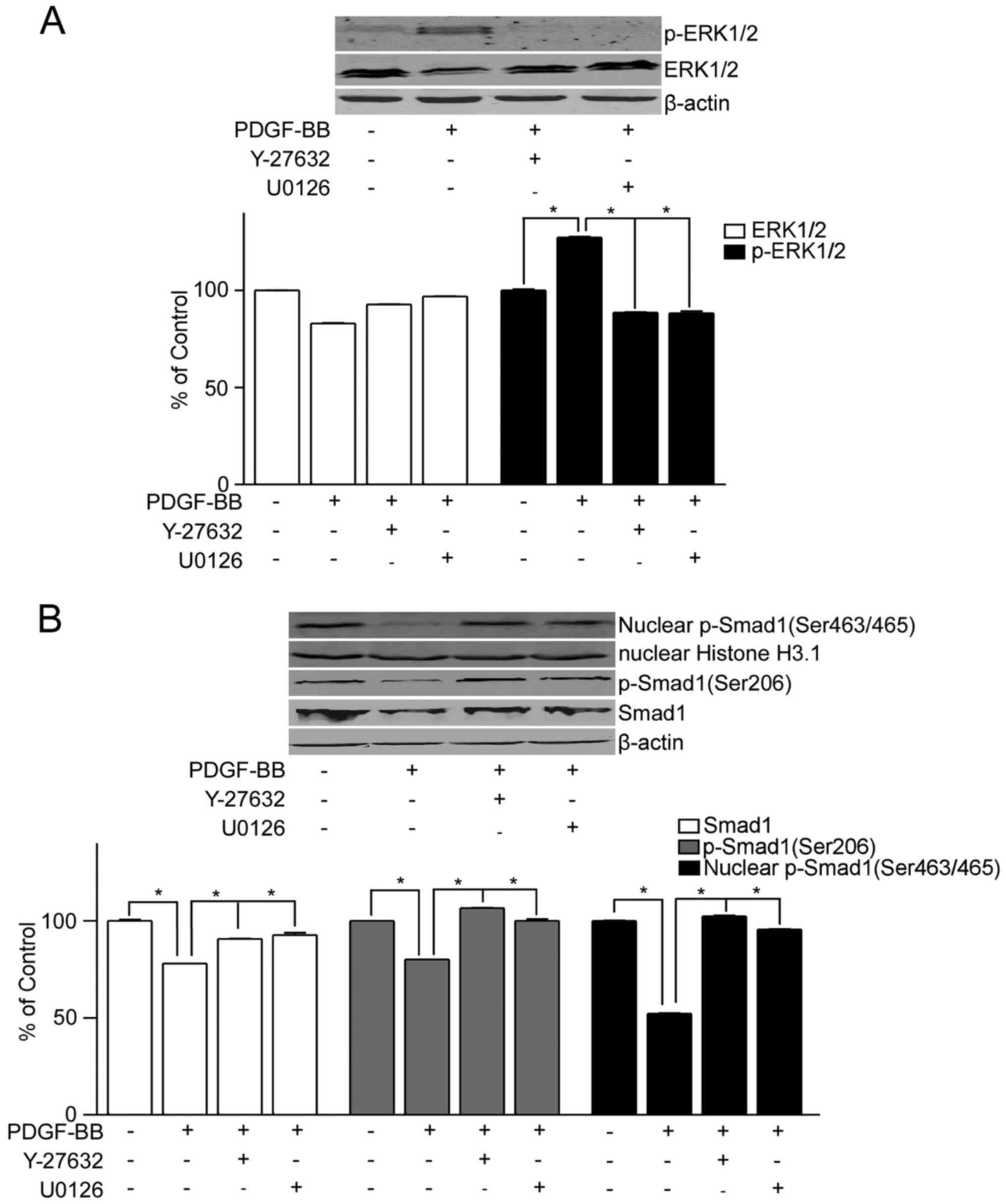
To further confirm the role of the RhoA/Rho
signaling in the regulation of Smad1, the PASMCs from control rats
were treated with PDGF-BB, with or without Y-27632 or U0126 (the
inhibitors of ROCK or MEK1/2, respectively) to activate or inhibit
the RhoA/Rho signaling, respectively. Phosphorylation of ERK
increased following treatment with PDGF-BB compared with the
untreated control, but not following co-treatment with Y-27632 or
U0126 (Fig. 2A). Furthermore,
decreased expression levels of Smad1 and p-Smad1 (Ser206) were
observed in PASMCs treated with PDGF-BB compared with the untreated
control, but not in cells treated with PDGF-BB + Y-27632 or U0126
(Fig. 2B). Decreased expression
levels of nuclear p-Smad1 (Ser463/465) were also observed in PASMCs
treated with PDGF-BB compared with the untreated control cells, but
not in PASMCs treated with PDGF-BB + Y-27632 or U0126 (Fig. 2B).
