Introduction
Pancreatic cancer is the fifth leading cause of
cancer-associated mortalities in the world (1). The five-year survival rate of affected
patients is poor (<10%) and in the majority of cases, pancreatic
cancer is considered unresectable with a median survival of 6–11
months (2,3). The poor prognosis is mainly based on
difficulties in diagnosing pancreatic cancer at early stages, while
no curative treatments are currently available for patients
diagnosed at the late stage (4,5).
Surgical treatment and anti-neoplastic therapies combined with
neoadjuvant approaches are the most common treatments for patients
with resectable non-metastatic pancreatic cancer (6,7). Total
resection combined with strategies encompassing primary
chemotherapy and radiation is performed in 15–20% of patients with
borderline resectable, non-metastatic disease, which appears to be
promising (8). A previous study
reviewed the treatments for pancreatic cancer, including
chemotherapy, radiotherapy and/or neoadjuvant therapy, by
performing a meta-analysis of the available data and summarized the
toxicities and clinical benefits of these treatments (9). However, the survival rate of patients
with pancreatic cancer remains poor due to rapid metastasis and
malignant features of pancreatic carcinoma cells. Therefore, while
considerable research effort has been made to unveil the
pathogenesis, the molecular mechanisms underlying the metastasis
and systemic progression of pancreatic carcinoma have remain to be
fully elucidated (10).
Maternally expressed gene-3 (MEG-3) is homologous
with the mouse maternally imprinted gene Gtl2, which was identified
as an imprinted gene first mapped on mouse distal chromosome 12 and
human chromosome 14q (11). MEG-3
encodes a long non-coding RNA (lncRNA) that has been evidenced to
be a tumor suppressor whose downregulation is associated with the
progression of various cancer types (12). Previous studies have indicated that
MEG-3 functions through interacting with cyclic adenosine
monophosphate, murine double minute 2/p53 and growth
differentiation factor 15 to regulate tumor cell proliferation and
metastasis (13,14). Previous studies have also
demonstrated that MEG-3 is downregulated in most of human cancer
cell types, including melanoma, non-small cell lung cancer,
meningioma, melanoma, colon cancer, leukemia, nasopharyngeal
carcinoma and pancreatic cancer, while it is highly expressed in
normal human tissue (13,15,16). In
addition, a crosstalk interaction between MEG-3 and tumor
suppressor p53 signaling pathway has been reported in the
regulation of the growth of testicular germ cell tumors (17). However, the molecular mechanisms of
MEG-3 in pancreatic cancer have remained to be fully
elucidated.
Small non-coding RNA (ncRNAs) have an essential role
in tumor progression (18,19). They are a class of functional RNA
molecules in the regulation of growth, aggressiveness, apoptosis
and prognosis as well as immunoregulatory function in patients with
cancer (20–22). These functional RNA molecules include
small nucleolar RNAs, ribosomal RNAs, microRNAs (miRNAs/miRs),
transfer RNAs, Piwi-interacting RNAs and lncRNAs (23,24).
Aberration of ncRNA levels is thought to have a critical role in
cancer-associated cellular physiological processes (25). Furthermore, miRNAs exert significant
regulatory functions in carcinogenesis, growth and invasion via
regulating their upstream and downstream molecules (26).
The present study investigated the association
between the methylation of the gene encoding miR-148a and its
expression in pancreatic cancer cells. The inhibitory effects and
molecular mechanisms of miR-148a and MEG-3 in human pancreatic
cancer cell lines were examined. Activation of miR-148a methylation
was found to inhibit tumor formation, proliferation and invasion of
pancreatic cancer cells in vitro as well as in vivo.
miR-148a transduction is likely to induce MEG-3 via regulation of
the Wnt/β-catenin signaling pathway. The results clarified the
association between miR-148a and MEG-3 in pancreatic cancer, which
may contribute to the development of novel treatments for
pancreatic cancer.
Materials and methods
Ethics statement
The present study was performed in strict accordance
with the recommendations in the Guide for the Care and Use of
Laboratory Animals of the National Institutes of Health. During
experimental procedures and euthanasia, all efforts were made to
minimize suffering. This study was approved by the Institutional
Review Board and Ethics Committee of Wenzhou Medical University
(Wenzhou, China; approval no. 0812-10421C4).
