Introduction
Diabetic mellitus (DM), a major health problem due
to its high prevalence and concomitant risks of cardiovascular
disease, has been identified as a major factor associated with
cardiac mortality. There is currently rapid growth in the number of
diabetic patients worldwide, which will potentially reach 439
million by 2030 (1); clinical,
pathological and experimental studies all indicate this level of
increase (2,3). Diabetic cardiomyopathy (DCM), as an
independent cardiovascular complication of DM, is characterized by
myocardial dysfunction in the absence of valvular heart disease,
hypertension and coronary heart disease. Previous studies have also
revealed that myocardial inflammation, lipid accumulation,
oxidative stress, apoptosis and fibrosis are associated with the
pathophysiology of DCM (4,5). Although the pathogenesis of DCM is
multifactorial, oxidative stress has been recognized as a major
activating factor, and it serves a pivotal role in the pathology of
DCM (1–8).
Protein kinase C (PKC) enzymes comprise a family of
at least 12 serine/threonine protein kinases, which serve important
roles in signal transduction and intracellular crosstalk (9). A number of previous studies have
demonstrated the association of overexpression or activation of
PKCβ2 in the development and progression of DCM
(10,11). The Shc adaptor protein family
consists of P66shc, P52shc and
P46shc, but only P66shc has been indicated to
serve an important role as a redox enzyme implicated in
mitochondrial reactive oxygen species (ROS) generation and the
translation of oxidative signals into apoptosis (12). Hyperglycemic and hydrogen peroxide
stress may activate the PKCβ2 isoform to induce
phosphorylation of P66shc at ser36, which can result in
the transfer of phosphorylated (p)-P66shc from the
cytosol to the inner mitochondrial membrane, where
p-P66shc may amplify oxidative stress and catalyze ROS
production via cytochrome oxidation (13–15).
Consequently, it may be hypothesized that the
PKCβ2/P66shc signaling pathway is associated
with the pathogenesis of DCM.
Carvedilol, as a non-selective third-generation
β-adrenoceptor and selective α-adrenoceptor blocker, has been
widely used in the treatment of heart failure. In particular,
previous studies have demonstrated that carvedilol exerts
vasodilatatory, anti-oxidative stress and anti-inflammatory effects
(16–18).
In the present study, the aim was to investigate
whether the PKCβ2/P66shc oxidative
stress-signaling pathway is associated with DCM, and to investigate
the role and mechanisms of carvedilol in preserving cardiac
function.
Materials and methods
Animals
A total of 70 male 7 week old Wistar rats weighing
180–200 g were purchased from the Laboratory Animal Center of North
China University of Science and Technology (Tangshan, China). Prior
to the experiments, all rats were fed on a basal diet (BD) for 1
week. All experimental protocols were approved by the Animal Care
and Use Committee of North China University of Science and
Technology and in compliance with the Guide for the Care and Use of
Laboratory Animals (19).
All rats were housed at 22°C under a 12-h light-dark
cycle. Diabetes was induced by feeding animals a high-energy diet
(HD) during the entire experimental period. Another group (n=20) of
rats fed on BD (75% corn, 20% wheat bran, 3% fish meal, 1.5% farina
and 0.5% salt) served as a control group. To create the high-energy
diet, 20% (w/) sucrose and 20% (w/) lard were added to the BD.
Following 4 weeks, diabetes was induced in rats in the HD group by
a single intraperitoneal injection of streptozotocin (STZ; Abcam,
Cambridge, UK) at a dose of 45 mg/kg dissolved in 100 mM citrate
buffer (pH 4.5), whereas the control group rats were administered
an equivalent volume of citrate buffer. Blood glucose levels were
examined 1 week following STZ injection using a hand-held
glucometer (Roche Diagnostics GmbH, Mannheim, Germany) by tail vein
puncture blood sampling. Rats exhibiting a blood glucose value
≥16.7 mmol/l were used for the study (n=45). The diabetic rat group
(DM, n=15) and the other 30 diabetic rats were treated with
carvedilol [low dosage (n=15), 1 mg/kg/d (CarL); high dosage
(n=15), 10 mg/kg/d (CarH); Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany] or vehicle (the control group received a basal diet
without any drugs at this time) for 20 weeks. All rats were
provided with water and food ad libitum.
Measurement of left ventricular
function
Trans-thoracic echocardiographic analysis was
performed using a Vevo770 imaging system (VisualSonics, Inc.,
Toronto, ON, Canada) equipped with a high-frequency transducer
(frequency band, 12 MHz). Images were obtained from rats
anesthetized with pentobarbital sodium (1.5%; 30 mg/kg; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China). Left
ventricular end-diastolic diameter (LVEDd), left ventricular
end-systolic diameter (LVEDs) and left ventricular ejection
fraction (LVEF) were measured to evaluate the function of the
heart. Mean measurements were calculated for five consecutive
cardiac cycles and repeated three times. Blood N-terminal
pro-B-type natriuretic peptide (NT-proBNP) levels were determined
with an auto-biochemical analysis system (Roche
Cobas®C311; Roche Diagnostics, Indianapolis, IN,
USA).
Measurement of biochemical factors and
heart-to-body weight ratio
Blood glucose and total cholesterol were determined
with the auto-biochemical analysis system (Roche
Cobas®C311). Following analysis, rats were sacrificed
and decapitated, and the heart was immediately removed from each
rat to determine heart-to-body weight ratio (HW/BW%).
Electron microscopy detection
Tissue (~1 mm3) was obtained from the
left ventricle and fixed 4°C in 2.5% glutaraldehyde for 2 h, then
prepared by the steps described previously (20). The ultrastructural changes in the
heart tissue were observed by transmission electron microscopy (FEI
Tecnai G212).
Measurement of myocardium cell
size
The left ventricular tissue sections were embedded
in paraffin and stained with hematoxylin and eosin (H&E) as
described by Frustaci (21), and
were then used for the measurement of myocardium cell size. The
short-axis diameter of cardiac myocytes was measured for 10
myocytes selected per field at magnification, ×400 via light
microscopy. Mean values were obtained from the data on each set of
10 myocytes.