Taken together, the above data indicated that
activation of the RhoA/Rho signaling may lead to the degradation of
Smad1 and the decrease in the nuclear translocation of p-Smad1,
suggesting that the RhoA/Rho signaling may be involved in PAH
through regulating Smad1.
RhoA/Rho activation inhibits
BMP-2-induced nuclear translocation of Smad1
To determine the dynamic interactions between the
RhoA/Rho and BMP-2 signaling, immunofluorescent staining of p-Smad1
was performed in PAH-PASMCs under various drug treatments.
Treatment with BMP-2 significantly increased the levels of nuclear
p-Smad1 (Ser463/465), which was observed from 30 to 120 min after
treatment with BMP-2 compared with the untreated PAH-PASMCs, the
maximum increase was at 60 min and markedly decreased at 150 min
following treatment with BMP-2 for 210 min (Fig. 3A and B). Furthermore, an increase in
the expression of p-Smad1 (Ser463/465) and p-Smad1 (Ser206), but
not the total Smad1 protein was observed in the cytoplasm and
nucleus of the PAH-PASMCs treated with BMP-2, compared with
untreated PAH-PASMCs (Fig. 3C).
Pretreatment with PDGF-BB (12 h before adding BMP-2) decreased the
magnitude of nuclear expression of p-Smad1 (Ser463/465) induced by
BMP-2 (Fig. 3A and B), as well as
decreased the expression of p-Smad1 (Ser206) and total Smad1
protein, compared with PAH-PASMCs treated with BMP-2 (Fig. 3C). Furthermore, pretreatment with
