Introduction
Lung cancer is a common malignancy which causes one
million worldwide mortalities each year (1). Non-small cell lung cancer (NSCLC)
accounts for 70–85% lung cancer cases, among which 40% patients
reach late stage prior to diagnosis (2). The multidisciplinary paradigm of
therapies focused on surgery, chemotherapy and radiotherapy has
advanced greatly in recent decades; however, the 5-year survival
rate has remained almost constant (3–5).
Mesothelioma developed from lung and breast cancers is also a
malignant disease associated with aggressive disease progress and
extensive economic burden (6,7). As the
median overall survival in late stage was only 1 year, both NSCLC
and mesothelioma are refractory to standard chemotherapy and only a
marginal proportion of patients survive (8–10).
Therefore, novel therapies are required.
Previously, adoptive immunotherapy demonstrated
promise for prolonging the survival with minimum toxicity (11). The antitumor activity of adoptive
therapies was exerted by lymphokine-activated killer cells (LAKs),
tumor-infiltrating lymphocytes (TILs), cytokine-induced killer
cells (CIKs), dendritic and cytokine-induced killer cells
(DC-CIKs), natural killer (NK) cells, engineered T cells and
chimeric antigen receptor T cells (CAR-T cells), among which CAR-T
cell therapy has achieved remarkable efficacy in acute
lymphoblastic leukemia (ALL) (12).
CAR-T cells recognize surface antigens independently from major
histocompatibility complex restriction, mostly via single chain
variable fragments (scFvs), which are derived from tumor
antigen-reactive antibodies (13).
When targeting tumor surface antigens, the cluster of
differentiation (CD)3ζ chain domain and CD28 and/or 4-1BB
costimulatory domains will be activated to enhance T cell
proliferation, cytokine secretion, resistance to apoptosis and
in vivo persistence (13–15).
Nevertheless, ‘on target, off tumor’ toxicity is a major challenge
in CAR-T therapy, in which the antigen is also expressed in normal
tissues (16). Therefore,
constructing CAR-T cells that target tumor tissues with negligible
off-tumor toxicity is of critical importance.
Mesothelin (MSLN) is an immunogenic glycoprotein
that is abundant in ovarian cancers, NSCLC and mesotheliomas
(17). Due to its low expression in
normal mesothelial cells, MSLN is an ideal candidate for targeted
immunotherapy in mesotheliomas (18). In the present study,
second-generation CAR-T cells targeting MSLN, the scFvs, which have
affinities to intracellular domain of co-stimulatory factor CD28,
4-1BB and CD3ζ, were constructed. In both ex vivo and in
vivo experiments, this approach was demonstrated to exert
potent effects on tumor clearance. At the cellular level, the CAR-T
cells constructed from healthy individuals seemed to have more
potent effect than those derived from patients, indicating the
potential advantage of allogenic CAR-T therapy. The significantly
elevated targeting of CAR-T cells can be achieved with a 0.5:1
effector to target (E:T) ratio, and the antitumor effect of CAR-T
cells increase rapidly with increases of the E:T ratio. When it
reached 40:1, 78% cells were damaged. In an in vivo mouse
model, the difference in growth rate of tumor size was significant
at day 5, after which both groups became synchronized in growth of
tumor size. These findings suggest that CAR-T cells targeting MSLN
could inhibit tumor growth both in vivo and ex vivo,
although a sophisticated methodology that enhances the effect of
CAR-T cells is required to continuously suppress the tumor.
Materials and methods
Construct of pCAR-MSLN recombinant
lentiviral expression vector and viral production
Genetic synthesis of CAR targeting MSLN was
outsourced to iCARTab Biomed (Suzhou, China). The whole-gene
sequence was sub-cloned to lentiviral vector pCAR-puro following
cleavage via EcoRI-XbaI restriction enzyme (Takara
Bio, Inc., Otsu, Japan). pCAR-puro, which contains CD28 and 4-1BB
signaling modules, was developed specifically for CAR-T therapy
studies. The cloned sequence of MSLN CAR in the resulting
recombinant vector pCAR-MSLN was confirmed by Sanger sequencing
(19), followed by extraction using
a Qiagen Plasmid Maxi kit (Qiagen GmbH, Hilden, Germany). The
pCAR-MSLN vectors were then transfected into 293T cells (Cell Bank
of the Chinese Academy of Sciences, Shanghai, China) as previously
described (20). Titration of
lentivirus was performed by quantitative polymerase chain reaction
using woodchuck hepatitis virus posttranscriptional regulatory
element (WPRE) and albumin (ALB) genes as reference. Premix
Taq™ kit (cat. no. R004Q; Takara Bio, Inc.) was used for
the qPCR assay, according to the manufacturer's protocol. The
sequences of the primers and probes for WPRE and ALB are shown in
the Table I. PCR was performed under
the following conditions: 50 cycle denaturation at 95°C for 15 sec,
annealing at 60°C for 60 sec and elongation at 72°C for 30 sec. The
Cq value of lentivirus carrying pCAR-MSLN (Table II) was calculated using the
2−ΔΔCq method (21). The
vector copy number and titer were calculated as follows:
 | Table I.Primer and probe sequences used for
the quantitative polymerase chain reaction analysis. |
Table I.
Primer and probe sequences used for
the quantitative polymerase chain reaction analysis.
| Gene | Sequence
(5′-3′) |
|---|
| WPRE |
|
| Forward
primer |
GGCACTGACAATTCCGTGGT |
| Reverse
primer |
AGGGACGTAGCAGAAGGACG |
|
Probe |
FAM-ACGTCCTTTCCATGGCTGCTCGC-BHQ |
| ALB |
|
| Forward
primer |
GCTGTCATCTCTTGTGGGCTGT |
| Reverse
primer |
ACTCATGGGAGCTGCTGGTTC |
|
Probe |
FAM-CCTGTCATGCCCACACAAATCTCTCC-BHQ |
| Table II.Cq value of lentivirus carrying
pCAR-MSLN. |
Table II.
Cq value of lentivirus carrying
pCAR-MSLN.
| Sample | WPRE | ALB |
|---|
| Lentivirus | 23.30 | 23.71 | 27.29 | 27.62 |
Vector copy number=Quantity mean of WPRE
sequenceQuantity mean of Alb sequencex2Titer=Number of target cells
x mumber of copies per cell of the sampleVolume of
supernatant(ml)
T cell sampling and preparation of
CAR-T cells
A total of 5 patients (4 male and 1 female; age,
64.80±2.77) and one 45-year-old female healthy control were
recruited to provide T cells. Blood was obtained at Shanghai Chest
Hospital (Shanghai, China) in December 2015. T cells derived from
the healthy control and patients were used in separate experiments.
Blood samples (50 ml) were obtained from each donor. T cells were
isolated by Lymphoprep (StemCell Technologies, Vancouver, Canada).
The blood sample was added to the upper layer of the Lymphoprep and
then centrifuged at 800 × g for 20 min at room temperature to
obtain a mononuclear cell layer. Then separated mononuclear cells
were added to a new centrifuge tube with 50 µl/ml sorted magnetic
beads mix (EasySep™ Human T Cell Enrichment kit;
StemCell Technologies) and incubated for 10 min at room
temperature. The tube was then inserted into a magnetic pole
(EasySep™ Magnet; cat. no. 18000; Stem Cell
Technologies) and allowed to stand at room temperature for 5 min.