Analysis of inflammatory cytokines in
serum
Following the echocardiography measurements, blood
samples were collected from the abdominal artery and serum was
separated immediately using centrifugation at a speed of 3,000 × g
at 4°C for 5 min. The serum levels of tumor necrosis factor (TNF)-α
(cat. no. H052) and interleukin (IL)-1β (cat. no. H002) were
measured with ELISA kits (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China) according to the manufacturer's
instructions.
Catalase (CAT), superoxide dismutase
(SOD) and malondialdehyde (MDA) activity assays
The CAT (cat. no. A007), SOD (cat. no. A007) and MDA
(cat. no. A003) activities in heart tissue were measured via
colorimetric analysis using a spectrophotometer with the
corresponding detection kits (Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocols.
Detection of myocardium fibrosis
The cardiac tissue in paraffin-embedded sections was
stained with Masson's trichrome stain. In brief, the paraffin
sections (4 µm) were dewaxed and rehydrated through gradient
alcohol into water. The sections were then washed with tap water
and then distilled water, and Regaud's hematoxylin was then used to
dye nuclei for 5–10 min at room temperature. The sections were
washed with distilled water, then treated with Masson's stain for
5–10 min at room temperature. Following staining, the sections were
dipped in 2% glacial acetic acid solution, then treated with 9.1%
phosphomolybdate solution for differentiation for 3–5 min at room
temperature. Following a further 5 min and without water washing,
the sections were directly stained with aniline blue at room
temperature, then dipped again in 0.2% glacial acetic acid
solution, followed by dehydration with 95% and absolute alcohol,
xylene clearing and neutral gum sealing. Positive staining with
Masson's trichrome (fibrotic area) was observed using a light
microscope at a magnification of ×200, and quantified with a color
image analyzer (Leica Microsystems, Ltd., Milton Keynes, UK). The
ratio of fibrotic to total area was calculated to determine the
extent of myocardial fibrosis.
Immunohistochemistry
Immunohistochemical analysis was performed as
described previously (22), using
antibodies against caspase-3 at a dilution of 1:50 (cat. no.
ab13847; Abcam). The staining for caspase-3 was quantified using
Image-Pro plus 6 software (Media Cybernetics, Inc., Rockville, MD,
USA).
Western blot analysis
The left ventricle tissues were lysed in
radioimmunoprecipitations assay buffer (20 mM Tris, pH 7.5; 150 mM
NaCl; 1 mM EDTA; 1 mM EGTA; 1% TritonX-100; 1 mM
Na3VO4; 1 mg/ml aprotinin, leupeptin and
pepstatin; and 1 mM phenylmethylsulfonyl fluoride) and then
centrifuged at 10,000 × g for 15 min at 4°C. The bicinchoninic acid
protein assay (Beyotime Institute of Biotechnology, Haimen, China)
was used to examine the protein concentration. Equal quantities of
protein (20 µg/lane) were separated using 13% SDS-PAGE and
electro-transferred onto polyvinylidene difluoride membranes (Roche
Diagnostics, Indianapolis, IN, USA). The membranes were blocked
with 5% non-fat milk at room temperature for 1 h and then incubated
with primary antibodies against the following proteins:
PKCβ1 (cat. no. sc-8049; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), p-PKCβ1 (cat. no. ab75657; Abcam),
PKCβ2 (cat. no. sc-13149), p-PKCβ2 (cat. no.
sc-365463), P66shc (cat. no. sc-967),
p-P66shc (cat. no. sc-81520), caspase-3, collagen I
(cat. no. sc-293182) and β-actin (cat. no. sc-47778) at a dilution
of 1:1,000 at 4°C overnight. Following primary incubation, the
membranes were incubated with peroxidase-labeled affinity-purified
anti-rabbit/mouse IgG (H+L) secondary antibodies (cat. no.
074-1506/074-1806; Kirkegaard & Perry Laboratories, Inc.,
Gaithersburg, MD, USA; 1:5,000) for 1 h at 37°C. Blots were
developed with an enhanced chemiluminescence detection kit (Pierce;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Band intensities
were quantified with a densitometer analysis system using Image
Lab™ software version 5.11 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Statistical analysis
The statistical analyses were performed using the
SPSS software package, version 18.0 (SPSS, Inc., Chicago, IL, USA);
data were presented as the mean ± standard deviation. Comparisons
of the data were performed using one-way analysis of variance
followed by a post-hoc Bonferroni test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Animal characteristics
The metabolic characteristics of the rats used in
the present study are provided in Table
I. In the DM group, there were significantly higher blood
glucose and cholesterol levels compared with in the control group
(P<0.05), but there were no significant differences between the
treated and untreated diabetic rats (P>0.05). HW/BW ratio was
used to reflect the size of the heart. In the DM group, rats
exhibited significantly lower body weight and a higher HW/BW ratio
than those in the control group (P<0.05). Compared with the DM
group, both the DM+CarL and DM+CarH groups exhibited significantly
greater body weight and lower HW/BW ratios (P<0.05).
 | Table I.Metabolic characteristics of
experimental animals. |
Table I.
Metabolic characteristics of
experimental animals.
| Group | Glucose
(mmol/l) | TC (mmol/l) | TG (mmol/l) | BW (g) | HW/BW (mg/g) |
|---|
| NC | 5.30±1.20 | 1.35±0.62 | 0.70±0.16 | 530±48 | 2.66±1.13 |
| DM |
21.50±7.10a |
2.76±1.32a |
7.18±2.21a | 256±59a |
4.15±0.78a |
| DM+CarL |
20.90±4.30a |
2.73±0.41a |
7.11±3.19a | 325±45a,b |
3.11±0.35a,b |
| DM+CarH |
17.60±6.50a |
2.69±0.75a |
7.15±2.72a | 398±67a–c |
3.07±0.42a–c |
| F-value | 20.221 | 11.437 | 18.490 | 44.662 | 7.426 |
Carvedilol attenuates left ventricular
dysfunction in DCM rats
Echocardiographic parameters and measured NT-proBNP
levels are presented in Table II.
