Introduction
Nasopharyngeal carcinoma (NPC) is a common cancer
type worldwide, with >60,000 new cases reported in China each
year (1). The treatment of NPC
largely relies on early detection and a combination of chemotherapy
and radiotherapy (2,3). However, only a small proportion of NPC
cases are detected sufficiently early to be eligible for curative
treatment (2). Understanding the
pathogenesis of NPC is critical in order to identify novel
treatment targets for this disease.
The initiation and development of NPC is a multistep
process that is manipulated by cumulative genetic and epigenetic
alterations, as well as microenvironmental cues (2). Atypical chemokine receptors (ACKRs) are
chemokine receptors that bind to their ligands with a high
affinity, but cannot induce downstream signal transduction
(4,5). Although ACKRs are structurally unable
to initiate signal transduction, they serve a non-redundant role in
keeping the homeostasis of inflammatory responses by efficiently
internalizing chemokines and maintaining chemotactic gradients
(4,5). As a new member of the ACKR family,
ACKR4 (also known as C-C motif chemokine receptor-like 1) is
usually downregulated in cancer tissues compared with normal
tissues (6–8). In breast, cervical and colorectal
cancer, low expression of ACKR4 was correlated with poor prognosis
(6,8,9). ACKR4
binds to homeostatic chemokines, including C-C motif chemokine
ligand (CCL)19, CCL21, CCL25 and C-X-C motif chemokine ligand
(CXCL)13, which are the major ligands of CC receptor (CCR)7, CCR9
and CXC receptor (CXCR)5, thereby tuning up the function of these
chemokine axes (4,5).
Inflammatory responses have been reported to
regulate the development of NPC (10). It is well established that the
CCL21/CCR7 axis promoted cancer development via enhancing the
proliferation, migration and invasion of tumor cells (11,12).
ACKR4-mediated scavenging of dermal-derived CCL21/CCR7 is critical
during inflammation, Thus, the effect of ACKR4 on NPC cells during
inflammation, facilitates CCR7-dependent cell trafficking by
scavenging CCL21 (12). However, the
effects of ACKR4 in regulating inflammation-mediated NPC
development, particularly the CCL21/CCR7 chemokine-axis related,
remain unclear. In the present study, it is hypothesized that loss
of ACKR4 in NPC is an important promoting factor of the
CCL21-mediated tumor growth and invasion.
Materials and methods
Cell culture and transfection
The mouse NPC cell line SUNE-1 was purchased from
the American Type Culture Collection (Manassas, VA, USA). Cells
were cultured in Dulbecco's modified Eagle's medium with high
glucose, supplemented with 10% fetal bovine serum, 100 IU/ml
penicillin G and 100 mg/ml streptomycin (all from Thermo Fisher
Scientific, Inc., Waltham, MA, USA), at 37°C in a humidified
incubator containing 5% CO2.
For the overexpression of ACKR4 in the SUNE-1 cell
line, Human ACKR4 cDNA plasmids (obtained from OriGene
Technologies, Inc., Rockville, MD, USA) were transfected into 293
cells (1×106/well) for 48 h to generate a lentivirus
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
viral supernatant was subsequently collected and used to infect the
SUNE-1 cells. Further analysis was performed 72 h
post-transfection.
SUNE-1 Cells were transfected with specific siRNA
(5′-GUUCUGCAGCAACAUUUAAUU-3′) against ACKR4 using Lipofectamine
2000 (Invitrogen; Thermo Fischer Scientific, Inc.) according to the
manufacturer's Protocol. Cells were seeded into 6-well plates and
cultured for an additional 24 h at 37°C. The medium was refreshed
with serum-free RPMI 1640 medium (Thermo Fisher Scientific, Inc.)
and siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA)
according to the protocol, and were added into each well. Following
6 h, the medium was replaced with normal RPMI 1640 medium
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.). Cells were harvested for subsequent analysis 48
h following transfection in order to collect total RNAs or
proteins. siRNAs were synthesized by Boute Biotech Co. Ltd.,
(Wuhan, China). The empty backbone vector was used as a
transfection control. Flow cytometry was then conducted to validate
the upregulation or downregulation of membrane ACKR4
expression.
Patients and tissues
A total of 30 patients (age, 30–75 years;
male:female, 14:16) who were diagnosed with nasopharyngeal
carcinoma and received radical surgery for this at the Department
of Head and Neck of The Third Affiliated Hospital of Kunming
Medical University (Kunming, China) were included in the present
study. All patients were admitted between January 2015 and January
2016 and were not treated with chemotherapy or radiotherapy prior
to surgery. Patients were included in the present study if they: i)
Were pathologically diagnosed with NPC; ii) were aged 18–80 years
old; iii) signed the written informed consent form provided by the
hospital medical ethics committee of the Third Affiliated Hospital
of Kunming Medical University. Cancerous tissues and matched
adjacent non-cancerous tissues (3 cm away from the tumor) were
harvested from each patient following surgery. The present study
was approved by the Research Ethics Committee of Third Affiliated
Hospital of Kunming Medical University. Tumor tissues were fixed
using 10% formalin solution for 12 h at 4°C, paraffin embedded and
sliced to 4 µm thick sections. Epitope retrieval was then performed
for further analysis.
Flow cytometry analysis
Flow cytometry was used to measure ACKR4 expression
in human fresh NPC patient samples and SUNE-1 cells. Briefly, tumor
and adjacent normal tissues were collected, and digested with 1
µg/ml type IV collagenase and 20 units/ml DNase type IV (both from
Thermo Fisher Scientific, Inc.) in Hank's balanced salt solution.
Subsequently, the isolated single cells from the tissues were
stained with fluorochrome-conjugated anti-ACKR4 antibody (1:500;
cat. no. TA340600; Origene technologies, Inc.) without
permeabilization and then incubated for 2 h at room temperature
with mouse anti goat H&L secondary antibodies (1:1,000; cat.
no. ab7064; Abcam, Cambridge, UK) Samples were analyzed by a BD
LSRFortessa™ cell analyzer (BD Biosciences, Franklin Lakes, NJ,
USA). The stained cells were analyzed using a Becton Dickinson
FACScan flow cytometer using WinMD Isoftware (version 2.9; BD
Biosciences).