Following the incubation, the cells were removed and then
re-suspended in RPMI-1640 supplemented with 10% FBS (both Gibco;
Thermo Fisher Scientific Inc., Waltham, MA, USA) and 100 U/ml
interleukin (IL)-2 (R&D Systems, Inc., Minneapolis, MN, USA).
When the cell density reached 1×106 cells/ml, a mixture
of 100 U/ml IL-2, 100 ng/ml anti-CD3 antibodies (OKT3; cat. no.
14-0037-82) and 250 ng/ml anti-CD28 antibodies (cat. no.
14-0289-82; both eBioscience; Thermo Fisher Scientific Inc.) was
added to the medium. Following 48 h of culture at 37°C,
1.83×108 TU/ml lentivirus carrying pCAR-MSLN was added
to the cell culture, together with 8 µg/ml polybrene
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and then incubated
for 24 h at 37°C and 50% CO2. The mixture of cell
culture and virus was centrifuged at 250 × g for 10 min at room
temperature and the supernatant containing additional viruses was
removed. The T cells were then re-suspended in fresh medium, and
incubated at the same condition for 3–6 days to produce CAR-T
cells.
The present study was approved by the Institutional
Review Board of the Shanghai Chest Hospital (Shanghai, China) and
was conducted in accordance with the Declaration of Helsinki and
Good Clinical Practice guidelines. All patients provided written
informed consent prior to any study-related procedure.
Detection of MSLN-CAR expression in
recombinant CAR-T cells
As protein L was able bind to the light chain of
mouse antibody (22), the affinity
system comprised of biotin-tagged protein L and phycoerythrin
(PE)-conjugated streptavidin could be used to detect the expression
of MSLN CAR in CAR-T cells. CAR-T cells were continuously cultured
in RPMI-1640 medium for 2–5 days at 37°C. Collected cells were
adjusted to a density of 1×106/ml and centrifuged at 500
× g for 5 min at 37°C. The cell pellets was washed 3 times with PBS
and centrifuged at 500 × g for 5 min at 37°C. A total of 100 µl
Protein L (500 ng; ACROBiosystems, Inc., Newark, DE, USA) in PBS
was added to the cell pellet and incubated for 30 min at room
temperature. Cells were washed 3 times with PBS and centrifuged at
500 × g for 5 min at 37°C then incubated with PE-streptavidin
(eBioscience, Thermo Fisher Scientific Inc.) for 30 min in the dark
at room temperature, washed 3 times with PBS and centrifuged at 500
× g for 5 min at 37°C. The CAR-T cells were then resuspended in 500
µl PBS and analyzed using a BD Accuri C6 flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA). The data was analyzed by
Flowjo software (version 10; FlowJo, LLC, Ashland, OR, USA).
Ex vivo cytotoxicity assay
Cytotoxicity assay was performed by measuring the
percentage of cell lysis using the LDH Assay kit (Promega
Corporation, Madison, WI, USA). CAR-T cells were washed with
sterile PBS (Gibco; Thermo Fisher Scientific, Inc.), resuspended
with serum-free RPMI-1640 and mixed into target cells, namely HeLa
cells or CHO-K1 cells (both Cell Bank of the Chinese Academy of
Sciences) overexpressing MSLN (CHO-K1-MSLN cells) at a gradient
ratio of effector to target (E:T ratios). T cells were used as a
control. A total of 9 wells were used for triplicate experiment to
measure the spontaneous lysis of target and effector cells and the
maximum number of lysis using lysis agent CytoTox 96
Non-Radioactive Cytotoxicity assay (cat. no. G1780; Promega
Corporation). The ‘Target Spontaneous’ and ‘Target Maximum’ wells
were seeded with 5×104 target tumor cells, and ‘Effector
Spontaneous’ wells were seeded with CAR-T MSLN cells according to
different E:T ratios. Following culturing for 6 h at 37°C, 10X
lysis agent was added to the ‘Target Maximum’ well and incubated at
37°C and 50% CO2 for 45 min. Following complete lysis of
target cells in ‘Target Maximum’ wells, the plate was centrifuged
at 1,200 × g at room temperature for 5 min, and the 50-µl
supernatant of each well was transferred to another plate. Assay
buffer was mixed with substrate mix and aliquoted to each well.
Following termination with stop solution, the absorbance of the
mixture at an optical density of 490 nm was measured via a
microplate reader. The percentage of lysis in experimental and
control well was calculated as follows:
Lysis%=Experimental (or control) -
Effector Spontaneous - Target SpontaneousTarget Maximum - Target
Spontaneousx100
Construct of CHO-K1-MSLN
The MSLN transcript NM_005823.5 was synthesized by
GenScript Biotech Corp (Nanjing, China) and subcloned into
Lenti-CMV-Puro vectors (iCARTab Biomedical. Co. Ltd.) as previously
described (23). Following Sanger
confirmation as previously described (19), vectors were extracted and then
transduced into packaging cells using polyetherimide (Polyscience,
Inc., Warrington, PA, USA), and Lenti-MSLN viruses were isolated by
adding PBS supplemented with 20% sucrose to the culture medium of
the packed cells. Then, the mixture was centrifuged at 20,000 × g
for 2 h at 4°C; the viruses, which were in the precipitate, were
then removed. CHO-K1 cells were then transfected with Lenti-MSLN
viruses. Following the centrifugation of CHO-K1 culture mixed with
Lenti-MSLN viruses at 800 × g at room temperature for 30 min and
removal of the supernatant, CHO-K1-MSLN cells were re-suspended in
fresh medium and cultured for 5 days.
Flow cytometry detection of MSLN
HeLa or CHO-K1-MSLN cells were divided into two
groups, which were blocked by a FcR blocking reagent (Miltenyi
Biotec GmbH, Bergisch Gladbach, Germany) for 30 min at room
temperature were then incubated with either allophycocyanin
(APC)-MSLN antibodies (cat. no. FAB32652A; R&D Systems, Inc.)
or rat immunoglobulin G2A APC isotype control (cat. no. IC006A;
R&D Systems, Inc.) for 30 min at room temperature. The cells
were then suspended in 500 µl PBS and analyzed using a BD Accuri C6
flow cytometer. The data was analyzed by Flowjo software.
In vivo validation of antitumor
effect
A total of 15 male NPG mice (weight, 18–22 g;
Beijing Biocytogen Co., Ltd., Beijing, China) aged 3–4 weeks were
housed in used in ventilated cages (5 mice/cage) at 20–26°C with
30–70% humidity and alternate lighting according to 12 h intervals.
The cages were ~300×180×150 mm. Dried granule food was sterilized
by radiation irradiation. The mice had free access to the food and
sterile water. A small section of patients' tumor tissue was
isolated. Collagenase type II (cat. no. 17101015; Thermo Fisher
Scientific Inc.) digested the tumors into a single cell suspension
and blocked by a FcR blocking reagent (Miltenyi Biotec GmbH) for 30
min at room temperature. The cells were then incubated with
APC-conjugated MSLN antibodies (cat. no. FAB32652A; 1:100; R&D
Systems, Inc.) for 30 min at room temperature and then washed 3
times with PBS. The expression of MSLN was analysed on the membrane
surface of tumor-derived cells from patients using flow cytometry
and the data was analysed by Flowjo software (version 10; FlowJo,
LLC). Following the irradiation of the mice with 0.8 Gy cobalt-60
for 24 h, NSCLC tissues (diameter, 2–3 mm) from 5 patients with
high MSLN expression were subcutaneously inoculated into the right
hackle of NPG mice. The tumor grew rapidly following transplant,
and following 26 days growth, the size of tumor reached a mean of
20–30 mm3. The mice were then randomly allocated to
control and experimental groups based on the tumor volume
(n=7/group). One mouse was not used in the current study. A total
of 8×106 CAR-T cells or T cells were administered via
tail vein infusion to the experimental and control groups,
respectively. The size of tumors was measured by vernier calipers
every 5 days, for a consecutive 15 days. The volume of the tumor
was calculated as: Volume (mm3)=(AxB2)/2,
where A represents the long diameter of tumor tissue and B
represents the short diameter.