The results indicated increases in LVEDs, LVEDd and NT-proBNP, and
a decrease in EF% in the DM group compared with the control group
(P<0.05). The DM+CarL and DM+CarH groups exhibited a decrease in
LVEDs, LVEDd and NT-proBNP, and an increase in EF% when compared
with the DM group (P<0.05). In turn, compared with the DM+CarL
group, the DM+CarH group exhibited lower LVEDs, LVEDd and
NT-proBNP, and higher EF% (P<0.05). Collectively, these changes
demonstrated that the diabetic rats developed cardiac dysfunction,
and that carvedilol could attenuate DM-induced left ventricular
dysfunction in rats; furthermore, CarH appeared to have a more
marked effect than CarL.
| Table II.Analysis of cardiac function in
experimental animals. |
Table II.
Analysis of cardiac function in
experimental animals.
| Group | LVEDs (mm) | LVEDd (mm) | LVEF (%) | NT-proBNP
(ng/ml) |
|---|
| NC | 1.23±0.23 | 2.83±0.04 | 81.2±1.8 | 0.259±0.061 |
| DM |
2.75±0.72a |
3.77±0.27a |
51.7±3.3a |
1.385±0.014a |
| DM+CarL |
1.87±0.41b |
3.11±0.15b |
62.5±4.1b |
0.973±0.027b |
| DM+CarH |
1.42±0.38b,c |
2.73±0.10b,c |
75.6±4.8b,c |
0.511±0.032b,c |
| F-value | 20.752 | 14.062 | 18.811 | 70.841 |
Carvedilol alleviates myocardium cell
hypertrophy and ultrastructural changes
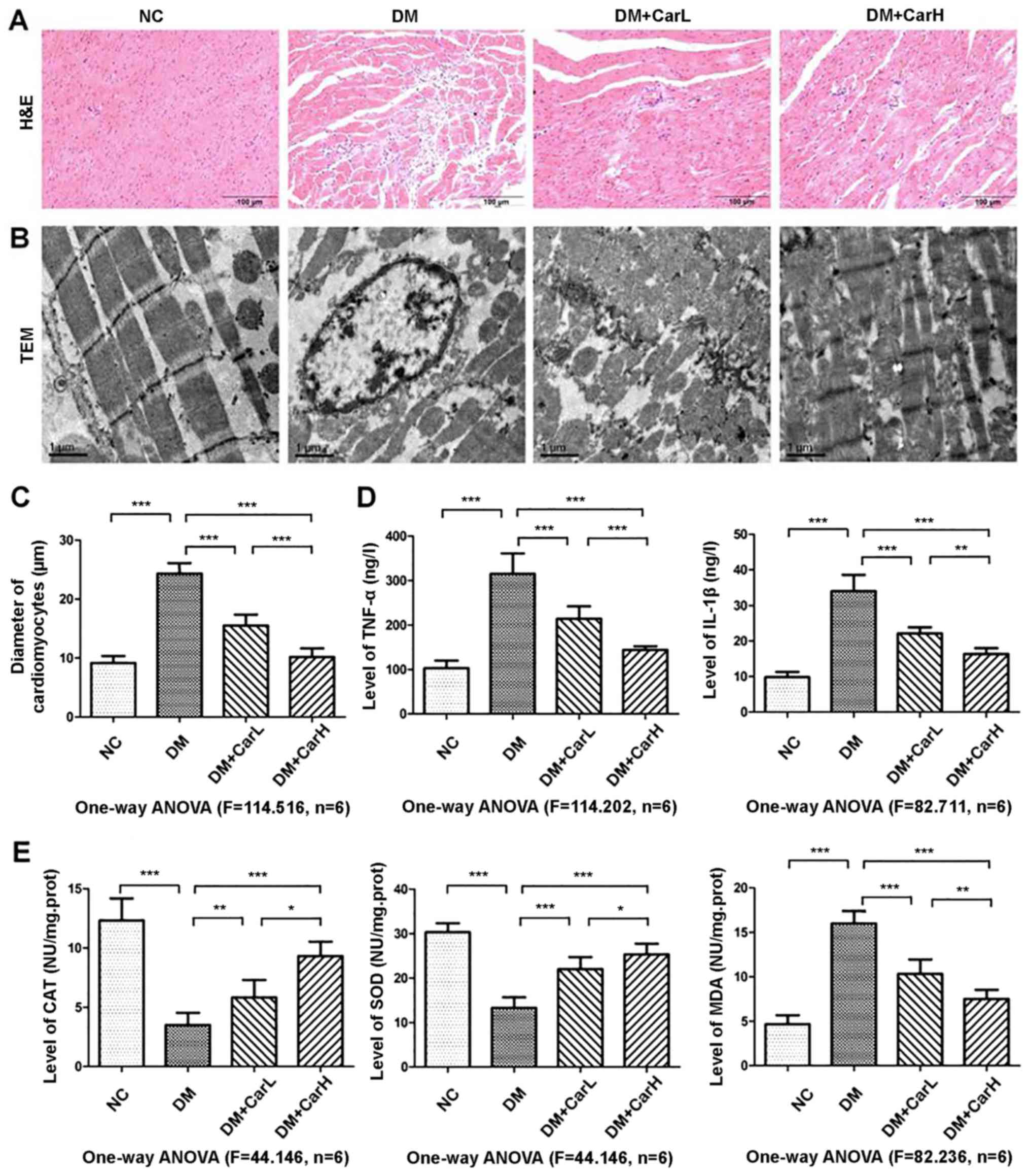
The myocardial structure was detected by H&E
staining and transmission electron microscopy, as presented in
Fig. 1A and B, respectively. As
demonstrated by transmission electron microscopy, in the control
group, typical symmetric myofibrils comprised of Z lines with
sarcomeres exhibited an organized arrangement. Additionally, packed
mitochondria adjacent to the fibers were evident. However, in the
DM group rats, swollen mitochondria and an increased level of
glycogen lysis were observed. Myofibrils appeared destroyed,
resulting in a reduction in sarcomere units, and vacuolization was
observed in the perinuclear space. Carvedilol treatment attenuated
these alterations in the mitochondria, myofilaments, nuclei and Z
lines, as well as glycogen lysis. The diameter of cardiomyocytes
was significantly increased in the DM group compared with in the
control group (P<0.05). In turn, the low and high (1 or 10
mg/kg) doses of carvedilol alleviated the diabetes-induced
cardiomyocyte hypertrophy in DM rats (P<0.05; Fig. 1C).