Invasion assay
Transwell chambers coated with Matrigel (Corning,
Inc., Corning, NY, USA) with an 8-µm polycarbonate filter membrane
were used to perform the invasion assay. Briefly, SUNE-1 cells
cultured in 6 well pates (3×106/well) with different
ACKR4 levels were added into the upper chamber along with
serum-free medium. The lower chamber was filled with 5% serum
medium, and the plate was incubated under normal conditions for 24
h. Subsequently, the cells remaining on the top surface of the
filter were removed, and the cells that had invaded into the lower
part of the filter were fixed for 20 min in paraformaldehyde at
room temperature and stained with 0.1% crystal violet for 20 min at
room temperature. The stained membrane was then washed with PBS for
three times. To quantify the amount of invading cells, cell count
was performed on each membrane.
Cell counting assay
Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, inc., Kumamoto, Japan) was used to estimate the cell
number according to the manufacturer's protocol. Briefly, the same
number of SUNE-1 cells (5×105/well) different levels of
ACKR4 expression was seeded in 96-well plates. The number of cells
in each well was counted at 0, 6, 12, 24 and 48 h after seeding. At
these time points, CCK-8 solution was added and incubated for 30
min at 37°C, and then the absorbance at 450 nm was measured by an
MRX II microplate reader (Dynex Technologies, Chantilly, VA,
USA).
Animal model
A total of 32 subcutaneous NPC model was established
using 5-week-old female NUDE mice (weight, 18–20 g; Shanghai SLAC
Laboratory Animal Co., Ltd., Shanghai, China) with SUNE-1 cells
expressing different levels of ACKR4 achieved by transfection with
wild-type, empty control, knockdown and overexpression vectors,
respectively. All mice were bred under a specific pathogen-free
environment with a controlled environment (temperature, 23±1°C;
humidity, 50–60%) with an artificial simulation of 12-h light-dark
cycle, standard food, and free access to autoclaved water. Mice
were acclimatized prior to experimentation for 3 days. A total of
5×105 SUNE-1 cells were inoculated subcutaneously to the
flanks of the mice, and the tumor size and body weight were
measured daily. The tumor volume was calculated according to the
following formula: Tumor volume=length × width2 ×0.5. In
total, 8 mice were included in each group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
To measure the expression levels of various
epithelial-mesenchymal transition (EMT)-associated genes in SUNE-1
cells, the EMT PCR Array for RT-qPCR analysis (Qiagen, Hilden,
Germany) was used. Initially, the mirVana miRNA Isolation kit
(Thermo Fisher Scientific, Inc.) was used to extract total RNA from
the cells, which was then reverse transcribed to cDNA using the
miScript Reverse Transcription kit (Qiagen). Next, qPCR was
performed according to the protocol described in the EMT PCR Array
kit. The primers were designed as follows: Snail family
transcriptional repressor 1 (SNAI-1) forward,
5′-CAAAGGTGGATCAGATTCAAG-3′ and reverse,
5′-GGTGAGCATTATCACCCAGAA-3′; Fibronectin 1 (FN1) forward,
5′-CAAAGGTGGATCAGATTCAAG-3′ and reverse,
5′-GGTGAGCATTATCACCCAGAA-3′; Variant in Methylation (VIM) forward,
5′-AGAAACCGGCAGAGTGCTCTTA-3′ and reverse,
5′-GTACCCAGGCAACAGAATCCA-3′; CAV2 forward,
5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3; β-actin forward,
5′-AGAAAATCTGGCACCACACC-3′ and reverse, 5′-TAGCACAGCCTGGATAGCAA-3′.
The thermocycling conditions were as follows: 95°C for 5 min,
followed by 35 cycles at 95°C for 20 sec, 58°C for 20 sec and 72°C
for 20 sec, with a final extension at 72°C for 5 min. β-actin was
used as the internal reference gene. The top four differentially
expressed genes were selected and plotted in a barplot. Relative
gene expression was determined by the comparative quantitative
cycle (Cq) method (13).
Furthermore, to detect lymphatic metastasis in the
animal model following inoculation for 50 days, the human 18srRNA
level in the tumor-draining lymph nodes of the mice was measured by
RT-qPCR assay as described previously, with the following primers:
Forward, 5′-GGCCCTGTAATTGGAATGAGTC-3′, and reverse,
5′-CCAAGATCCAACTACGAGCTT-3′. The tumor-draining lymph nodes with
positive human 18srRNA expression were considered to be tumor
metastasis-positive.
ELISA
An enzyme-linked immunosorbent assay (ELISA) was
used to measure the concentrations of CCL19 (cat. no. ab100601;
Abcam, Cambridge, UK), CCL21 (cat. no. ab208985; Abcam), CCL25
(cat. no. ab100645; Abcam), CXCL13 (cat. no. ab179881; Abcam),
matrix metalloproteinase (MMP)2 (cat. no. ab100606; Abcam) and MMP9
(cat. no. ab100610; Abcam) in the cell lysate or tumor tissues. The
samples from mice were collected after inoculation for 50 days and
samples from cells were collected after inoculation for 48 h. All
ELISA kits were purchased from Thermo Fisher Scientific, Inc.
(eBioscience), and the experiments followed the manufacturer's
protocol. The total protein concentration was normalized to the
concentration in the samples prior to incubation with vectors or to
inoculated animals.
Statistical analysis
GraphPad Prism software (Version 7.0; GraphPad
Software, Inc., La Jolla, CA, USA) was used for statistical
analysis and data visualization. Comparisons were analyzed by
t-test if the results were presented as quantitative data.
χ2 analysis was used for frequency data. The
Kaplan-Meier method and log-rank test were used for survival
analysis and for evaluating the difference between different
cohorts, respectively. A two-tailed P-value of <0.05 was
considered to be an indicator of a statistically significant
difference.