Y-27632 or U0126 (12 h before treatment with PDGF-BB) rescued the
BMP-2 induced nuclear translocation of p-Smad1 (Ser463/465) that
was inhibited by PDGF-BB (Fig.
3A-C). In addition, Y-27632 and U0126 rescued the
PDGF-BB-decreased expression of p-Smad1 (Ser206) and total Smad1
(Fig. 3C).
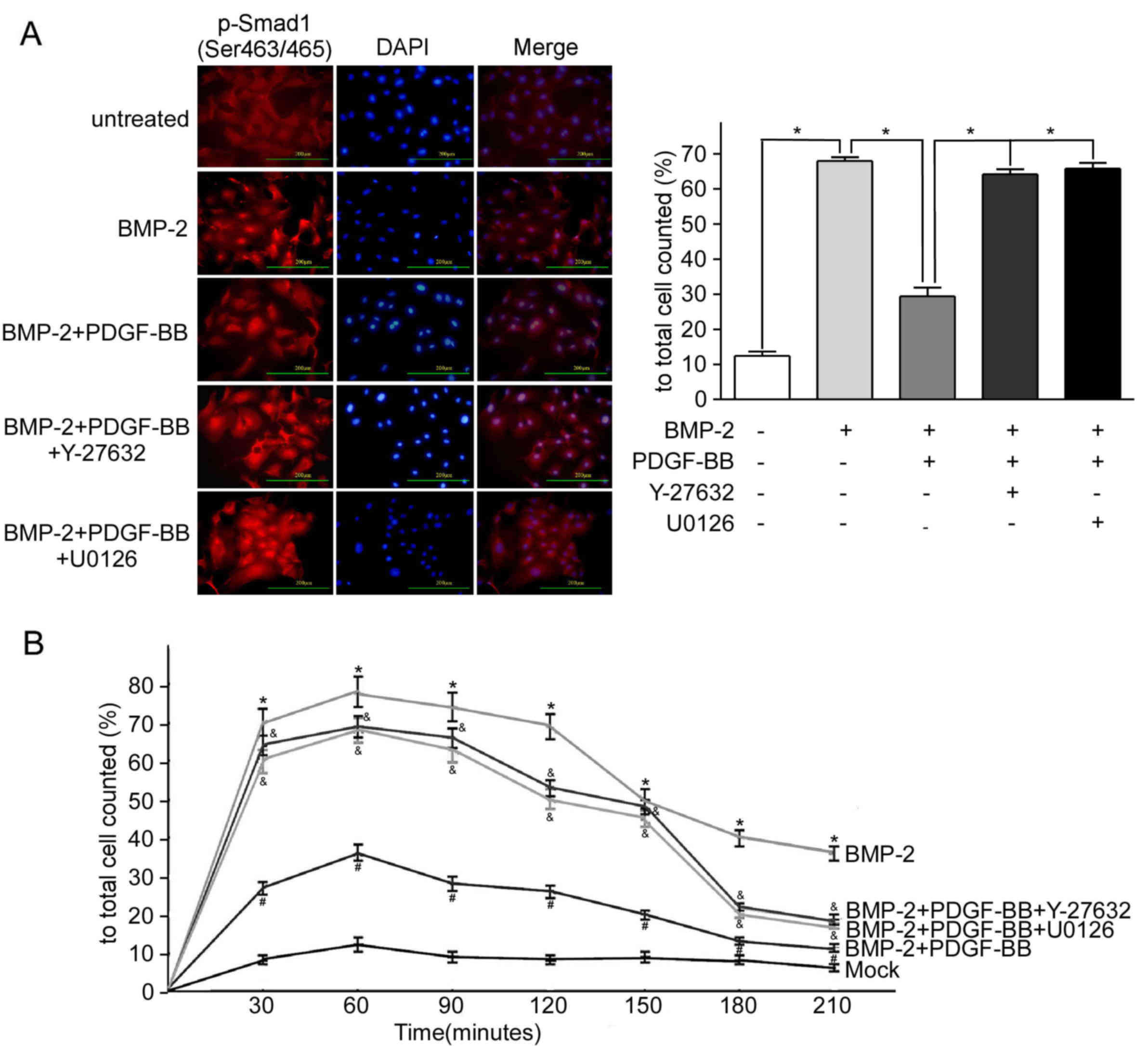 | Figure 3.Activation of the Rho signaling
pathway inhibits BMP-2-induced Smad1 expression in PAH. PAH-PASMCs
were cultured and treated with 10 ng/ml Rho activator PDGF-BB, 2
ng/ml Smad1 activitor BMP-2, 10 µM ROCK inhibitor Y-27632 and 20 µM
MEK1 inhibitor U0126. Immunofluorescence staining results were
measured at 0–210 min of the experiment. Or 12 h following
treatment with final BMP-2, total and nuclear proteins were
extracted, respectively. (A) Representative confocal
immunofluorescent staining micrographs of distribution of p-Smad1
(Ser463/465) 60 min after exposure to activator and inhibitor in
PASMCs. The results were analyzed quantitatively. *P<0.05; Scale
bar, 200 µm. (B) Changes of the percentage of cells positive for
p-Smad (Ser463/465) nuclear accumulation after activator and
inhibitor exposure in PAH-PASMCs. *P<0.05. the BMP-2 group vs.
the untreated group; #P<0.05 the BMP-2+PDGF-BB group
vs. the BMP-2 group; &P<0.05 the
BMP-2+PDGF-BB+Y27632 or the BMP-2+PDGF-BB+U0126 group vs. the
BMP-2+PDGF-BB group. (C) Total Smad1, total p-Smad1 (Ser206) and
nuclear p-Smad1 (Ser463/465) protein levels following activation of