Carvedilol inhibits myocardial
inflammation and oxidative stress
The inflammatory factors TNF-α and IL-1β were
elevated in the myocardium of the DM group compared with the
control group (P<0.05); whereas, decreased TNF-α and IL-1β
levels were detected in the carvedilol-treated groups compared with
the DM group (Fig. 1D; Table III). CAT, SOD and MDA are
indicators of oxidative stress. As presented in Fig. 1E and Table IV, compared with the control group,
there was an increased level of MDA activity and decreased levels
of SOD and CAT activities in the DM group (all P<0.05).
Carvedilol treatment of diabetic rats significantly inhibited MDA
activity and upregulated the activity of CAT and SOD (all P<0.05
vs. DM group).
| Table III.Levels of myocardial inflammatory
factors in experimental animals. |
Table III.
Levels of myocardial inflammatory
factors in experimental animals.
| Group | TNF-α (ng/l) | IL-1β (ng/l) |
|---|
| NC | 121.78±14.50 | 11.82±2.18 |
| DM |
294.38±56.98a |
26.07±7.94a |
| DM+CarL |
239.75±44.78b |
21.53±0.81b |
| DM+CarH |
208.64±35.25b,c |
16.82±0.73b,c |
| F-value | 31.102 | 14.335 |
| Table IV.Oxidative stress parameters of
experimental animals. |
Table IV.
Oxidative stress parameters of
experimental animals.
| Group | MDA (nmol/g
protein) | SOD (U/g
protein) | CAT(U/g
protein) |
|---|
| NC | 6.73±1.21 | 0.41±0.05 | 0.38±0.03 |
| DM |
15.89±2.93a |
0.21±0.03a |
0.17±0.04a |
| DM+CarL |
12.24±1.57b |
0.31±0.06b |
0.29±0.05b |
| DM+CarH |
8.62±0.99b,c |
0.22±0.05b,c |
0.21±0.02b,c |
| F-value | 48.723 | 37.221 | 33.772 |
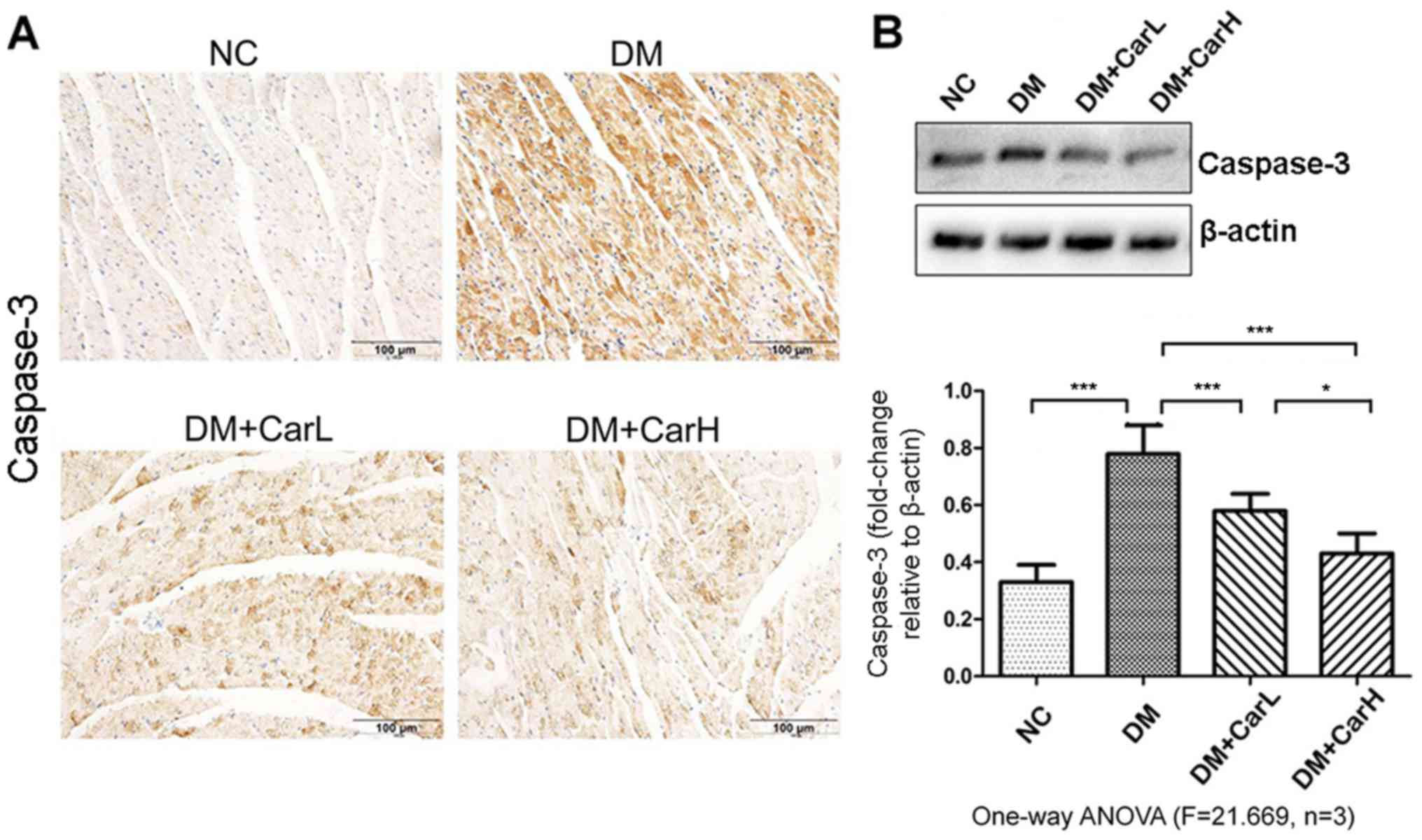
Carvedilol attenuates cardiomyocyte
apoptosis
The expression of proapoptotic protein caspase-3 was
assessed by immunohistochemistry and western blotting, as depicted
in Fig. 2. Enhanced expression of
caspase-3 was identified in the DM group compared with that in the
control group. Notably, the immunohistochemical staining revealed
that carvedilol treatment downregulated the expression of
caspase-3, which was further demonstrated by the western blot
analysis.