Results
ACKR4 downregulation in NPC
tissues
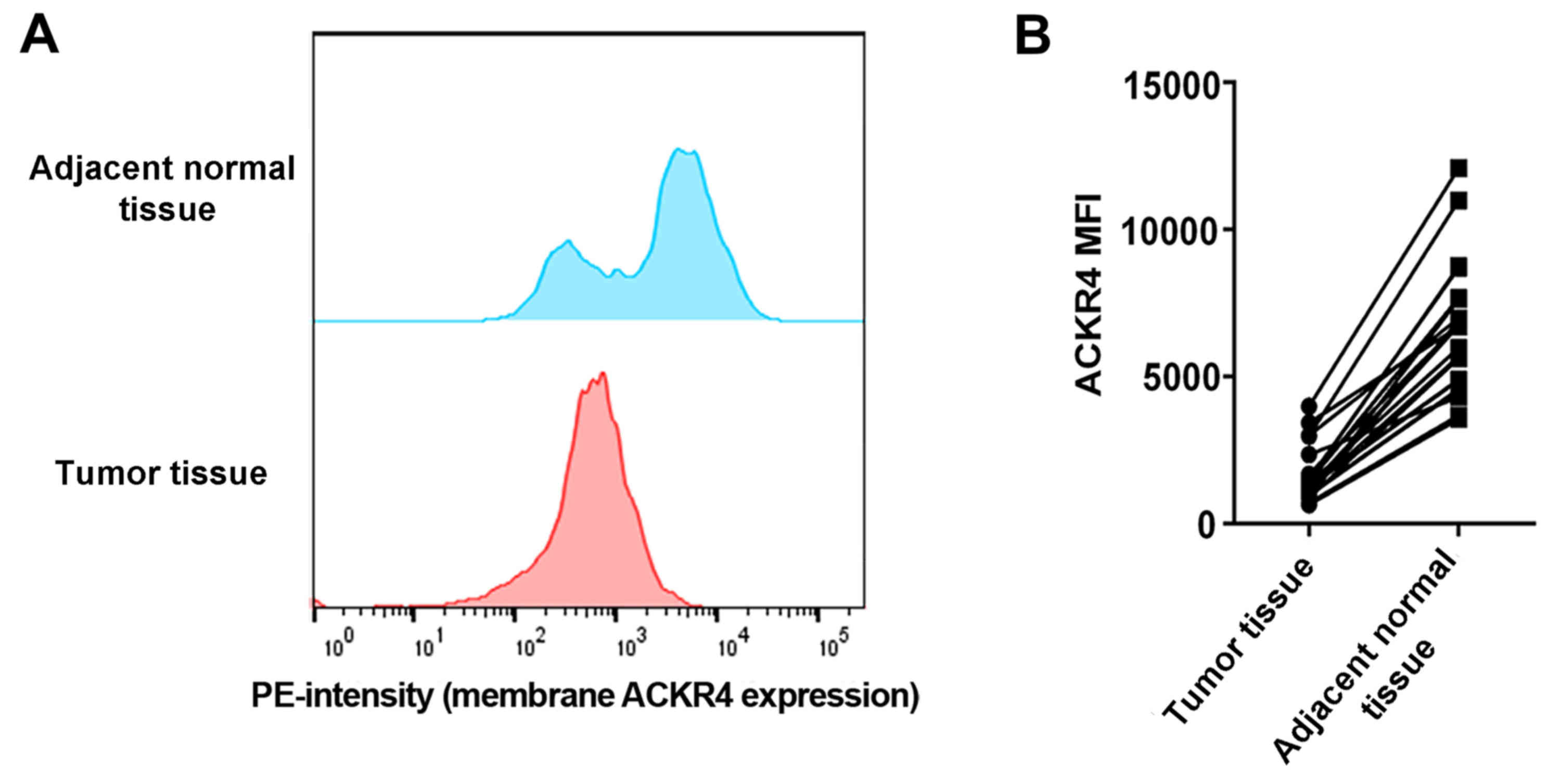
To understand the role of ACKR4 in NPC development,
the current study first investigated whether ACKR4 was dysregulated
in human NPC cancer tissue compared with its level in adjacent
normal liver tissue. Via flow cytometry analysis, it was observed
that the expression of ACKR4 was lower in NPC tissues as compared
with that in adjacent normal tissues (Fig. 1A and B). These data suggested that
ACKR4 was abnormally downregulated in NPC cells, providing the
rationale to manipulate ACKR4 expression in NPC cell lines and
animal models.
Loss of ACKR4 promotes tumor
development and lymph node metastasis in an NPC animal model
ACKR4 expression was upregulated or downregulated in
SUNE-1 cells (Fig. 2A and B).
Subsequently, a subcutaneous tumor animal model, in which different
levels of ACKR4 were expressed, was established by inoculation with
these cells (Fig. 2C). As shown in
Fig. 2C, downregulation of ACKR4
significantly accelerated the tumor growth rate, while tumors with
high ACKR4 expression exhibited delayed tumor growth (Fig. 2C). Lymph node metastasis of these
tumors was further investigated, and loss of ACKR4 was observed to
promote tumor cell lymph node metastasis (Fig. 2D). Taken together, the present in
vivo study indicated that the loss of ACKR4 promoted NPC
development and lymph node metastasis.
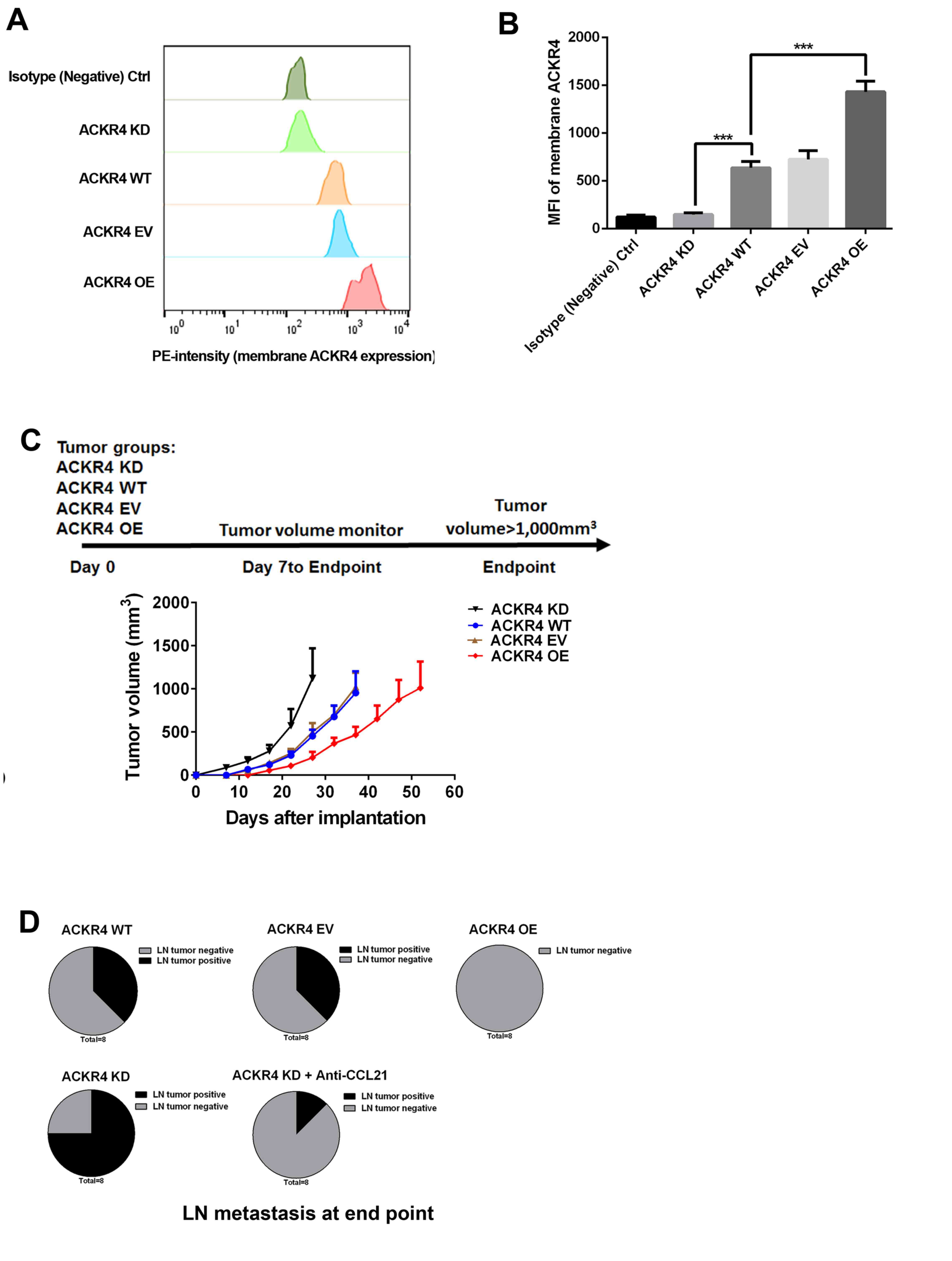 | Figure 2.ACKR4 expression and nasopharyngeal
carcinoma development in a subcutaneous SUNE-1 tumor animal model