the Rho and BMP-2 signaling pathways in PASMCs from rat models of
PAH were measured by western blotting. *P<0.05. Data are
presented as the mean ± standard deviation, n=3. PAH, pulmonary
artery hypertension; PASMC, pulmonary artery smooth muscle cell;
BMP-2, bone morphogenetic protein 2; PDGF-BB, platelet derived
growth factor-BB; Smad1, mothers against decapentaplegic homolog 1;
p, phosphorylated. |
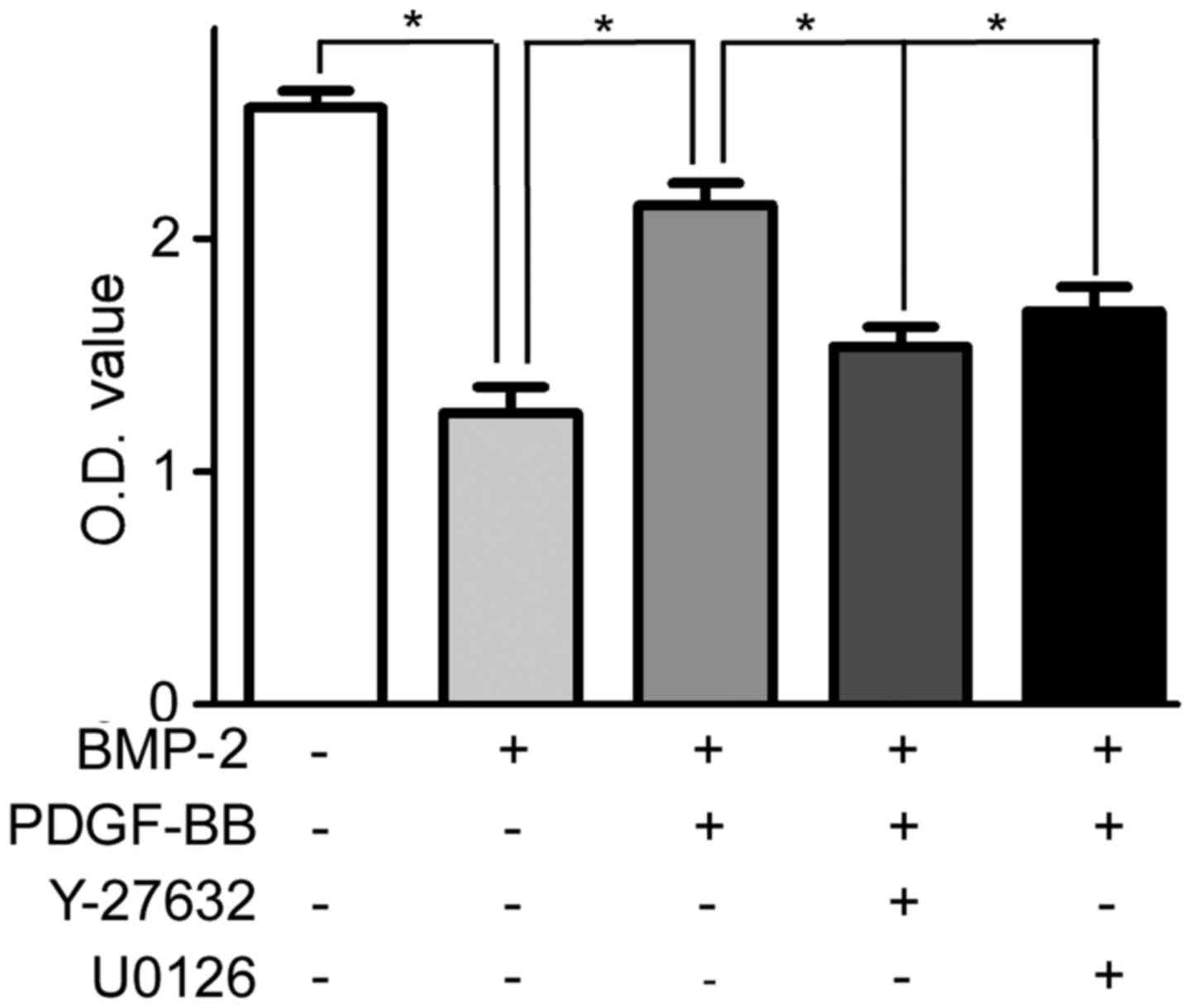
RhoA/Rho activation rescues
BMP-2-inhibited proliferation of PAH-PASMCs
Compared with the untreated PAH-PASMCs, treatment
with BMP-2 significantly decreased the viability of PAH-PASMCs
(Fig. 4). Pretreatment with PDGF-BB
(12 h after adding BMP-2) for 12 h rescued the decreased viability
of PAH-PASMCs (Fig. 4). Furthermore,
pretreatment (12 h before adding PDGF-BB) with ROCK and ERK1/2
inhibitors, Y-27632 and U0126, counteracted the effect of PDGF-BB;
the viability of PAH-PASMCs decreased to the comparable level to
the group of cells treated with BMP-2 only (Fig. 4).
Discussion
The results of the present study indicated that
activation of the Rho signaling pathway decreased Smad1 and nuclear
translocation of p-Smad1 in PAH-PASMCs. PDGF-BB, an activator of
the Rho signaling pathway, interfered with the BMP-2-stimulated
nuclear translocation of p-Smad1 via a Rho/ROCK/MEK/ERK-dependent
mechanism (Fig. 5). PDGF-BB
stimulated PASMC proliferation inhibited by BMP-2.