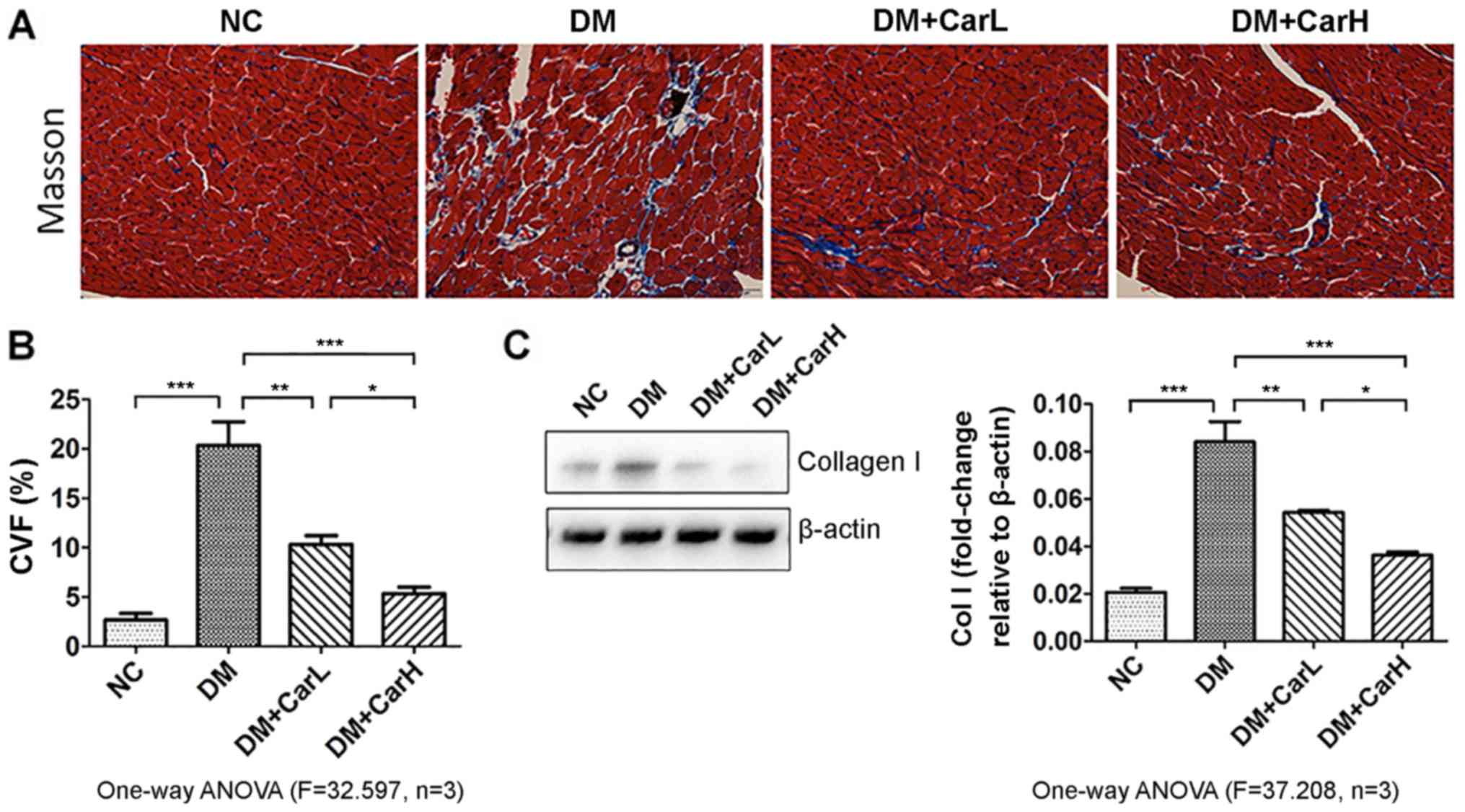
Carvedilol prevents myocardial
fibrosis
As presented in Fig.
3A, positive staining with Masson's trichrome revealed apparent
fibrosis in the DM group, with a diminished and disorganized
collagen network structure in the myocardium, as well as the
interstitial and perivascular areas. However, following treatment
with carvedilol, it was observed that the dose of either 1 or 10
mg/kg was equally effective in markedly alleviating the fibrotic
changes in the heart. As depicted in Fig. 3B, collagen volume fraction (CVF) was
analyzed using the Image-Pro-Plus system (with CVF defined as the
ratio of the positive staining area to the total myocardium area on
visual inspection). Collagen type I protein is an important
indicator of myocardial fibrosis. Western blot analysis of collagen
I is presented in Fig. 3C. In the DM
group, there was an increase of collagen I compared with the
control group. Meanwhile, the diabetic rats treated with carvedilol
exhibited a significantly reduced level of collagen I (P<0.05
vs. DM group).
Carvedilol suppresses the
PKCβ2/P66shc signaling pathway
The results of western blotting demonstrated that
p-PKCβ2 and p-P66shc were upregulated in the
DM group (both P<0.05 vs. control group). No marked differences
were observed in the levels of p-PKCβ1 between the four
groups. By contrast, 1 or 10 mg/kg carvedilol induced a significant
decrease in the phosphorylation of PKCβ2 and
P66shc in the myocardium (P<0.05; Fig. 4). Therefore, it was demonstrated that
carvedilol inhibited PKCβ2 activation, and thus
PKCβ2-mediated P66shc activation, in the
myocardium of DM-induced rats. This indicated that the
PKCβ2/P66shc signaling pathway was associated
with DCM in the present in vivo model.