Statistical analysis
Data were analyzed by SPSS 18.0 (SPSS, Inc.,
Chicago, IL, USA). All data are presented as mean ± standard
deviation. Statistical analysis of ex vivo tumor cell lysis
was performed with Wilcoxon matched pairs signed rank test, and the
in vivo experiment was analyzed with independent sample
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Successful construction of pCAR-MSLN
recombinant lentiviral expression vector
Second generation CAR molecules were designed for
the present study. The lentiviral vector pCAR-MSLN integrated with
anti-MSLN CAR also contains co-stimulator, CD28 and 4-1BB. The
vectors were excised by EcoRI-XbaI, and
electrophoresis demonstrated that they were ~2,200 bp in length,
which was close to 2,171 bp, as calculated by adding together the
number of base pairs of DNA expressing the anti-MSLN scFv peptide,
CD28 and 4-1BB retrieved from the NCBI databse (www.ncbi.nlm.nih.gov). The pCAR-MSLN vectors were
amplified and confirmed by Sanger sequencing. No mutations were
detected in the recombinant lentiviral vector pCAR-MSLN (data not
shown).
Titration of recombinant lentivirus
containing pCAR-MSLN
The pCAR-MSLN vectors were transfected to packaging
293T cells. qPCR was performed to titrate the virus. WPRE oligos
amplified the WPRE sequence present in almost all later generation
lentiviral vectors. ALB oligos were used to normalize the genomic
DNA. Based on the Cq value of WPRE and ALB in pCAR-MSLN-containing
293T cells, two experimental replicates yielded the following
number of lentivirus copies: WPRE, 23.30 and 23.71 and ALB, 27.29
and 27.62 (Table II). The titer of
pCAR-MSLN in 293T was quantified as 1.83×108 TU/ml (data
not shown).
MSLN CAR expression in recombinant
CAR-T cells
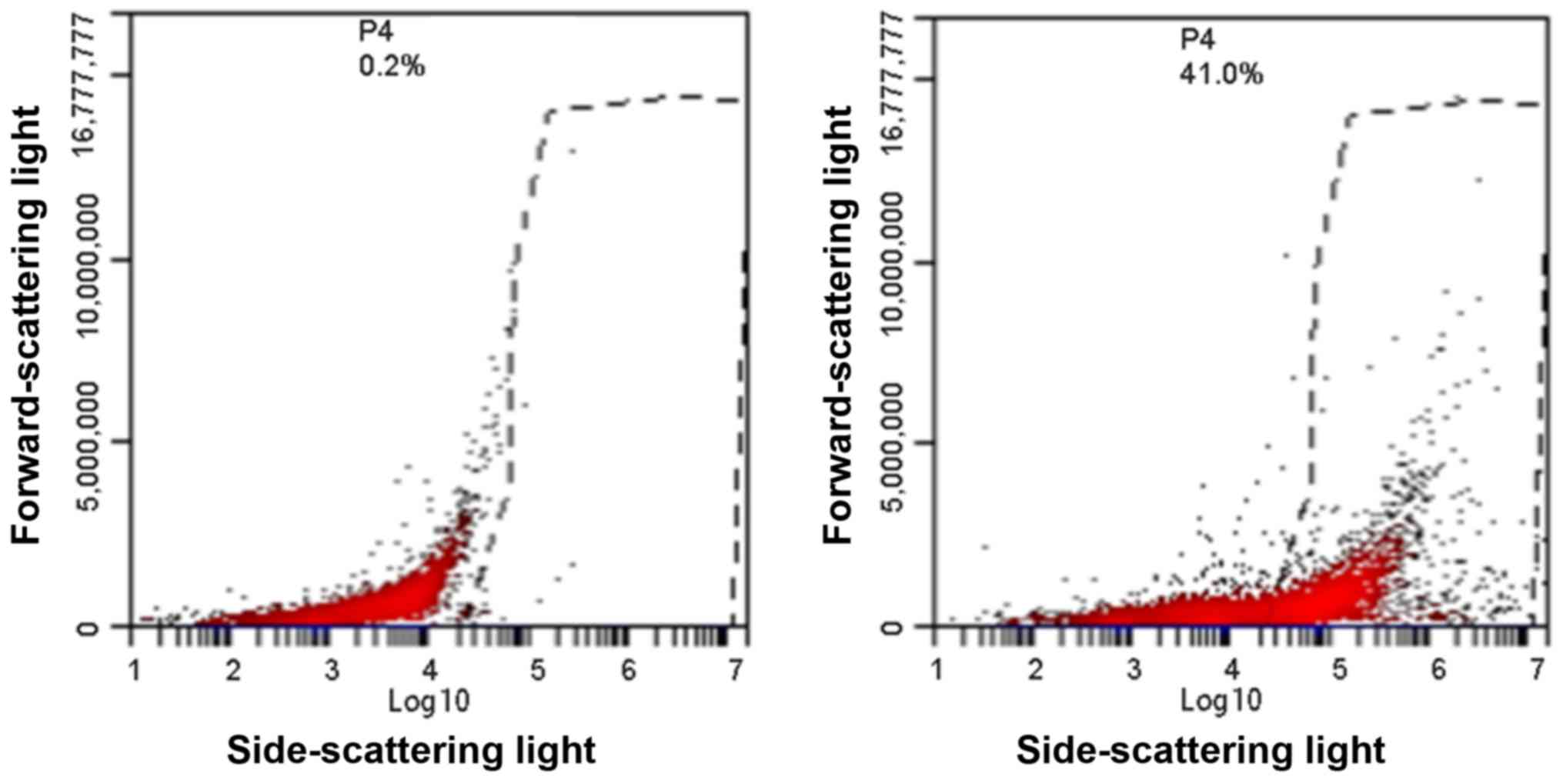
MSLN CAR expression was detected by flow cytometry.
The output graph of flow cytometry indicated a markedly difference
in MSLN CAR between CAR-T cells and control T cells (Fig. 1), suggesting the successful
construction of MSLN CAR-T cells by transfecting recombinant
lentiviruses to primary T cells.