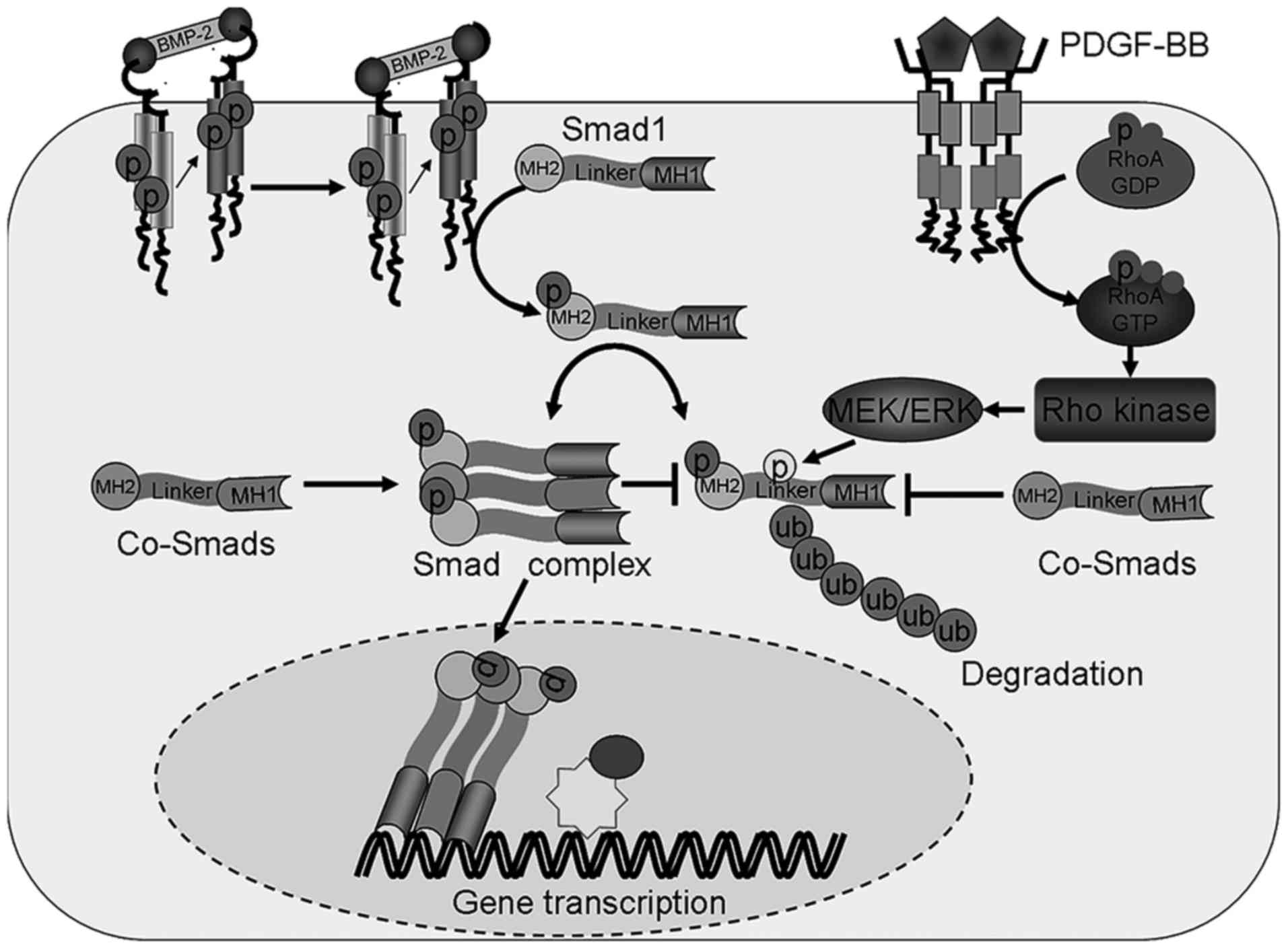 | Figure 5.Crosstalk between the Rho kinase and
BMP-2 signaling pathways. Crosstalk between the Rho kinase and
BMP-2 signaling pathways occur via the phosphorylation of the Smad1
linker region, which leads to the degradation or phosphorylation of
its C-terminal, resulting in its translocation into the nucleus.
Linker region phosphorylation following the activation of the RHO
kinase signaling pathway leads to Smad1 ubiquitination and
degradation. The C-terminus phosphorylation of Smad1 induced by
BMP-2 leads to nuclear the translocation of this protein. PDGF-BB,
platelet derived growth factor-BB; Smad1, mothers against
decapentaplegic homolog 1; BMP-2, bone morphogenetic protein 2; p,
phosphorylated; ub, ubiquitination; RhoA, transforming protein
RhoA; GTP, guanosine triphosphate; GDP, guanosine diphosphate;
Co-Smads, common mediator mothers against decapentaplegic homologs;
MEK, mitogen-activated protein kinase kinase; ERK,
mitogen-activated protein kinase; MH, mad homology domain. |
The present study demonstrated that PDGF-BB induced
degradation of Smad1 through phosphorylation of Smad1 at Ser206,
which reduced the nuclear translocation of this protein induced by
BMP-2-mediated phosphorylation at Ser463/465. These results suggest
a novel molecular mechanism by which the Rho kinase signaling may
intercept the BMP signaling and stimulate the proliferation of
PAH-PASMCs. This implies that p-Smad1 may be a novel marker or a
treatment target in PAH therapy.