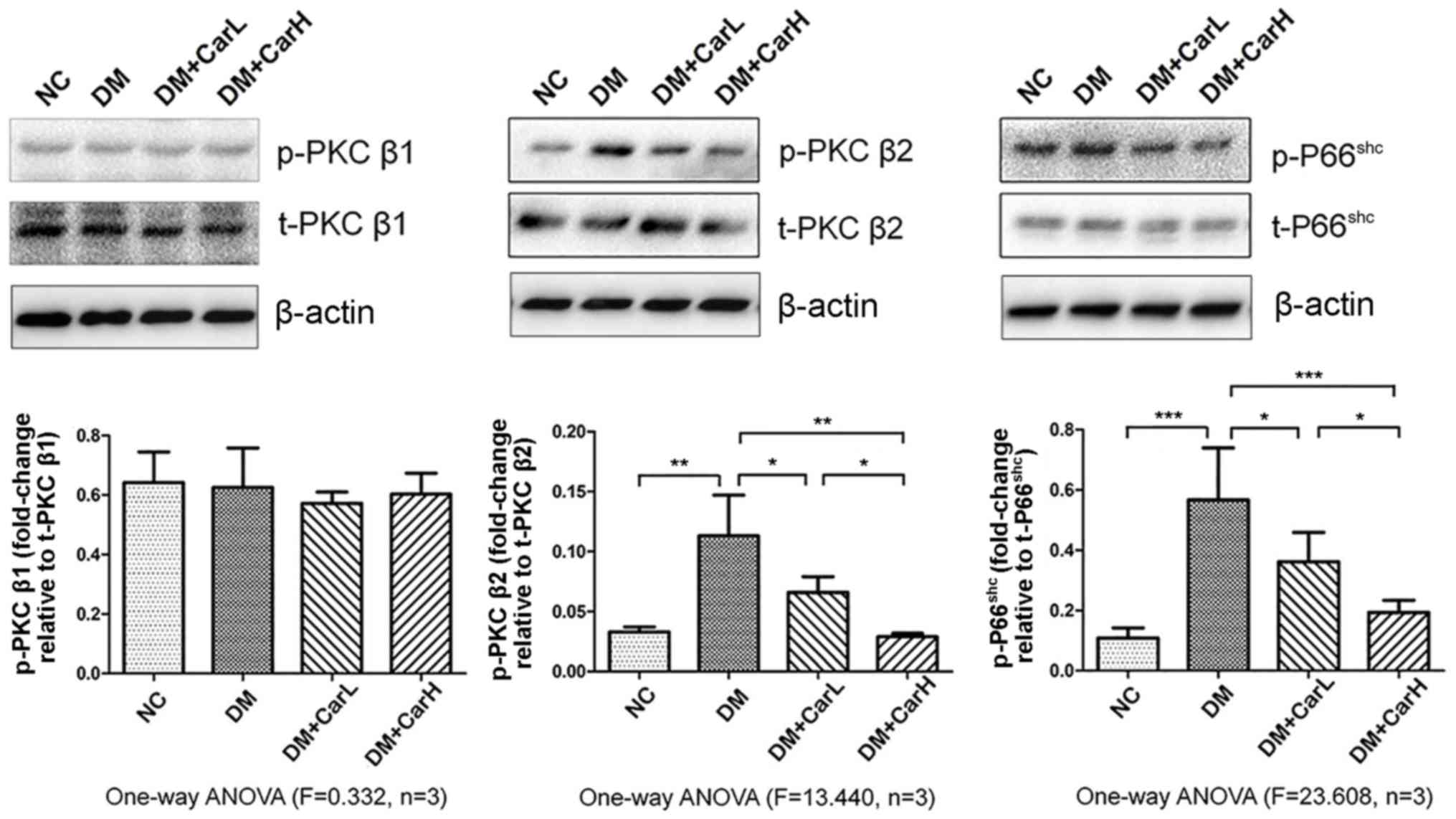 | Figure 4.Carvedilol suppressed the
PKCβ2/P66shc signaling pathway. Western blot
analysis of p-PKCβ1, t-PKCβ1,
p-PKCβ2, t-PKCβ2, p-P66shc and
t-P66shc in myocardial tissue protein expression.
*P<0.05, **P<0.01 and ***P<0.01. PKC, protein kinase C; p,
phosphorylated; t, total; NC, normal control; DM, diabetes
mellitus; CarL, carvedilol low dosage; CarH, carvedilol high
dosage; ANOVA, analysis of variance. |
Discussion
Results of the present study suggested that the
PKCβ2/P66shc mitochondrial oxidative stress
signaling pathway may be associated with the development of DCM.
Activated PKCβ2 has been indicated to serve a crucial
role in the pathogenesis of DCM (23). Carvedilol, a novel third-generation
non-selective β-blocker with α-receptor blockade activity, does not
exhibit side-effects on glucose or lipid metabolism, which is
contrasting to the first and second generation β-blockers (24). Previous studies have demonstrated
that carvedilol exerts marked improvements in cardiac performance
and reduces mortality in humans (25,26). In
recent years, there has been increasing interest in the application
of carvedilol for the treatment of diabetes-associated
complications.
DCM has been defined as left ventricular systolic
and diastolic dysfunction in the absence of coronary heart disease,
hypertension and valvular heart disease (27), and occurs in both humans and animals.
In the present study, a DCM rat model was successfully established,
which was characterized by severe disorder of metabolism,
cardiomyocyte hypertrophy, fibrosis, cardiac dysfunction, excessive
oxidative stress and subsequent inflammation and apoptosis. These
characteristics are consistent with a previous study (10). Furthermore, it was demonstrated that
long-term carvedilol administration attenuated the development of
the aforementioned characteristic alterations in DCM. The present
data supported the protective effect of carvedilol against DCM.
First, carvedilol treatment alleviated the increase in HW/BW ratio
and relatively increased body weight; however, had no effect on the
levels of blood glucose and cholesterol. Second, carvedilol
appeared to protect cardiac function as evaluated by
echocardiography and the serum level of NT-proBNP. Third, as is
well established, an important hallmark of DCM is fibrosis
(28). Morsy et al (29) reported that carvedilol may protect
STZ-induced early diabetic nephropathy fibrosis in rats via
antioxidant as well as anti-inflammatory activities. The current
results demonstrated that carvedilol markedly ameliorated cardiac
fibrosis and ultrastructural changes in DCM rats. Fourth, oxidative
stress is defined as the imbalance between the production and
elimination of oxygen-free radicals, which serve an essential role
in the development of heart failure in DCM (30). Hyperglycemia accelerates the glucose
oxidation response and mitochondrial generation of ROS, which
results in damage of DNA fragments and contributes to exacerbated
apoptosis. The activation of PKCβ2 may serve a crucial
role in the occurrence of oxidative stress injury in DCM.