CAR-T cells are detrimental to tumor
cells
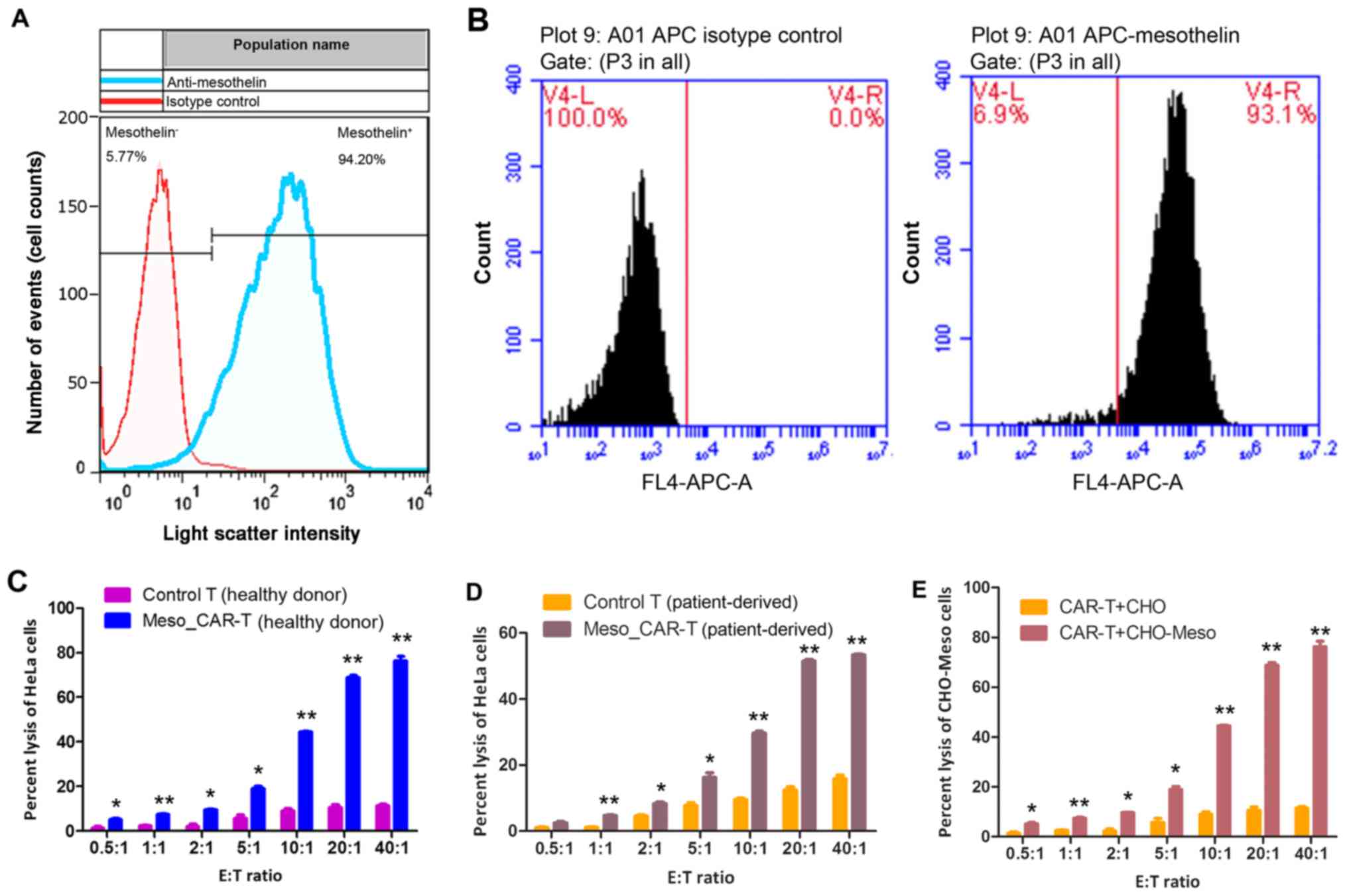
HeLa cells were chosen as target cells to validate
the effect of MSLN CAR-T cells. To confirm the targetability of
HeLa cells, the expression of MSLN was measured. Flow cytometry
demonstrated that 94.20% cells express MSLN, and 5.77% HeLa cells
were MSLN negative (Fig. 2A). As the
whole HeLa cell culture exhibited high expression of MSLN, such
discrepancy within HeLa cells may be due to heterogeneity of cancer
cells. Similarly, recombinant CHO-K1-MSLN exhibited abundant MSLN
expression, where 93.1% CHO-K1-MSLN cells overexpressed MSLN, and
6.9% of them carried low content of MSLN (Fig. 2B).
Following the confirmation of targetability of HeLa
cell and CHO-K1-MSLN cells, the antitumor effect of CAR-T cells was
verified by in vitro experiments. When the E:T ratio reached
0.5:1, the antitumor effect of CAR-T cells was significantly higher
than control T cells (P<0.05; Fig. 2C
and D), as indicated by LDH assay of tumor cells. The CAR-T
cells constructed from the healthy donor and patients exhibited
significantly more potent antitumor effects compared with their
respective T cells (all P<0.05; Fig.
2C and D).
To confirm that CAR-T cells could exert the same
effect on other types of cells, recombinant CHO-K1-MSLN
overexpressing MSLN was used as a target of CAR-T cells constructed
from healthy individual. In accordance with HeLa cells, the
significantly elevated targeting of CAR-T cells was achieved with
0.5:1 E:T ratio, and the antitumor effect of CAR-T cells increased
rapidly with increases of the E:T ratio (P=0.04). When this reached
40:1, 78% cells were lysed (Fig.
2E).
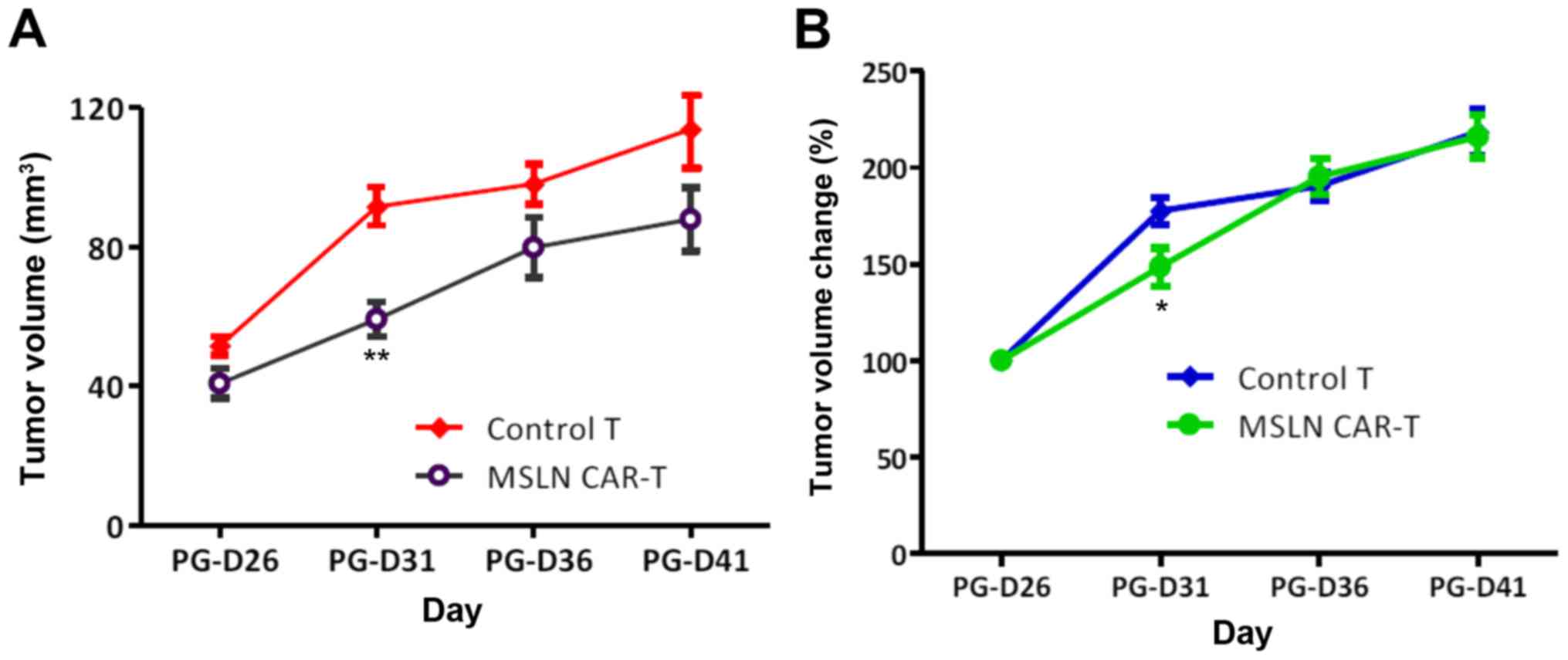
The in vivo antitumor effect of CAR-T
cells
With the effective E:T ratio obtained from in
vitro experiments, NPG mice were used to validate in
vivo antitumor activity. All tumors grew following tail vein
injection, whereas those infused with CAR-T cells grew slower. The
difference in growth rate of tumor size was significant at PG-D31
(P=0.03), whereas subsequently, both groups gradually synchronized
in tumor growth rate without continuous injection (Fig. 3). This result suggests that a
sophisticated methodology that enhances the effect of CAR-T cells
is required to continuously suppress the tumor.