Via flow cytometry analysis, (A) quantitative analysis of
expression of ACKR4 (B) ACKR4 expression level was modulated by its
expression vector and small hairpin RNA in SUNE-1 cells to achieve
overexpression and knockdown, respectively. The membrane ACKR4
expression was measured and plotted. (C) Experimental plan of the
animal model and tumor growth curves. (D) At the endpoint, tumor
metastasis in tumor-draining lymph nodes was measured, with the
ACKR4 knockdown group exhibiting the highest tumor lymph node
metastasis frequency. ***P<0.001. ACKR4, atypical chemokine
receptor 4; MFI, mean fluorescence intensity; WT, wild-type; EV,
empty vector; KD, knockdown; OE, overexpression; CCL21, C-C motif
chemokine ligand 21. |
Loss of ACKR4 leads to CCL21
accumulation in NPC tissues
ACKR4 exerts its biological function by scavenging
CCL19, CCL21, CCL25 and CXCL13 (12). Among these chemokines, ACKR4 exhibits
the highest affinity for CCL21 (12). The concentrations of CCL19, CCL21,
CCL25 and CXCL13 in the patient tumor tissue lysates were measured.
As shown in Fig. 3A, loss of ACKR4
in NPC tumors led to significant accumulation of CCL21 in the tumor
tissue. By contrast, overexpression of ACKR4 markedly reduced CCL21
concentration in NPC tumor tissue (Fig.
3A). However, the level of ACKR4 expression did not regulate
the concentrations of CCL19, CCL25 and CXCL13 in the NPC tumor
model (Fig. 3B-D). These data
suggested that CCL21 mediated the tumor development caused by ACKR4
downregulation.