Cells and reagents
PANC-1 and Aspc-1 pancreatic cancer cell lines were
purchased from the American Type Tissue Collection (Manassas, VA,
USA) and were cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with
10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific,
Inc.). Human normal pancreatic cells (human primary pancreatic
microvascular endothelial cells; Hengfei Bioscience, Shanghai,
China; http://boyaobio.biomart.cn) were
cultured in minimum essential medium (Thermo Fisher Scientific,
Inc.) with 10% FBS. All cells were maintained at 37°C in a
humidified atmosphere containing 5% CO2. Pancreatic
tumor and adjacent normal tissues from a patient (female, 43 years,
recruited in March 2014) were obtained from the First Affiliated
Hospital of Wenzhou Medical University (Wenzhou, China). Patient
consent was obtained.
Transduction of lentivirus in
pancreatic cancer cell lines
A mixture of pWPXL-miR-148a (5 µg) or pWPXL-control
(5 µg) plasmids (Invitrogen; Thermo Fisher Scientific, Inc.) were
transfected into PANC-1 or Aspc-1 cells using Lipofectamine 2000
reagent (Thermo Fisher Scientific, Inc.) to generate cell lines
stably transfected with miR-148a expression or control vector.
PANC-1 or Aspc-1 cells were transfected with the recombinant
lentivirus-transducing units plus 6 mg/ml Polybrene (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) according to a previous protocol
(27). PANC-1 and Aspc-1 cells were
further transfected with pWPXL-miR-148a (5 µg) in combination with
G-A-T-A-base (GATA)-1, GATA2, catalytic domain protein-C region
(CDP-CR) or upstream stimulatory factor 1 (USF1; all 100 ng; all
Promega Corporation, Madison, WI, USA) as an internal control. At
72 h following transfection, a dual luciferase assay kit
(0000060417; Promega Corporation) was used to measure luciferase
activities according to manufacturer's instructions. Results were
normalized to Renilla.
MTT cytotoxicity assay
PANC-1 and Aspc-1 cells were transfected with
pWPXL-miR-148a or pWPXL-control in 96-well plates for 96 h in
triplicate for each condition. Subsequently, 20 µl MTT (5 mg/ml;
Sigma-Aldrich; Merck KGaA) in PBS was added to each well and the
cells were further incubated for 4 h. The entire medium was removed
and 100 µl dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) was added
to the wells to solubilize the crystals. The optical density was
measured using an ELISA reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) at a wavelength of 450 nm.
Colony formation assay
For each group, 4 ml DMEM containing
5×104 cells/ml was added to a 6-well plate. Following
culturing at 37°C at 5% CO2 for 14 days, the supernatant
was discarded and cells were washed with PBS (3X). Cells were then
fixed with 4% paraformaldehyde for 15 min at room temperature.
Colonies were counted under an inverted microscope (magnification,
×40; Nikon Corporation, Tokyo, Japan).
Tumor cell migration and invasion
assays
PANC-1 and Aspc-1 cells transfected with
pWPXL-miR-148a or pWPXL-control for 48 h. For the invasion assay,
PANC-1 and Aspc-1 (5×104 in 200 µl serum-free DMEM) were
added to the upper chambers of BD BioCoat Matrigel Invasion
Chambers (BD Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer's instructions. For the migration assay, a control
insert (BD Biosciences) was used instead of the Matrigel Invasion
Chamber. Cells were incubated for 48 h at 37°C. The tumor cell
invasion and migration were determined in at least three randomly
selected microscopic fields of the stained lower side of each
membrane.
Flow cytometric analysis
An Annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI) analysis system apoptosis detection kit (BD
Biosciences) was used to evaluate apoptosis of PANC-1 and Aspc-1
cells. Cells were incubated with pWPXL-miR-148a or pWPXL-control
for 12 h at 37°C and then collected. A total of 1×106
cells were suspended in binding buffer containing Annexin V-FITC
and PI according to the manufacturer's instructions. Fluorescence
was detected with a BD FACScan flow cytometer and analyzed using BD
FACSChorus™ Software 1.2 (BD Biosciences).