Discussion
The first immunotherapy used clinically was the
injection of Streptococcus erysipelas and the Bacillus
prodigiosus to treat inoperable sarcoma, in which the
anticancer effect was observed and drew extensive research
interests (1). There are numerous
therapeutic methodologies designed to activate the immune system to
kill tumor cells, which are divided into the following four
categories based on the underlying mechanisms (24): Adoptive cell therapy, tumor vaccine,
monoclonal antibody and other non-specific cytokines. Adoptive cell
therapy has long been established in cancer treatment, which
involves transferring in vitro cultured lymphocytes back to
cancer patients. Adoptive cell therapy could remedy the immune
inactivity following radio-chemotherapy (25). Currently, the most used lymphocyte
subgroups in clinical settings include: LAK, TIL, CIK, DC-CIK, NK
cells, γδT cells, CAR-T cells and TCR-T cells (26). Both LAK and TIL were activated by
IL-2, yet the antitumor potency of TIL was 50–100-fold higher than
LAK as TIL were isolated from tumor infiltrating lymphocytes
(27). They were successfully
applied in treating sarcoma (28),
but time spent in culturing and the difficulties in separating TIL
constitute the present challenge in improving the efficacy. DC
cells could recognize tumor antigens and CIK cells secrete
cytotoxic factors, and combination of DC and CIK was demonstrated
to improve efficacy of chemotherapy, mitigate side effects and
prolong life expectancy of patients (29). However, immune tolerance and immune
escape are major barriers for DC-CIK therapy (30). NK cell-based therapy has achieved
marked efficacy in NSCLC, myeloma, breast cancer, renal cell
carcinoma and colorectal carcinoma (31), and γδT cell-based therapy has been
effective in treating renal cell carcinoma and prostatic cancer
(32). However, the targetability
and tumor-killing capacity of both methods fell short of
expectations (33).
Engineered T cells that target a tumor antigen via
T-cell receptors (TCRs) or a CAR exhibited promise in rapid
stimulation of tumor immunity and reducing tumor burdens (34,35).
TCRs are restricted to leukocyte antigen, limiting their
application as a mainstream therapeutic strategy, whereas CARs may
be engineered to directly target proteins, carbohydrates or
glycolipids on the cell surface, providing design flexibility and
diversity (36). Compared with
first-generation CARs, which contains CD3ζ cytoplasmic domain,
second-generation CARs integrate intracellular signaling domains
from various costimulatory factors, such as CD28, 4-1BB or
inducible T cell costimulator, to augment the activation signal by
CD3ζ and promote amplification of T cells (37). Such dual signaling may eliminate
deficiency of T cells and enhance the persistence and function of T
cells (36). For example, CD28 can
bind to phosphoinositide 3-kinase (PI3K) via YMNM cytoplasmic
domain, thereby initiating the PI3K-protein kinase B pathway to
promote proliferation of T cells (38); 4-1BB can be transiently induced by
TCR and CD28 signaling through extracellular signal-regulated
kinase and c-Jun N-terminal kinase signaling pathways, resulting in
fast proliferation and durable functioning of CD4+ and
CD8+ T cells (36).
In the present study, second-generation CAR-T cells
targeting MSLN were constructed, of which the scFvs have affinities
to the intracellular domains of co-stimulatory factor CD28, 4-1BB
and CD3ζ (39). In ex vivo
and in vivo experiments, this approach was demonstrated to
exert a potent effect on tumor clearance. The lentiviral vector was
used to deliver engineered DNAs, which demonstrated high
transduction efficiency. Another advantage of employing lentiviral
vectors is that it avoids the integration of foreign genomics into
non-dividing human primary cells, which has been a concern for
retroviral vectors, therefore eliminating the undesired risk of
insertional oncogenesis (40–42).
Although CAR-T therapy has great potential for
killing tumors, the ‘on target, off tumor’ toxicity poses a major
concern. An approach to minimize such toxicity is to engineer
additional antibodies targeting specific antigens that are
differentially, if not exclusively, expressed in tumor than normal
tissues (43). CD19 is ubiquitously
expressed in malignant and normal B cells, but the normal B cells
expressing CD19 are hematopoietic or approaching cell death
(44). Therefore, CD19 is a nearly
ideal target for B cell malignancies. Recently, treatment of B cell
malignancies achieved a breaking advancement with CAR-T cell
therapy: Multiple clinical trials have revealed that CAR-T cells
targeting CD19 could treat refractory lymphoma with response rates
over 50% (45,46), and in myeloma, CAR-T cells engineered
to target CD19+ demonstrated efficacy in eradicating the
disease (47). Other target antigens
include tumor-associated glycoprotein 72 for metastatic colorectal
cancer (48), folate receptor-α for
ovarian cancer (49), L1-cell
adhesion molecule for metastatic neuroblastoma (50), and CD22 for ALL (51), in which 5 targets have entered phase
2 trials: GD2 (NCT02765243), CD22 (NCT03196830), CD20
(NCT03196830), CD30 (NCT03196830) and carcinoembryonic antigen
(NCT01723306) (52). Although these
studies envisage great potentials of second generation CAR-T cell
therapy, antigens that are rarely expressed in normal cells but
abundant in malignancies are still rare (53). Two outstanding tumor targets for
solid tumor are ERBB2 and MSLN, which were applied in 8 and 6
cancer types, respectively (54).
MSLN is a glycoprotein anchored to the plasma
membrane, which has minimal expression in normal tissue but
abundant expression in solid tumors, including mesothelioma,
ovarian cancer, pancreatic cancer and lung cancer (18,54–56)
Multiple studies (57–59) have suggested that MSLN expression is
correlated with poor prognosis. It can activate nuclear factor-κB,
mitogen-activated protein kinase and PI3K intracellular pathways
that contribute to cell proliferation and resistance to apoptosis
(60–62). In addition, overexpression of MSLN
could lead to excessive expression of matrix metalloproteinase-9,
promoting migration and infiltration (63). The initial clinical MSLN-specific
CAR-T therapy was conducted in 2 patients affected with malignant
pleural mesotheliomas and pancreatic cancer, respectively (64). Antitumor potency was achieved by
infusion of mRNA-engineered CAR-T MSLN cells with acceptable safety
despite the transient nature of CAR-T cells and the absence of
pretreated lymphodepletion.