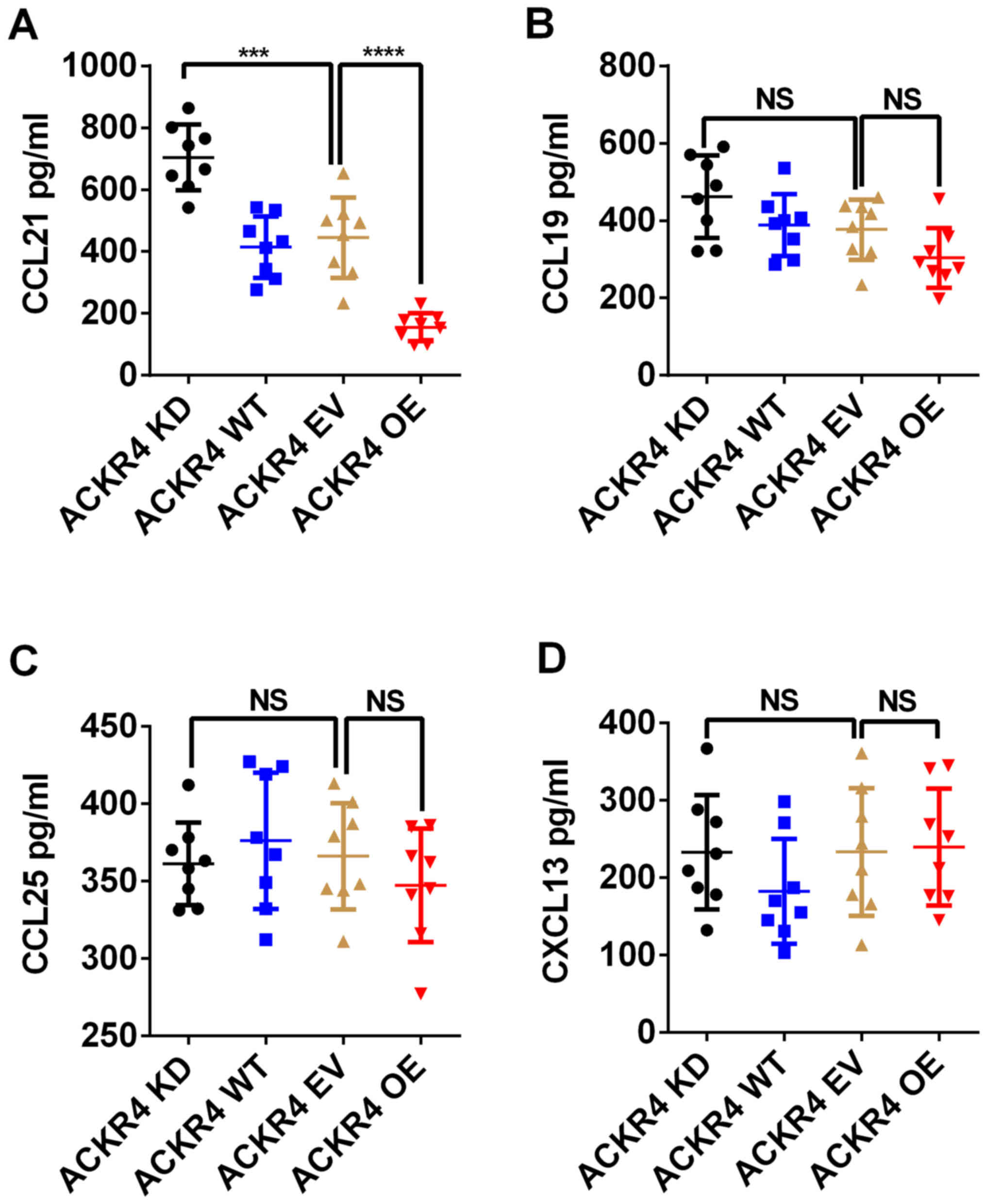 | Figure 3.ACKR4 regulates CCL21 level in NPC
tumor tissue. The intratumoral levels of (A) CCL21, (B) CCL19, (C)
CCL25 and (D) CXCL13 were measured in tumors with different levels
of ACKR4 expression. The concentration of CCL21 was evidently
reduced by ACKR4 overexpression in NPC tumor tissue. ***P<0.001
and ****P<0.0001. NS, no significant difference. ACKR4, atypical
chemokine receptor 4; NPC, nasopharyngeal carcinoma; WT, wild-type;
EV, empty vector; KD, knockdown; OE, overexpression; CCL, C-C motif
chemokine ligand; CXCL, C-X-C motif chemokine ligand. |
Loss of ACKR4 enhances SUNE-1 cell
proliferation in vitro
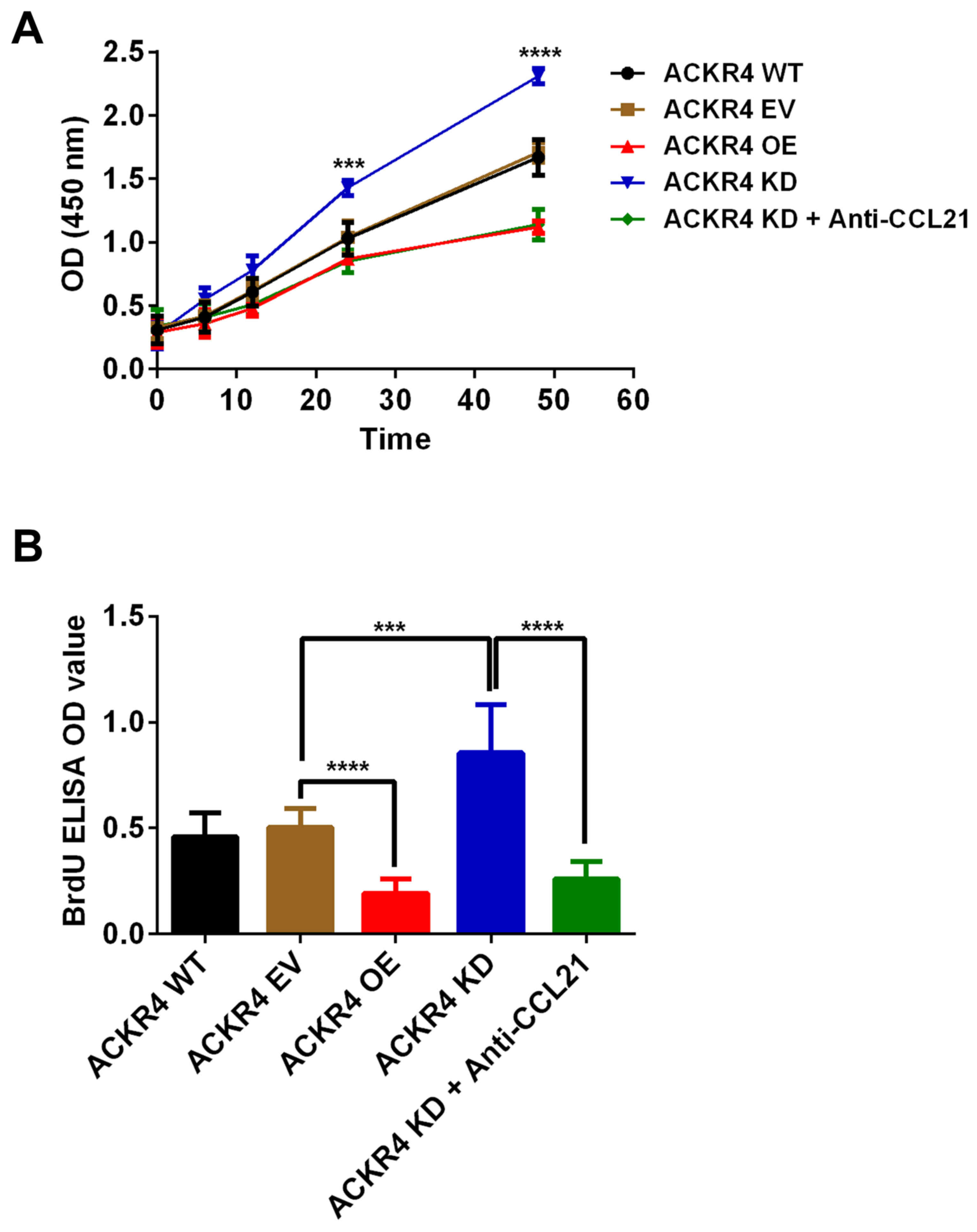
Given that the loss of ACKR4 promoted SUNE-1 tumor
development in vivo, the present study further hypothesized
that loss of ACKR4 may directly enhance SUNE-1 cell proliferation.
In vitro assays revealed that SUNE-1 cells with
downregulated ACKR4 expression grew much faster in comparison with
SUNE-1 cells with wild-type ACKR4 expression (Fig. 4A). An ELISA assay was also performed
to validate that the cell proliferation rate was changed due to
ACKR4 expression (Fig. 4B). SUNE-1
cells with downregulated ACKR4 expression had the highest BrdU
incorporation (Fig. 4B). Notably,
when CCL21 was blocked by its neutralizing antibody, the
proliferation rate of SUNE-1 cells with downregulated ACKR4 was
significantly reduced (Fig. 4A and
B). Taken together, these data supported that loss of ACKR4
accelerated SUNE-1 cell proliferation via a CCL21-depedent
mechanism.