Previous studies indicated that the Rho kinase and
BMP signaling pathways serve important counter-regulatory roles in
pulmonary vascular hypertension (3–7,26–28). As
an important molecule, activated Rho kinase pathway participates in
the pathogenesis of hypoxia-, monocrotaline- and high pulmonary
blood flow-induced PAH; treatment with ROCK inhibitors reduces
pulmonary artery pressure and attenuates pulmonary arterial lesions
in animal models of PAH (18,19,26).
In clinical trials, activated Rho kinase was also detected in
patients with PAH, and Rho kinase inhibitor fasudil could decrease
pulmonary artery pressure and pulmonary artery resistance (27,28). By
contrast, the BMP signaling pathway exhibits meaningful effects on
PAH. For example, Luo et al (29) demonstrated that APN suppressed the
proliferation of PAH-PASMCs by regulating the AMPK/BMP/Smad
pathway. Furthermore, Zhang et al (30) indicated that BMP-2 participates in
the regulation of Ca(2+) signaling in PAH-PASMCs,
reducing cell proliferation and migration (29,30).
Mutations in BMPR2 or reduced expression levels of BMPR2 and Smad1
were observed in patients with PAH and animal models of this
disease (3–7).
Activation of Rho kinase and disruption of BMP
signaling both have been demonstrated to promote the proliferation
of PASMCs (31,32). In the present study, PDGF-BB
stimulated proliferation of PASMCs by interfering with nuclear
accumulation of Smad1, suggesting that the Rho kinase signaling and
BMP signaling converge on Smad1.
Protein expression levels were determined by western
blotting to elucidate the mechanism of PDGF-BB-mediated inhibition
of nuclear accumulation of Smad1. PDGF-BB inhibited BMP-2-induced
nuclear accumulation of Smad1 through certain mechanisms, inducing
degradation. Furthermore, PDGF-BB stimulated the Rho signaling
pathway and increased Smad1 phosphorylation at Ser206, leading to
the degradation of Smad1 and the interruption of nuclear
translocation. Rho kinase has been demonstrated to mediate the
mitogenic effect of PDGF-BB on PASMCs in PAH (33). MAPK was demonstrated to prevent
nuclear translocation of Smads by phosphorylating the linker region
(14). In the present study, ROCK
inhibitor or MEK1/2 inhibitor reversed the PDGF-BB-mediated
suppression of nuclear translocation of Smad1. It may therefore be
hypothesized that PDGF-BB phosphorylates the linker region of
p-Smad1 via the Rho kinase/MEK/ERK pathway, leading to its
degradation and decreased nuclear translocation. Increased
expression levels of p-Smad1 (Ser206) were observed in the
PAH-PAMSCs treated with BMP-2 compared with the untreated
PAH-PAMSCs; however, treatment with PDGF-BB decreased the
expression levels of p-Smad1 (Ser206) compared with untreated
cells. PDGF-BB treatment increased p-Smad1 (Ser206), leads to its
degradation. However, BMP-2 affected p-Smad1 (Ser463/465), but not
p-Smad1 (Ser206). The reason for the increase of p-Smad1 (Ser206)
may be that BMP-2 is short-acting and decreases its effect. The
results of the present study indicate that Rho kinase
phosphorylates p-Smad1 at the linker region and BMP-2 phosphorylate
Smad1 at the C-terminal; these two signaling pathways crosstalk at
p-Smad1 and exert counter regulatory effects on the cell fate.
Future studies are required to further confirm this hypothesis,
including those to detect the phosphorylation of truncations, which
contain different lengths and locations of Smad1.