In the present study, it was demonstrated that CAR-T
cells derived from healthy individuals exhibited better effects
than those derived from patients, indicating that allogenic T cells
may be more effective in suppressing malignancies than autologous T
cells at the cell level. The T cells derived from patients may have
undergone exhaustion, which is a state of dysfunction that commonly
arise from chronic infections and cancers (65). The mechanisms underlying T cell
exhaustion comprise elevated multiple inhibitory signaling,
including programmed cell death protein 1, lymphocyte activation
gene 3, CD160, T cell immunoglobulin and mucin-domain containing-3,
T cell immunoreceptor with Ig and ITIM domains and cytotoxic T
lymphocyte-associated protein 4 (66–70),
resulting in loss of T cell effector functions, altered metabolism
and a parallel but ineffective transcriptional program (71). Allogenic T cells circumvent the
exhaustion conditioning of T cells, and thus are more effective
towards tumor cells (72,73).
Compared with autologous CAR-T cells, allogenic
approach allows for expanded manufacturing of ‘off-the-shelf’ CAR-T
cells for numerous recipients. Although the cellular level of
antitumor efficacy of allogenic CAR-T cells are more potent than
autologous CAR-T cells, graft-versus-host disease (GVHD) remains a
major impediment to the successful adoption of allogenic CAR-T
cells. Recently, Ghosh et al (74) demonstrated that alloreactive T cells
expressing CD28-costimulated CD19 CARs produced enhanced
stimulation, leading to overt mitigation of effector function and
clonal deletion, and significantly decreased occurrence of GVHD. A
recent case of reducing GVHD in CAR-T cell therapy was conducted on
an infant with CD19+ ALL for whom autologous T cells
could not be obtained, yet the endogenous TCR was deleted to
prevent GVHD (75).
There are also a number of limitations in the
present study, which will be improved upon in further
investigations. For example, the cell line used was HeLa, a
cervical cancer cell line, which is a considerable confounding
factor. The single cell line applied in the present study may not
be strong enough evidence to support the targetability of MSLN
CAR-T cells, therefore cells with higher MSLN expression may be
more suitable. In vivo cytotoxicity assay is required to
complement the in vitro assay of the present study. Apart
from the suppression on the growth of tumor, the tumor elimination
effect of MSLN CAR-T cells will be helpful in addressing the
stability of MSLN CAR-T cell therapy. The use of the 293T cell line
constitutes another limitation: Stepanenko and Dmitrenko recently
raised a concern for the use of this cell line (76), as it demonstrated no evident
tissue-specific gene expression signature, which may compromise the
resembling certain tissue-origin tumors. Furthermore, the compound
phenotype and unstable, heterogeneous karyotype made it difficult
to allow consistent and rigorous comparison between different
experimental groups.
The lack of blank control made it difficult to
examine the specificity of MSLN CAR-T cells. Case-control
comparison was performed for elucidating cytotoxicity and tumor
suppressing effect of MSLN targeted CAR-T cells via using
un-engineered T cells as control, and MSLN-abundant HeLa cells as
target. Results suggested that MSLN CAR-T cells have more potent
cytotoxicity than T cells, yet this advantageous effect was due to
the cumulative influence of MSLN targeting T cells and the
targetability of MSLN antigen. This finding is primarily sufficient
to support the conclusion that MSLN CAR-T cells are superior than T
cells in killing MSLN expressing tumors. However, the specificity
was not addressed clearly for a lack of blank control such as
non-MSLN expressing cells or non-tumor epithelial cells. This is a
major limitation of the present study. CAR-T cells were initially
designed to improve the specificity of T cells, which has been
demonstrated to be successful (77,78),
although specificity remains problematic in a CAR-T therapy study
(79). Improving specificity is a
major issue in CAR-T cell engineering, which can be addressed by
improving the targetability of chimeric antigen receptors, or
adding other tumor-specific antigen receptors. Unlike clinical
trials, the present study was limited to and focused on exploring
the therapeutic potential of MSLN CAR-T cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by Western Medicine
Guidance Project of Shanghai Science and Technology Commission
(grant no. 11411961300).
Availability of data and materials
All data generated or analyzed during the current
study are included in this published article.
Authors' contributions
XF performed the major experiments. LY analyzed the
data and was a major contributor in writing the manuscript. YL
performed the statistical analysis and provided important
suggestions in manuscript writing. LL designed the experiment and
provided advice during manuscript revision. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the Shanghai Chest Hospital (Shanghai, China) and
all patients provided written informed consent.
Patient consent for publication
All patients provided written informed consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kirkwood JM, Butterfield LH, Tarhini AA,
Zarour H, Kalinski P and Ferrone S: Immunotherapy of cancer in
2012. CA Cancer J Clin. 62:309–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goldstraw P, Chansky K, Crowley J,
Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P,
Mitchell A and Bolejack V: International Association for the Study
of Lung Cancer Staging and Prognostic Factors Committee, Advisory
Boards, and Participating Institutions, et al The IASLC lung
cancer staging project: Proposals for revision of the TNM stage
groupings in the forthcoming (eighth) edition of the TNM
Classification for lung cancer. J Thorac Oncol. 11:39–51. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ricciardi GR, Russo A, Franchina T,
Ferraro G, Zanghì M, Picone A, Scimone A and Adamo V: NSCLC and
HER2: Between lights and shadows. J Thorac Oncol. 9:1750–1762.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnson DB, Peng C and Sosman JA:
Nivolumab in melanoma: Latest evidence and clinical potential. Ther
Adv Med Oncol. 7:97–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kumarakulasinghe NB, van Zanwijk N and Soo
RA: Molecular targeted therapy in the treatment of advanced stage
non-small cell lung cancer (NSCLC). Respirology. 20:370–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weill H, Hughes JM and Churg AM: Changing
trends in US mesothelioma incidence. Occup Environ Med. 61:438–441.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Robinson BW and Lake RA: Advances in
malignant mesothelioma. N Engl J Med. 353:1591–1603. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiller JH, Harrington D, Belani C,
Langer CP, Sandler A, Krook J, Zhu J and Johnson DH; Eastern
Cooperative Oncology Group, : Comparison of four chemotherapy
regimens for advanced non-small-cell lung cancer. N Engl J Med.
346:92–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S,
Manegold C, et al: Phase III study of pemetrexed in combination
with cisplatin versus cisplatin alone in patients with malignant
pleural mesothelioma. J Clin Oncol. 21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosenberg SA, Yang JC and Restifo NP:
Cancer immunotherapy: Moving beyond current vaccines. Nat Med.
10:909–915. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weber JS: At the bedside: Adoptive cell
therapy for melanoma-clinical development. J Leukoc Biol.