Presently, the inhibition of PKCβ2 activation by
carvedilol appeared to attenuate DCM-associated myocardial injury
at least partly via reduction of P66shc.
P66shc is a redox enzyme that is implicated in
mitochondrial ROS generation, mitochondrial membrane potential
change, and the translation of oxidative signals into apoptosis
(31). The in vivo
experiments of the current study have demonstrated that excessive
P66shc activation is associated with PKCβ2
activation. To the best of our knowledge, the present study is the
first to demonstrate an association between PKCβ2 and
P66shc in DCM.
PKCβ1 and β2 are two of the
classic isoforms (among the α, β and γ-types) of PKC (32). Previous studies have indicated that
PKCβ2 is preferentially overexpressed in the myocardium
of patients and animals with diabetes (33,34). In
the present study, it was demonstrated that the primary activated
isoform of PKC in DCM-related oxidative stress was
PKCβ2, and not PKCβ1. Thus, it is possible
that a primary effect of carvedilol in DCM is inhibiting the
activation of PKCβ2. The current study also demonstrated
that inhibition of PKCβ2 activation by carvedilol
attenuated the overexpression and phosphorylation of
P66shc in DCM. The results also demonstrated that
carvedilol inhibited the accumulation of MDA (an indicator of lipid
peroxidation) and increased the hyperglycemia-induced
downregulation of SOD and CAT (two typical primary ROS scavenging
enzymes), which demonstrated that carvedilol could inhibit
oxidative stress. Furthermore, the present data demonstrated that
the PKCβ2/P66shc oxidative stress signaling
pathway may be associated with the process of DCM. It was also
observed that hyperglycemia-induced myocardial injury markedly
increased the serum levels of TNF-α and IL-1β (two typical
pro-inflammatory cytokines), which suggested that a severe
myocardial inflammation response was induced during the development
of DCM. Carvedilol administration inhibited this increase in the
serum concentration of TNF-α and IL-1β. Finally, in addition to
ameliorating cardiac inflammation, carvedilol treatment also
appeared to reduce cardiomyocyte apoptosis. Cardiomyocyte apoptosis
is an important pathological change that contributes to cardiac
dysfunction in DCM (35). Caspase-3,
an established pro-apoptotic protease, serves an important role in
the formation of apoptotic bodies and induction of cell death. In
the untreated DM group, caspase-3 was markedly elevated, while in
the carvedilol treatment group, cardiomyocyte caspase-3 activity
and subsequent apoptosis were reduced.
Taken together, the present study results
demonstrate that the principal activated isoform of PKC in DCM may
be PKCβ2, and not PKCβ1; furthermore,
carvedilol-mediated alleviation of myocardial injury and systemic
inflammation in DCM may occur through its inhibition of
P66shc-mediated oxidative stress and subsequent
apoptosis. Considering these present findings, carvedilol may
represent a novel, clinically applicable therapy for DCM.
Acknowledgements
The authors would like to thank The North China
University of Science and Technology for technical support during
the experiments.
Funding
The current study was supported by Hebei Province
Key Projects of Medical Science Research Grants 2018 (grant no.
20181253).
Availability of data and materials
All data are available without restriction.
Authors' contributions
WZ performed the study and wrote the manuscript. WZ
and DL contributed to data analysis and interpretation. WZ, DL, XG
and WZ contributed materials and analysis tools. BOR reviewed the
manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Animal Care and Use Committee of North China University of Science
and Technology, and in compliance with Guidelines for the Care and
Use of Laboratory Animals Published by the National Academy Press
(NIH publication no. 85-23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shaw JE, Sicree RA and Zimmet PZ: Global
estimates of the prevalence of diabetes for 2010 and 2030. Diabetes
Res Clin Pract. 87:4–14. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bell DS: Heart failure: The frequent,
forgotten, and often fatal complication of diabetes. Diabetes Care.
26:2433–2441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fang ZY, Prins JB and Marwick TH: Diabetic
cardiomyopathy: Evidence, mechanisms, and therapeutic implications.
Endocr Rev. 25:543–567. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Westermann D, Walther T, Savvatis K,
Escher F, Sobirey M, Riad A, Bader M, Schultheiss HP and Tschöpe C:
Gene deletion of the kinin receptor B1 attenuates cardiac
inflammation and fibrosis during the development of experimental
diabetic cardiomyopathy. Diabetes. 58:1373–1381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Falcão-Pires I and Leite-Moreira AF:
Diabetic cardiomyopathy: Understanding the molecular and cellular
basis to progress in diagnosis and treatment. Heart Fail Rev.
17:325–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aksakal E, Akaras N, Kurt M, Tanboga IH,
Halici Z, Odabasoglu F, Bakirci EM and Unal B: The role of
oxidative stress in diabetic cardiomyopathy: An experimental study.
Eur Rev Med Pharmacol Sci. 15:1241–1246. 2011.PubMed/NCBI
|
|
7
|
Halliwell B: Biochemistry of oxidative
stress. Biochem Soc Trans. 35:1147–1150. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matés JM and Sánchez-Jiménez F:
Antioxidant enzymes and their implications in pathophysiologic
processes. Front Biosci. 4:D339–D345. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mellor H and Parker P: The extended
protein kinase C superfamily. Biochem J. 332:281–292. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gurusamy N, Watanabe K, Ma M, Zhang S,
Muslin AJ, Kodama M and Aizawa Y: Inactivation of 14-3-3 protein
exacerbates cardiac hypertrophy and fibrosis through enhanced
expression of protein kinase C beta 2 in experimental diabetes.