Loss of ACKR4 induces EMT in SUNE-1
cells
EMT is an initial event of tumor cell invasion and
metastasis. As the results presented earlier indicated that loss of
ACKR4 promoted tumor cell lymph node metastasis (Fig. 1G), the study further investigated
whether the loss of ACKR4 induced EMT. The EMT gene signature was
measured, and the top four genes with altered expression were
plotted (Fig. 5). The EMT-associated
genes, including VIM, FN1 and SNAI1, were upregulated in ACKR4
knockdown SUNE-1 cells, as compared with their levels in ACKR4
wild-type SUNE-1 cells (Fig. 5A-C).
However, CAV2, a gene associated with epithelial function, was
upregulated in ACKR4 overexpression cells (Fig. 5D). Furthermore, in the ACKR4
overexpression cells, the expression levels of three EMT-associated
genes were suppressed (Fig. 5A-C).
Notably, when CCL21 was blocked, the EMT gene expression was
reduced in the ACKR4 knockdown SUNE-1 cells, suggesting that loss
of ACKR4 induced EMT via CCL21-depedent mechanisms.
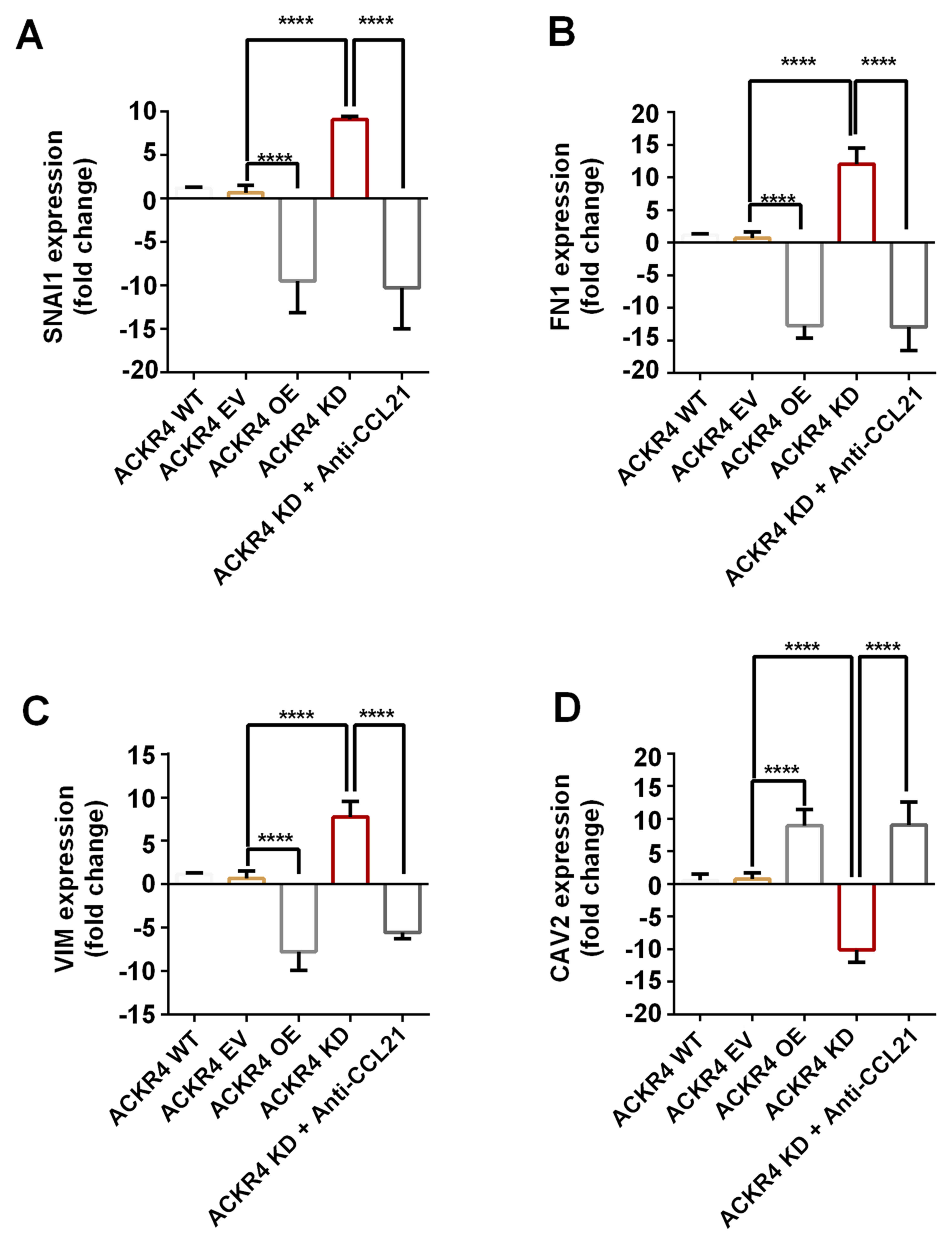 | Figure 5.Expression of EMT-associated genes in
nasopharyngeal carcinoma cells with different ACKR4 expression
levels. The four plotted genes exhibited the highest fold change
between the ACKR4 OE and ACKR4 KD cells out of all the
EMT-associated genes examined. (A) SNAIL1, (B) FN1 and (C) VIM were
upregulated in ACKR4 KD cells, but were downregulated in ACKR4 OE
cells. (D) CAV2 gene exhibited the opposite trend. ****P<0.0001.
EMT, epithelial-mesenchymal transition; ACKR4, atypical chemokine
receptor 4; WT, wild-type; EV, empty vector; KD, knockdown; OE,
overexpression; CCL21, C-C motif chemokine ligand 21. VIM, Variant
in Methylation; FN1, Fibronectin 1; SNAI1, snail family
transcriptional repressor 1. |
Loss of ACKR4 enhances SUNE-1 cell
invasion in vitro
Since ACKR4 knockdown upregulated the EMT genes in
SUNE-1 cells, the role of ACKR4 on SUNE-1 cell invasion was further
examined. As shown in Fig. 6, loss
ACKR4 expression improved the invasion ability of SUNE-1 cells,
while overexpression of ACKR4 reduced invasion. MMP2 and MMP9 are
the two key MMPs that mediate tumor cell invasion. As expected,
loss of ACKR4 induced MMP2 and MMP9 expression in SUNE-1 cells
(Fig. 6C and D), supporting that the
loss of ACKR4 was able to improve SUNE-1 cell invasion ability.