95:875–882. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maude SL, Frey N, Shaw PA, Aplenc R,
Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et
al: Chimeric antigen receptor T cells for sustained remissions in
leukemia. N Engl J Med. 371:1507–1517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carpenito C, Milone MC, Hassan R, Simonet
JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF,
Albelda SM, et al: Control of large, established tumor xenografts
with genetically retargeted human T cells containing CD28 and CD137
domains. Proc Natl Acad Sci USA. 106:3360–3365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kreitman RJ, Hassan R, FitzGerald DJ and
Pastan I: Phase I Trial of continuous infusion anti-mesothelin
recombinant immunotoxin SS1P. Clin Cancer Res. 15:5274–5279. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adusumilli PS, Cherkassky L,
Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR and
Sadelain M: Regional delivery of mesothelin-targeted CAR T cell
therapy generates potent and long-lasting CD4-dependent tumor
immunity. Sci Transl Med. 6:261ra1512014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Glienke W, Esser R, Priesner C, Suerth JD,
Schambach A, Wels WS, Grez M, Kloess S, Arseniev L and Koehl U:
Advantages and applications of CAR-expressing natural killer cells.
Front Pharmacol. 6:212015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pastan I and Hassan R: Discovery of
mesothelin and exploiting it as a target for immunotherapy. Cancer
Res. 74:2907–2912. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hassan R, Bera T and Pastan I: Mesothelin:
A new target for immunotherapy. Clin Cancer Res. 10:3937–3942.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng YM, Spirason N, Iannello P, Jelley L,
Lau H and Barr IG: A simplified Sanger sequencing method for full
genome sequencing of multiple subtypes of human influenza A
viruses. J Clin Virol. 68:43–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hollyman D, Stefanski J, Przybylowski M,
Bartido S, Borquez-Ojeda O, Taylor C, Yeh R, Capacio V, Olszewska
M, Hosey J, et al: Manufacturing validation of biologically
functional T cells targeted to CD19 antigen for autologous adoptive
cell therapy. J Immunother. 32:169–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akerstrom B and Bjorck L: Protein L: An
immunoglobulin light chain-binding bacterial protein.
Characterization of binding and physicochemical properties. J Biol
Chem. 264:19740–19746. 1989.PubMed/NCBI
|
|
23
|
Taghian DG and Nickoloff JA: Subcloning
strategies and protocols. Methods Mol Biol. 58:221–235.
1996.PubMed/NCBI
|
|
24
|
Topalian SL, Taube JM, Anders RA and
Pardoll DM: Mechanism-driven biomarkers to guide immune checkpoint
blockade in cancer therapy. Nat Rev Cancer. 16:275–287. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dudley ME, Wunderlich JR, Yang JC, Sherry
RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE,
Mavroukakis SA, et al: Adoptive cell transfer therapy following
non-myeloablative but lymphodepleting chemotherapy for the
treatment of patients with refractory metastatic melanoma. J Clin
Oncol. 23:2346–2357. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Currie GA: Eighty years of immunotherapy:
A review of immunological methods used for the treatment of human
cancer. Br J Cancer. 26:141–153. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosenberg SA: IL-2: The first effective
immunotherapy for human cancer. J Immunol. 192:5451–5458. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rosenberg SA and Restifo NP: Adoptive cell
transfer as personalized immunotherapy for human cancer. Science.
348:62–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Ren B, Li H, Yu J, Cao S, Hao X
and Ren X: Enhanced antitumor effects of DC-activated CIKs to
chemotherapy treatment in a single cohort of advanced
non-small-cell lung cancer patients. Cancer Immunol Immunother.
62:65–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pan Y, Tao Q, Wang H, Xiong S, Zhang R,
Chen T, Tao L and Zhai Z: Dendritic cells decreased the concomitant
expanded tregs and tregs related IL-35 in cytokine-induced killer
cells and increased their cytotoxicity against leukemia cells. PLoS
One. 9:e935912014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miller JS, Soignier Y,
Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna
D, Le C, Defor TE, Burns LJ, et al: Successful adoptive transfer
and in vivo expansion of human haploidentical NK cells in patients
with cancer. Blood. 105:3051–3057. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fisher JP, Heuijerjans J, Yan M,
Gustafsson K and Anderson J: γδ T cells for cancer immunotherapy: A
systematic review of clinical trials. OncoImmunology. 3:e275722014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smyth MJ, Godfrey DI and Trapani JA: A
fresh look at tumor immunosurveillance and immunotherapy. Nat
Immunol. 2:293–299. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ho WY, Blattman JN, Dossett ML, Yee C and
Greenberg PD: Adoptive immunotherapy: Engineering T cell responses
as biologic weapons for tumor mass destruction. Cancer Cell.
3:431–437. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sadelain M, Riviere I and Brentjens R:
Targeting tumours with genetically enhanced T lymphocytes. Nat Rev
Cancer. 3:35–45. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
van der Stegen SJ, Hamieh M and Sadelain
M: The pharmacology of second-generation chimeric antigen
receptors. Nat Rev Drug Discov. 14:499–509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Savoldo B, Ramos CA, Liu E, Mims MP,
Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, et al:
CD28 costimulation improves expansion and persistence of chimeric
antigen receptor-modified T cells in lymphoma patients. J Clin
Invest. 121:1822–1826. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garçon F, Patton DT, Emery JL, Hirsch E,
Rottapel R, Sasaki T and Okkenhaug K: CD28 provides T-cell
costimulation and enhances PI3K activity at the immune synapse
independently of its capacity to interact with the p85/p110
heterodimer. Blood. 111:1464–1471. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fesnak AD, June CH and Levine BL:
Engineered T cells: The promise and challenges of cancer
immunotherapy. Nat Rev Cancer. 16:566–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hacein-Bey-Abina S, Garrigue A, Wang GP,
Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse
E, Beldjord K, et al: Insertional oncogenesis in 4 patients after
retrovirus-mediated gene therapy of SCID-X1. J Clin Invest.
118:3132–3142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Durand S and Cimarelli A: The inside out
of lentiviral vectors. Viruses. 3:132–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Montini E, Cesana D, Schmidt M, Sanvito F,
Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A,
Di Serio C, et al: Hematopoietic stem cell gene transfer in a
tumor-prone mouse model uncovers low genotoxicity of lentiviral
vector integration. Nat Biotechnol. 24:687–696. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Srivastava S and Riddell SR: Engineering
CAR-T cells: Design concepts. Trends Immunol. 36:494–502. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cooper LJ, Al-Kadhimi Z, DiGiusto D, Kalos
M, Colcher D, Raubitschek A, Forman SJ and Jensen MC: Development
and application of CD19-specific T cells for adoptive immunotherapy
of B cell malignancies. Blood Cells Mol Dis. 33:83–89. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kochenderfer JN, Dudley ME, Kassim SH,
Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ,
Hughes MS, Sherry RM, et al: Chemotherapy-refractory diffuse large
B-cell lymphoma and indolent B-cell malignancies can be effectively
treated with autologous T cells expressing an anti-CD19 chimeric
antigen receptor. J Clin Oncol. 33:540–549. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Schuster SJ, Svoboda J, Nasta SD, Porter
DL, Chong EA, Landsburg DJ, Mato AR, Lacey SF, Melenhorst JJ, Chew
A, et al: Sustained remissions following chimeric antigen receptor
modified T cells directed against CD19 (CTL019) in patients with
relapsed or refractory CD19+ lymphomas. Blood.
126:1832015.
|
|
47
|
Garfall AL, Maus MV, Hwang WT, Lacey SF,
Mahnke YD, Melenhorst JJ, Zheng Z, Vogl DT, Cohen AD, Weiss BM, et
al: Chimeric antigen receptor T cells against CD19 for multiple
myeloma. N Engl J Med. 373:1040–1047. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hege KM, Bergsland EK, Fisher GA,
Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH
and Sherwin SA: Safety, tumor trafficking and immunogenicity of
chimeric antigen receptor (CAR)-T cells specific for TAG-72 in
colorectal cancer. J Immunother Cancer. 5:222017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kershaw MH, Westwood JA, Parker LL, Wang
G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S,
Rogers-Freezer L, et al: A phase I study on adoptive immunotherapy
using gene-modified T cells for ovarian cancer. Clin Cancer Res.