Immunohistochemical staining
Immunohistochemical staining was performed via the
avidin-biotin-peroxidase technique. Paraffin-embedded tumor tissue
sections were prepared. Sections were deparaffinized and rehydrated
with xylene, alcohol and tap water for 1 h at 37°C in each step.
Tissue sections were then incubated with hydrogen peroxide (3%) for
10–15 min and subsequently blocked with a regular blocking solution
for 10–15 min at 37°C. Finally, the sections were incubated with
anti-miR-148a (cat. no. 10302) or anti-MEG-3 (cat. no. 5122; all
1:500; Cell Signaling Technology, Inc., Danvers, MA, USA) antibody
at 4°C for 12 h. All sections were washed three times and incubated
with horseradish peroxidase-conjugated goat anti-mouse secondary
antibody (1:2,000; ab6785; Abcam, Cambridge, UK) for 1 h at 37°C.
An enhanced chemiluminescence substrate ECL Select™ (Bio-Rad
Laboratories, Inc.) was used to detect protein expression. The
results were observed using a fluorescent microscope
(magnification, ×40) and analyzed using Quantity One software
version 4.62 (Bio-Rad Laboratories, Inc.).
Methylation regions of miR-148a in
pancreatic cancer
The methylation rate of miRNA-148a in PANC-1 and
Aspc-1 cells was determined by quantitative methylation-specific
polymerase chain reaction (qMS-PCR). DNA was obtained from PANC-1,
Aspc-1 and human normal pancreatic cells using the PicoPure™ DNA
Extraction kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). A total of 10 ng of DNA was amplified using the AmpFLSTR™
SGM Plus™ PCR Amplification kit (Thermo Fisher Scientific, Inc.)
with the following primers: miRNA-148a, forward,
5′-TGGGTATTTGTTTTTGTTGATTG-3′ and reverse,
5′-ACTACACTTAAACCCCCTCTAACC-3′. Reactions were performed using the
following conditions: 90°C for 2 min, followed by 40 cycles of 95°C
for 20 sec, 60°C for 30 sec and 72°C for 15 sec and a final
extension of 10 min at 72°C. PCR products were diluted with water
(1:500) and 1 µl diluted product was subjected to qMS-PCR using
LightCycler® 480 SYBR-Green I Master mix (Thermo Fisher
Scientific, Inc.). Reverse transcription-qPCR was performed using
the High Capacity cDNA Reverse Transcription kit (Invitrogen;
Thermo Fisher Scientific, Inc.) according to manufacturer's
instructions. Primers were designed as follows:
Unmethylated-specific miRNA-148a forward,
5′-TATGATTTGTTTTATTATTGGTT-3′ and reverse,
5′-AACACTAACAACATCAACAACC-3′; methylated-specific miRNA-148a
forward, 5′-TGATTCGTTTTATTATCGGTC-3′ and reverse,
5′-AACACTAACGACATCGACG-3′; and β-actin forward,
5′-ACGGTCAGGTCATCACTATCG-3′ and reverse,
5′-GGCATAGAGGTCTTTACGGATG-3′. The reaction was conducted at 95°C
for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 60
sec. The methylation level was using the 2−ΔΔCq method
(28). The results are expressed as
the fold of the β-actin.
5-aza-2′-deoxycytidine treatment of
PANC-1 and Aspc-1
For treatment with DNA methylation inhibitor
5-aza-2′-deoxycytidine (5-aza-dC; 10 pM in PBS; Sigma-Aldrich;
Merck KGaA), PANC-1 and Aspc-1 cells (1×105) were seeded
into 6-well plates on day 0 and exposed to 5-aza-dC at a final
concentration of 5 µmol/l for 2 days at 37°C. Control samples were
incubated with an equal volume of PBS. Cells were harvested for
RT-qPCR to assess miRNA-148a expression as described above.
Cell lines stably expressing MEG3
To obtain a cell line stably expressing MEG3, the
pcDNA3.0-MEG3 (Thermo Fisher Scientific, Inc.) expression vector
was transfected into PANC-1 and Aspc-1 cells as described above.
Cells were used for further analysis after 48 h transfection. Cells
were screened with G418 (1,000 µg/ml, Sigma-Aldrich; Merck KGaA) as
described previously (29). RT-qPCR
was performed to examine the expression of MEG3 at the RNA level as
described above. The cell line with stably expressing empty vector
pcDNA3.0 was used as control.