Furthermore, by blocking CCL21, tumor invasion caused by ACKR4
knockdown was suppressed in SUNE-1 cells.
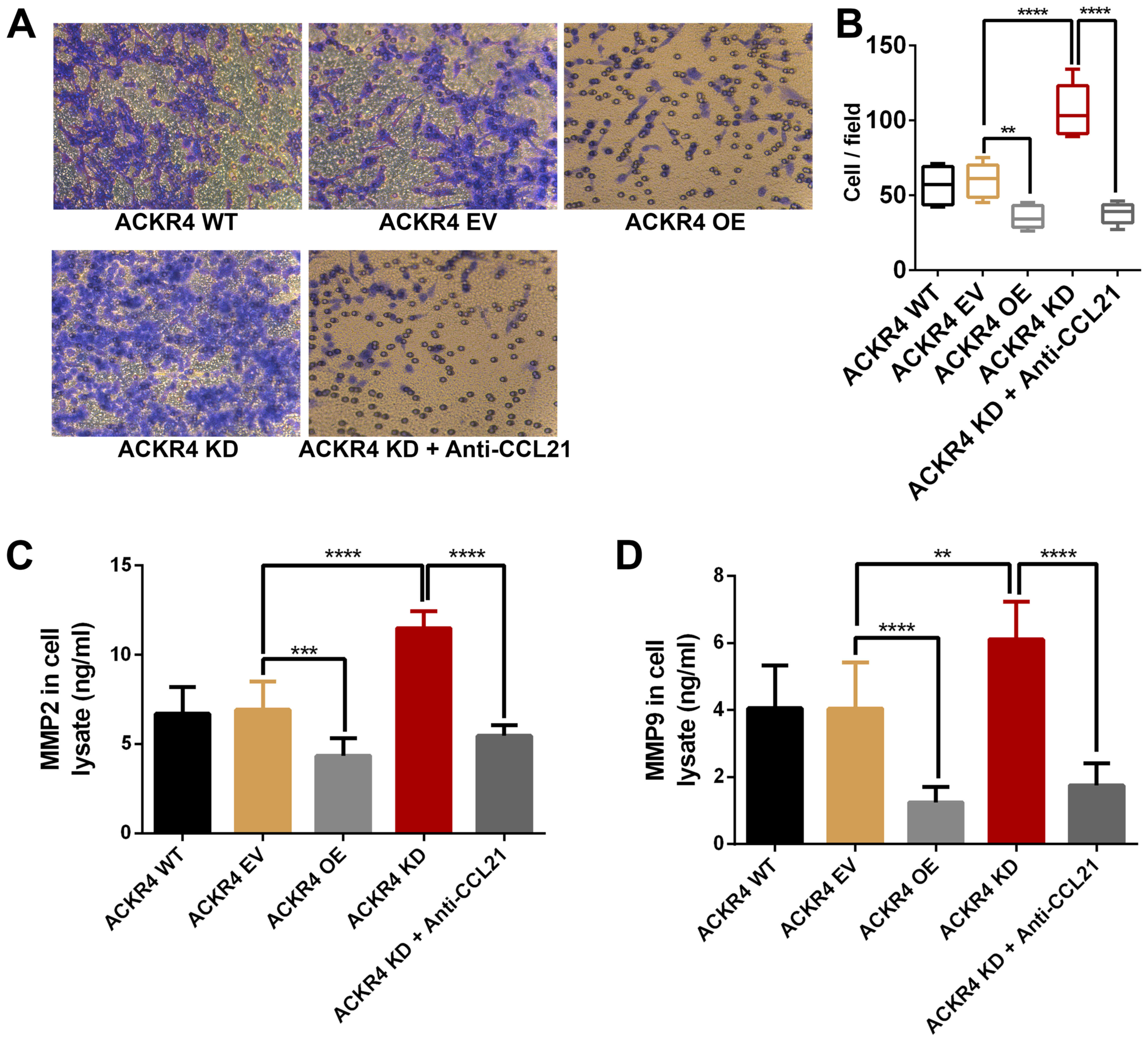 | Figure 6.ACKR4 expression and nasopharyngeal
carcinoma SUNE-1 cell invasion. (A) Representative figures of cell
invasion assay (magnification, ×200). (B) ACKR4 OE cells and
anti-CCL21-treated ACKR4 KD cells displayed decreased cell
invasion, whereas ACKR4 KD cells exhibited elevated cell invasion.
Expression levels of (C) MMP2 and (D) MMP9 were increased in ACKR4
KD cells. **P<0.01, ***P<0.001 and ****P<0.0001. ACKR4,
atypical chemokine receptor 4; WT, wild-type; EV, empty vector; KD,
knockdown; OE, overexpression; CCL21, C-C motif chemokine ligand
21; MMP, matrix metalloproteinase. |
Discussion
Chemokines are involved in almost all pathologies,
including cancer (14). In addition,
the inflammatory response has been recognized as a constitutive
hallmark of cancer (14). Over the
past two decades, ACKRs have been identified as a significant
component of the inflammatory response (4,5),
efficiently decreasing the chemokine concentration in inflamed
tissues via internalization (4,5). As a
member of the ACKR family, ACKR4 internalizes CCL19 and CCL21 with
a high efficiency once bound to them (4,5). CCL19
and CCL21 are also the two sole ligands to CCR7, which has a
critical impact on regulating tumor biology (15). However, there is limited information
on the pathological functions of ACKR4 in NPC.
To examine the effects of ACKR4 on NPC development,
the present study first measured ACKR4 expression in NPC clinical
samples. Notably, ACKR4 was significantly downregulated in the
tumor tissue, when compared with the adjacent normal tissue.
Meanwhile, knockdown of ACKR4 in NPC cells significantly increased
tumor growth and lymph node metastasis. These observations were in
line with those of previous studies in breast cancer, colorectal
cancer and NPC, which reported that ACKR4 was abnormally
downregulated in tumors (6,9,16).
Therefore, these findings suggested that loss of ACKR4 is a
promoting factor of NPC.