12:6106–6115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Park JR, Digiusto DL, Slovak M, Wright C,
Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg
JR and Jensen MC: Adoptive transfer of chimeric antigen receptor
re-directed cytolytic T lymphocyte clones in patients with
neuroblastoma. Mol Ther. 15:825–833. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Haso W, Lee DW, Shah NN, Stetler-Stevenson
M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ,
Barrett DM, et al: Anti-CD22-chimeric antigen receptors targeting
B-cell precursor acute lymphoblastic leukemia. Blood.
121:1165–1174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Arabi F, Torabi-Rahvar M, Shariati A,
Ahmadbeigi N and Naderi M: Antigenic targets of CAR T Cell Therapy.
A retrospective view on clinical trials. Exp Cell Res. 369:1–10.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mami-Chouaib F, Echchakir H, Dorothee G,
Vergnon I and Chouaib S: Antitumor cytotoxic T-lymphocyte response
in human lung carcinoma: Identification of a tumor-associated
antigen. Immunol Rev. 188:114–121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rump A, Morikawa Y, Tanaka M, Minami S,
Umesaki N, Takeuchi M and Miyajima A: Binding of ovarian cancer
antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol
Chem. 279:9190–9198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li M, Bharadwaj U, Zhang R, Zhang S, Mu H,
Fisher WE, Brunicardi FC, Chen C and Yao Q: Mesothelin is a
malignant factor and therapeutic vaccine target for pancreatic
cancer. Mol Cancer Ther. 7:286–296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ho M, Bera TK, Willingham MC, Onda M,
Hassan R, Fitzgerald DJ and Pastan I: Mesothelin expression in
human lung cancer. Clin Cancer Res. 13:1571–1575. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huang CY, Cheng WF, Lee CN, Su YN, Chien
SC, Tzeng YL, Hsieh CY and Chen CA: Serum mesothelin in epithelial
ovarian carcinoma: A new screening marker and prognostic factor.
Anticancer Res. 26:4721–4728. 2006.PubMed/NCBI
|
|
58
|
Han SH, Joo M, Kim H and Chang S:
Mesothelin expression in gastric adenocarcinoma and its relation to
clinical outcomes. J Pathol Transl Med. 51:122–128. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Thomas A, Chen Y, Steinberg SM, Luo J,
Pack S, Raffeld M, Abdullaev Z, Alewine C, Rajan A, Giaccone G, et
al: High mesothelin expression in advanced lung adenocarcinoma is
associated with KRAS mutations and a poor prognosis. Oncotarget.
6:11694–11703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bharadwaj U, Marin-Muller C, Li M, Chen C
and Yao Q: Mesothelin confers pancreatic cancer cell resistance to
TNF-α-induced apoptosis through Akt/PI3K/NF-κB activation and
IL-6/Mcl-1 overexpression. Mol Cancer. 10:1062011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chang MC, Chen CA, Chen PJ, Chiang YC,
Chen YL, Mao TL, Lin HW, Lin Chiang WH and Cheng WF: Mesothelin
enhances invasion of ovarian cancer by inducing MMP-7 through
MAPK/ERK and JNK pathways. Biochem J. 442:293–302. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chang MC, Chen CA, Hsieh CY, Lee CN, Su
YN, Hu YH and Cheng WF: Mesothelin inhibits paclitaxel-induced
apoptosis through the PI3K pathway. Biochem J. 424:449–458. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cristaudo A, Foddis R, Vivaldi A,
Guglielmi G, Dipalma N, Filiberti R, Neri M, Ceppi M, Paganuzzi M,
Ivaldi GP, et al: Clinical significance of serum mesothelin in
patients with mesothelioma and lung cancer. Clin Cancer Res.
13:5076–5081. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Beatty GL, Haas AR, Maus MV, Torigian DA,
Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al:
Mesothelin-specific chimeric antigen receptor mRNA-engineered T
cells induce anti-tumor activity in solid malignancies. Cancer
Immunol Res. 2:112–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pauken KE and Wherry EJ: Overcoming T cell
exhaustion in infection and cancer. Trends Immunol. 36:265–276.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wherry EJ, Ha SJ, Kaech SM, Haining WN,
Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL and Ahmed
R: Molecular signature of CD8+ T cell exhaustion during
chronic viral infection. Immunity. 27:670–684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Blackburn SD, Shin H, Haining WN, Zou T,
Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA and Wherry
EJ: Coregulation of CD8+ T cell exhaustion by multiple
inhibitory receptors during chronic viral infection. Nat Immunol.
10:29–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Paley MA, Kroy DC, Odorizzi PM, Johnnidis
JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner
SL and Wherry EJ: Progenitor and terminal subsets of
CD8+ T cells cooperate to contain chronic viral
infection. Science. 338:1220–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Johnston RJ, Comps-Agrar L, Hackney J, Yu
X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al:
The immunoreceptor TIGIT regulates antitumor and antiviral
CD8+ T cell effector function. Cancer Cell. 26:923–937.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Callahan MK and Wolchok JD: At the
Bedside: CTLA-4- and PD-1-blocking antibodies in cancer
immunotherapy. J Leukoc Biol. 94:41–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wherry EJ: T cell exhaustion. Nat Immunol.
12:492–499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Brown CE, Badie B, Barish ME, Weng L,
Ostberg JR, Chang WC, Naranjo A, Starr R, Wagner J, Wright C, et
al: Bioactivity and safety of IL13R 2-redirected chimeric antigen
receptor CD8+ T cells in patients with recurrent
glioblastoma. Clin Cancer Res. 21:4062–4072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gomez GG: Adoptive T cell therapies for
glioblastoma. J Cancer Clin Trials. 1:1–5. 2015.
|
|
74
|
Ghosh A, Smith M, James SE, Davila ML,
Velardi E, Argyropoulos KV, Gunset G, Perna F, Kreines FM, Levy ER,
et al: Donor CD19 CAR T cells exert potent graft-versus-lymphoma
activity with diminished graft-versus-host activity. Nat Med.
23:242–249. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Qasim W, Amrolia P, Samarasinghe S,
Ghorashian S, Zhan H, Stafford S, et al: First clinical application
of talen engineered universal car19 T cells in B-ALL. Blood.
126:20462015.
|
|
76
|
Stepanenko AA and Dmitrenko VV: HEK293 in
cell biology and cancer research: Phenotype, karyotype,
tumorigenicity, and stress-induced genome-phenotype evolution.
Gene. 569:182–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jena B, Dotti G and Cooper LJ: Redirecting
T-cell specificity by introducing a tumor-specific chimeric antigen
receptor. Blood. 116:1035–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Singh H, Manuri PR, Olivares S, Dara N,
Dawson MJ, Huls H, Hackett PB, Kohn DB, Shpall EJ, Champlin RE and
Cooper LJ: Redirecting specificity of T-cell populations for CD19
using the sleeping beauty system. Cancer Res. 68:2961–2971. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY,
Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, et al: Versatile
strategy for controlling the specificity and activity of engineered
T cells. Proc Natl Acad Sci USA. 113:e450–e448. 2016. View Article : Google Scholar : PubMed/NCBI
|