Rho-kinase appears to be a point of convergence of
various signal transduction pathways involved in the mechanism of
both primary and secondary PAH (31,34). The
results of the present study suggested that suppressed nuclear
translocation of Smad1 is one of the mechanisms used by the
activated Rho kinase to stimulate the proliferation of PASMCs in
PAH. In conclusion, PDGF-BB-activated Rho kinase suppressed
BMP-2-induced nuclear translocation p-Smad1 via MEK1/ERK1/2 in rat
PASMCs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Foundation
for Medical Program of Shandong Province (grant no. 2013WS0220) and
the Key Research and Development Plan of Shandong Province (grant
no. 2017G006042).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HW performed the experiments; DZ analyzed the data
and drafted the manuscript; LL aquired and analyzed the data; WX
conceived and designed the present study; FL designed the current
study, acquired data, analyzed and interpreted the data, and
drafted the manuscript critically for important intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal welfare and experimental procedures were
performed in accordance with the Guide for the Care and Use of
Laboratory Animals, published by the Ministry of Science and
Technology of China. The present study was also approved by the
Animal Ethics Committee of Shandong University (Shandong,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miyazono K, Kamiya Y and Morikawa M: Bone
morphogenetic protein receptors and signal transduction. J Bio
chem. 147:35–51. 2010.
|
|
2
|
Sieber C, Kopf J, Hiepen C and Knaus P:
Recent advances in bmp receptor signaling. Cytokine Growth Factor
Rev. 20:343–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Awad KS, West JD, de Jesus Perez V and
MacLean M: Novel signaling pathways in pulmonary arterial
hypertension. Pulm Circ. 6:285–294. 2015. View Article : Google Scholar
|
|
4
|
West J, Austin E, Fessel JP, Loyd J and
Hamid R: Rescuing the BMPR2 signaling axis in pulmonary arterial
hypertension. Drug Discov Today. 19:1241–1245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Soubrier F, Chung WK, Machado R, Grünig E,
Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH and
Humbert M: Genetics and genomics of pulmonary arterial
hypertension. J Am Coll Cardio. l62 25 Suppl:D13–D21. 2013.
View Article : Google Scholar
|
|
6
|
Ormiston ML, Upton PD, Li W and Morrell
NW: The promise of recombinant BMP ligands and other approaches
targeting BMPR-II in the treatment of pulmonary arterial
hypertension. Glob Cardiol Sci Pract. 2015:472015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Long L, Ormiston ML, Yang X, Southwood M,
Gräf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, et
al: Selective enhancement of endothelial BMPR-II with BMP9 reverses
pulmonary arterial hypertension. Nat Med. 21:777–785. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang J, Li X, Al-Lamki RS, Wu C, Weiss A,
Berk J, Schermuly RT and Morrell NW: Sildenafil potentiates bone
morphogenetic protein signaling in pulmonary arterial smooth muscle
cells and in experimental pulmonary hypertension. Arterioscler
Thromb Vasc Biol. 33:34–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Price LC, Montani D, Tcherakian C,
Dorfmüller P, Souza R, Gambaryan N, Chaumais MC, Shao DM, Simonneau
G, Howard LS, et al: Dexamethasone reverses monocrotaline-induced
pulmonary arterial hypertension in rats. Eur Respir J. 37:813–822.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Machado RD, Southgate L, Eichstaedt CA,
Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG,
Eyries M, et al: Pulmonary arterial hypertension: A current
perspective on established and emerging molecular genetic defects.
Hum Mutat. 36:1113–1127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Upton PD and Morrell NW: The transforming
growth factor-β-bone morphogenetic protein type signalling pathway
in pulmonary vascular homeostasis and disease. Exp Physiol.
98:1262–1266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wrana JL: Regulation of smad activity.
Cell. 100:189–192. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sapkota G, Alarcón C, Spagnoli FM,
Brivanlou AH and Massagué J: Balancing BMP signaling through
integrated inputs into the Smad1 linker. Mol Cell. 25:441–454.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kretzschmar M, Doody J and Massague J:
Opposing bmp and egfsignalling pathways converge on the tgf-beta
family mediator smad1. Nature. 389:618–622. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wilson JL, Yu J, Taylor L and Polgar P:
Hyperplastic growth of pulmonary artery smooth muscle cells from
subjects with pulmonary arterial hypertension is activated through
JNK and p38 MAPK. PLoS One. 10:e01236622015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin C and Guo J, Qiu X, Ma K, Xiang M, Zhu
X and Guo J: IGF-1 induces iNOS expression via the p38 MAPK signal
pathway in the anti-apoptotic process in pulmonary artery smooth
muscle cells during PAH. J Recept Signal Transduct Res. 34:325–331.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ziino AJ, Ivanovska J, Belcastro R,
Kantores C, Xu EZ, Lau M, McNamara PJ, Tanswell AK and Jankov RP:
Effects of rho-kinase inhibition on pulmonary hypertension, lung
growth, and structure in neonatal rats chronically exposed to
hypoxia. Pediatr Res. 67:177–182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yasuda T, Tada Y, Tanabe N, Tatsumi K and
West J: Rho-kinase inhibition alleviates pulmonary hypertension in
transgenic mice expressing a dominant-negative type II bone
morphogenetic protein receptor gene. Am J Physiol Lung Cell Mol
Physiol. 301:L667–L674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jasińska-Stroschein M, Owczarek J,
Sołtysiak U and Orszulak-Michalak D: Rosuvastatin intensifies the
beneficial effects of rho-kinase inhibitor in reversal of
monocrotaline-induced pulmonary hypertension. Arch Med Sci.