ACKR4 has been reported to be a scavenger of CCL19,
CCL21, CCL25 and CXLC13 (4,5). To determine the mechanism by which loss
of ACKR4 promotes NPC development, the scavenging ability of ACKR4
to its ligands was measured in the NPC cell condition. ACKR4 was
found to efficiently decrease CCL21 concentration in NPC tumor
tissue. It is known that overexpression of CCL21 promoted tumor
proliferation, invasion and immune suppression in tumor models
(17,18). In the present in vitro
experiments, when CCL21 was neutralized, knockdown of ACKR4 did not
exhibit any tumor promoting effects. Thus, in NPC, accumulation of
CCL21 in tumor tissue may be the major mechanisms through which
loss of ACKR4 promotes tumor development.
It has been reported that the CCL21/CCR7 signaling
pathway enhances transforming growth factor-β1-dependent EMT, which
is a major mechanism of tumor invasion and metastasis (19). In light of this fact, the current
study further explored the role of ACKR4 in reducing EMT in NPC.
Loss of ACKR4 significantly upregulated EMT gene expression and
tumor cell invasion in vitro. When ACKR4 was overexpressed
or CCL21 was neutralized, EMT genes were significantly
downregulated, and the invasion ability of tumor cells was reduced
in vitro. Altogether, these key observations supported that
overexpression of ACKR4 impaired CCL21/CCR7 mediated EMT and tumor
cell invasion.
In conclusion, the present study investigated the
effects of ACKR4 on tumor development in the NPC system. The
findings of this study have provided evidence that loss of ACKR4 in
NPC is prevalent and may promote NPC development via accumulating
CCL21 in tumor tissue. Therefore, neutralizing CCL21 in NPC with
low ACKR4 expression may be a novel treatment that requires further
investigation.
Acknowledgements
Not applicable.
Funding
The present study was supported by an internal grant
from the Third Affiliated Hospital of Kunming Medical University
(Kunming, China).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW and CS designed the present study and analyzed
the data; CS and YJ performed the experiments.
Ethics approval and consent to
participate
All animal procedures and the use of human tissues
were reviewed and approved by the Ethical Committee of Third
Affiliated Hospital of Kunming Medical University (kunming, China)
and all efforts were made to minimize the suffering of the
experimental mice.
Patient consent for publication
Informed consent was obtained from all patients
prior to enrollment
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chua ML, Wee JT, Hui EP and Chan AT:
Nasopharyngeal carcinoma. Lancet. 387:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blanchard P, Lee A, Marguet S, Leclercq J,
Ng WT, Ma J, Chan AT, Huang PY, Benhamou E, Zhu G, et al:
Chemotherapy and radiotherapy in nasopharyngeal carcinoma: An
update of the MAC-NPC meta-analysis. Lancet Oncol. 16:645–655.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ulvmar MH, Hub E and Rot A: Atypical
chemokine receptors. Exp Cell Res. 317:556–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Massara M, Bonavita O, Mantovani A, Locati
M and Bonecchi R: Atypical chemokine receptors in cancer: Friends
or foes? J Leukoc Biol. 99:927–933. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feng LY, Ou ZL, Wu FY, Shen ZZ and Shao
ZM: Involvement of a novel chemokine decoy receptor CCX-CKR in
breast cancer growth, metastasis and patient survival. Clin Cancer
Res. 15:2962–2970. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harata-Lee Y, Turvey ME, Brazzatti JA,
Gregor CE, Brown MP, Smyth MJ, Comerford I and McColl SR: The
atypical chemokine receptor CCX-CKR regulates metastasis of mammary
carcinoma via an effect on EMT. Immunol Cell Biol. 92:815–824.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hou T, Liang D, Xu L, Huang X, Huang Y and
Zhang Y: Atypical chemokine receptors predict lymph node metastasis
and prognosis in patients with cervical squamous cell cancer.
Gynecol Oncol. 130:181–187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu Y, Tang W, Liu Y, Wang G, Liang Z and
Cui L: CCX-CKR expression in colorectal cancer and patient
survival. Int J Biol Markers. 29:e40–e48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hernandez-Gea V, Toffanin S, Friedman SL
and Llovet JM: Role of the microenvironment in the pathogenesis and
treatment of hepatocellular carcinoma. Gastroenterology.
144:512–527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pang MF, Georgoudaki AM, Lambut L,
Johansson J, Tabor V, Hagikura K, Jin Y, Jansson M, Alexander JS,
Nelson CM, et al: TGF-β1-induced EMT promotes targeted migration of
breast cancer cells through the lymphatic system by the activation
of CCR7/CCL21-mediated chemotaxis. Oncogene. 35:748–760. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mo M, Zhou M, Wang L, Qi L, Zhou K, Liu
LF, Chen Z and Zu XB: CCL21/CCR7 enhances the proliferation,
migration, and invasion of human bladder cancer T24 cells. PLoS
One. 10:e01195062015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin
C and Flavell RA: Inflammation-induced cancer: Crosstalk between
tumours, immune cells and microorganisms. Nat Rev Cancer.
13:759–771. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Forster R, Schubel A, Breitfeld D, Kremmer
E, Renner-Muller I, Wolf E and Lipp M: CCR7 coordinates the primary
immune response by establishing functional microenvironments in
secondary lymphoid organs. Cell. 99:23–33. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi JY, Yang LX, Wang ZC, Wang LY, Zhou J,
Wang XY, Shi GM, Ding ZB, Ke AW, Dai Z, et al: CC chemokine
receptor-like 1 functions as a tumour suppressor by impairing
CCR7-related chemotaxis in hepatocellular carcinoma. J Pathol.
235:546–558. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xiong Y, Huang F, Li X, Chen Z, Feng D,
Jiang H, Chen W and Zhang X: CCL21/CCR7 interaction promotes
cellular migration and invasion via modulation of the MEK/ERK1/2
signaling pathway and correlates with lymphatic metastatic spread
and poor prognosis in urinary bladder cancer. Int J Oncol.
51:75–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Irshad S, Flores-Borja F, Lawler K,
Monypenny J, Evans R, Male V, Gordon P, Cheung A, Gazinska P, Noor
F, et al: RORγt+ Innate lymphoid cells promote lymph node
metastasis of breast cancers. Cancer Res. 77:1083–1096. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma H, Gao L, Li S, Qin J, Chen L, Liu X,
Xu P, Wang F, Xiao H, Zhou S, et al: CCR7 enhances TGF-β1-induced
epithelial-mesenchymal transition and is associated with lymph node
metastasis and poor overall survival in gastric cancer. Oncotarget.
6:24348–24360. 2015. View Article : Google Scholar : PubMed/NCBI
|