Biol Pharm Bull. 28:957–962. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Way KJ, Isshiki K, Suzuma K, Yokota T,
Zvagelsky D, Schoen FJ, Sandusky GE, Pechous PA, Vlahos CJ,
Wakasaki H and King GL: Expression of connective tissue growth
factor is increased in injured myocardium associated with protein
kinase C beta2 activation and diabetes. Diabetes. 51:2709–2718.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Migliaccio E, Giorgio M, Mele S, Pelicci
G, Reboldi P, Pandolfi PP, Lanfrancone L and Pelicci PG: The p66shc
adaptor protein controls oxidative stress response and life span in
mammals. Nature. 402:309–313. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nemoto S and Finkel T: Redox regulation of
forkhead proteins through a p66shc-dependent signaling pathway.
Science. 295:2450–2452. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pinton P, Rimessi A, Marchi S, Orsini F,
Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F,
Wieckowski MR, et al: Protein kinase C beta and prolyl isomerase 1
regulate mitochondrial effects of the life-span determinant p66Shc.
Science. 315:659–663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paneni F, Mocharla P, Akhmedov A,
Costantino S, Osto E, Volpe M, Lüscher TF and Cosentino F: Gene
silencing of the mitochondrial adaptor p66shc suppresses vascular
hyperglycemic memory in diabetes. Circ Res. 111:278–289. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arab HH and El-Sawalhi MM: Carvedilol
alleviates adjuvant-induced arthritis and subcutaneous air pouch
edema: Modulation of oxidative stress and inflammatory mediators.
Toxicol Appl Pharmacol. 268:241–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yasar A, Erdemir F, Parlaktas BS, Atilgan
D, Koseoglu RD, Saylan O and Firat F: The effect of carvedilol on
serum and tissue oxidative stress parameters in partial ureteral
obstruction induced rat model. Kaohsiung J Med Sci. 29:19–25. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feuerstein GZ, Poste G and Ruffolo RR Jr:
Carvedilol update III: Rationale for and use in congestive heart
failure. Drugs Today. 31:307–326. 1995. View Article : Google Scholar
|
|
19
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the care and use of laboratory animals.
(Eighth). Publication no. 85-23(rev). 327:963–965. 1996.
|
|
20
|
Maeda H, Nagai H, Takemura G,
Shintani-Ishida K, Komatsu M, Ogura S, Aki T, Shirai M, Kuwahira I
and Yoshida K: Intermittent-hypoxia induced autophagy attenuates
contractile dysfunction and myocardial injury in rat heart. Biochim
Biophys Acta. 1832:1159–1166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frustaci A, Kajstura J, Chimenti C,
Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B and Anversa P:
Myocardial cell death in human diabetes. Circ Res. 87:1123–1132.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu X, Li B, Wang W, Zhang C, Zhang M,
Zhang Y, Xia Y, Dong Z, Guo Y and An F: Effects of HMG-CoA
reductase inhibitor on experimental autoimmune myocarditis.
Cardiovasc Drugs Ther. 26:121–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lei S, Li H, Xu J, Liu Y, Gao X, Wang J,
Ng KF, Lau WB, Ma XL, Rodrigues B, et al: Hyperglycemia-induced
protein kinase C β2 activation induces diastolic cardiac
dysfunction in diabetic rats by impairing caveolin-3 expression and
Akt/eNOS signaling. Diabetes. 62:2318–2328. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang H, Shan J, Pan XH, Wang HP and Qian
LB: Carvedilol protected diabetic rat hearts via reducing oxidative
stress. J Zhejiang Univ Sci B. 7:725–731. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olsen SL, Gilbert EM, Renlund DG, Taylor
DO, Yanowitz FD and Bristow MR: Carvedilol improves left
ventricular function and symptoms in chronic heart failure: A
double-blind randomized study. J Am Coll Cardiol. 25:1225–1231.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Packer M, Bristow MR, Cohn JN, Colucci WS,
Fowler MB, Gilbert EM and Shusterman NH: The effect of carvedilol
on morbidity and mortality in patients with chronic heart failure.
N Engl J Med. 334:1349–1355. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tarquini R, Lazzeri C, Pala L, Rotella CM
and Gensini GF: The diabetic cardiomyopathy. Acta Diabetol.
48:173–181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou H, Li YJ, Wang M, Zhang LH, Guo BY,
Zhao ZS, Meng FL, Deng YG and Wang RY: Involvement of RhoA/ROCK in
myocardial fibrosis in a rat model of type 2 diabetes. Acta
Pharmacol Sin. 32:999–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morsy MA, Ibrahim SA, Amin EF, Kamel MY,
Abdelwahab SA and Hassan MK: Carvedilol ameliorates early diabetic
nephropathy in streptozotocin-induced diabetic rats. Biomed Res
Int. 2014:1052142014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rajesh M, Mukhopadhyay P, Bátkai S, Patel
V, Saito K, Matsumoto S, Kashiwaya Y, Horváth B, Mukhopadhyay B,
Becker L, et al: Cannabidiol attenuates cardiac dysfunction,
oxidative stress, fibrosis, and inflammatory and cell death
signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol.
56:2115–2125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Z, Wang G, Zhai X, Hu Y, Gao D, Ma L,
Yao J and Tian X: Selective inhibition of protein kinase C β2
attenuates the adaptor P66Shc-mediated intestinal
ischemia-reperfusion injury. Cell Death Dis. 5:e11642014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clarke M and Dodson PM: PKC inhibition and
diabetic microvascular complications. Best Pract Res Clin
Endocrinol Metab. 21:573–586. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simpson PC: Beta-protein kinase C and
hypertrophic signaling in human heart failure. Circulation.
99:334–337. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Connelly KA, Kelly DJ, Zhang Y, Prior DL,
Advani A, Cox AJ, Thai K, Krum H and Gilbert RE: Inhibition of
protein kinase C-beta by ruboxistaurin preserves cardiac function
and reduces extracellular matrix production in diabetic
cardiomyopathy. Circ Heart Fail. 2:129–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Devereux RB, Roman MJ, Paranicas M,
O'Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER and
Howard BV: Impact of diabetes on cardiac structure and function the
strong heart study. Circulation. 101:2271–2276. 2000. View Article : Google Scholar : PubMed/NCBI
|