12:898–905. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Y, Ren W, Warburton R, Toksoz D and
Fanburg BL: Serotonin induces Rho/ROCK-dependent activation of
Smads 1/5/8 in pulmonary artery smooth muscle cells. FASEB J.
23:2299–2306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen XY, Dun JN, Miao QF and Zhang YJ:
Fasudil hydrochloride hydrate, a rho-kinase inhibitor, suppresses
5-hydroxytryptamine-induced pulmonary artery smooth muscle cell
proliferation via jnk and erk1/2 pathway. Pharmacology. 83:67–79.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanabe T, Furuya H, Kanemoto N, Goto Y and
Hata J: Experimental study on monocrotaline induced pulmonary
hypertensive rats. (1) Effect of long-term injection of
immunosuppressant. Tokai J Exp Clin Med. 6:41–48. 1981.PubMed/NCBI
|
|
23
|
Li F, Xia W, Li A, Zhao C and Sun R:
Long-term inhibition of Rho kinase with fasudil attenuates high
flow induced pulmonary artery remodeling in rats. Pharmacol Res.
55:64–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
The Ministry of Science and Technology of
the People's Republic of China: Guidance Suggestions for the Care
and Use of Laboratory Animals. 2006.http://www.most.gov.cn/fggw/zfwj/zfwj2006/200609/t20060930_54389.htmSeptember
30–2006
|
|
25
|
Amano M, Nakayama M and Kaibuchi K:
Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell
polarity. Cytoskeleton (Hoboken). 67:545–554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gupta V, Gupta N, Shaik IH, Mehvar R,
McMurtry IF, Oka M, Nozik-Grayck E, Komatsu M and Ahsan F:
Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary
preferential vasodilation in pulmonary arterial hypertension. J
Control Release. 167:189–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiao JW, Zhu XY, Wang QG, Zhang DZ, Cui
CS, Zhang P, Chen HY and Meng LL: Acute effects of Rho-kinase
inhibitor fasudilon pulmonary arterial hypertension in patients
with congenital heart defects. Circ J. 79:1342–1348. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang X, Wang YF, Zhao QH, Jiang R, Wu Y,
Peng FH, Xu XQ, Wang L, He J and Jing ZC: Acute hemodynamic
response of infused fasudil in patients with pulmonary arterial
hypertension: A randomized, controlled, crossover study. Int J
Cardiol. 77:61–65. 2014. View Article : Google Scholar
|
|
29
|
Luo L, Zheng W, Lian G, Chen H, Li L, Xu C
and Xie L: Combination treatment of adipose-derived stem cells and
adiponectin attenuates pulmonary arterial hypertension in rats by
inhibiting pulmonary arterial smooth muscle cell proliferation and
regulating the AMPK/BMP/Smad pathway. Int J Mol Med. 41:51–60.
2018.PubMed/NCBI
|
|
30
|
Zhang Y, Lu W, Yang K, Xu L, Lai N, Tian
L, Jiang Q, Duan X, Chen M and Wang J: Bone morphogenetic protein 2
decreases TRPC expression, store-operated Ca(2+) entry, and basal
[Ca(2+)]i in rat distal pulmonary arterial smooth muscle cells. Am
J Physiol Cell Physiol. 304:C833–C843. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Watanabe H: Rho-kinase activation in
patients with pulmonary arterial hypertension. Circ J.
73:1597–1598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Morrell NW: Role of bone morphogenetic
protein receptors in the development of pulmonary arterial
hypertension. AdvExp Med Biol. 661:251–264. 2010. View Article : Google Scholar
|
|
33
|
Kamiyama M, Utsunomiya K, Taniguchi K,
Yokota T, Kurata H, Tajima N and Kondo K: Contribution of Rho A and
Rho kinase to platelet-derived growth factor-BB-induced
proliferation of vascular smooth muscle cells. J Atheroscler
Thromb. 10:117–123. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nunes KP, Rigsby CS and Webb RC:
Rhoa/rho-kinase and vascular diseases: What is the link? Cell Mol
Life Sci. 67:3823–3836. 2010. View Article : Google Scholar : PubMed/NCBI
|