Luciferase assays
3′-UTR sequences of miR-148a target genes containing
upstream and downstream 100 bp flanking sequences of the putative
miR148a target sites were amplified as described above using the
AmpFLSTR™ SGM Plus™ PCR Amplification kit (Thermo Fisher
Scientific, Inc.) from PANC-1 and Aspc-1 cells genomic DNA by PCR
and cloned into the pMIR-Report-Vector (Ambion, USA). Primers used
for PCR were as follow: Forward, 5′-TGGGTATTTGTTTTTGTTGATTG-3′ and
reverse, 5′-ACTACACTTAAACCCCCTCTAACC-3′. 3′-UTR luciferase reporter
assays were performed by calcium phosphate transient transfection
(100 pM) of PANC-1 and Aspc-1 cells (1×106) with 20 ng
of pMIR-reporter-3′-UTR and 200 ng of the vector control or
miR-148a expression clone and 10 ng pCMV-Renilla (internal
control). The Dual Luciferase assay (Promega Corporation, Madison,
WI, USA) was used to measure the luciferase activities after 72-h
transfection. All luciferase activities were normalized to
Renilla and the ratio is presented.
Confocal laser microscopy
PANC-1 and Aspc-1 cells grown on lysine-coated glass
coverslips were treated with pWPXL-miR-148a or pWPXL-control for 48
h. Subsequently, the cells were fixed with 4% paraformaldehyde,
followed by blocking in 1% bovine serum albumin and 0.1% Triton
X-100 in PBS for 60 min at 37°C. The pancreatic tumor cells were
then incubated with antibodies against MEG-3 (1:500 dilution; Cell
Signaling Technology, Inc., Danvers, MA, USA) for 2 h at 25°C in a
humidified atmosphere. Subsequently, the cells were incubated with
Alexa Fluor 488-conjugated anti-rabbit secondary antibody (1:400
dilution; cat. no. 4412; Cell Signaling Technology, Inc.) after
washing with PBS. Pancreatic tumor cell nuclei were stained with
DAPI (10 mg/ml) for 30 min at 25°C in a humidified atmosphere. The
PANC-1 and Aspc-1 cells were mounted in anti-fade mounting medium
and images were captured using a Zeiss Confocal Spectral microscope
(magnification, ×40; Carl Zeiss, Jena, Germany).
Western blot analysis
PANC-1 and Aspc-1 cells were treated with
pWPXL-miR-148a or pWPXL-control for 24 h at 37°C, homogenized in
lysate buffer containing protease-inhibitor and centrifuged at
6,000 × g at 4°C for 10 min. The supernatant of was used for
analysis of the total protein concentration using bicinchoninic
acid protein assay kit (Thermo Fisher Scientific, Inc.). For
detection, proteins (40 µg) were loaded and separated using 12%
SDS-PAGE gels, transferred to nitrocellulose membranes and
hybridized as previously described (30). Subsequent to blocking in 5% skimmed
milk, membranes were probed with primary antibodies MEG-3 (1:500;
5122 Cell Signaling Technology, Inc.), E-cadherin (1:1,000;
ab11512), Vimentin (1:1,000; ab92547), Snail2 (1:1,000; ab180714),
β-catenin (1:1,000; ab32572), C-myc (1:1,000; ab32072), Cyclin D1
(1:1,000; ab134175) and β-actin (1:1,000; ab8226) and incubated for
1 h at 37°C, followed by incubation with HRP-conjugated goat
anti-mouse secondary antibody (1:2,000; ab6785; all Abcam) for 24 h
at 4°C. The blots were visualized using a chemiluminescence kit
(Thermo Fisher Scientific, Inc.). Quantity of protein was analyzed
using Quantity One software version 4.62 (Bio-Rad Laboratories,
Inc.).
Pancreatic colonization assay of
PANC-1-miR-148a cells in nude mice
Six-week-old female BALB/c nude mice (n=40; weight,
20–25 g) were purchased from Beijing Vital River Laboratory Animal
Technology Co., Ltd (Beijing, China). All animals were reared under
specific pathogen-free conditions. Mice were maintained at a 12-h
light/dark cycle with free access to food and water. Two million
PANC-1-miR-148a or PANC-1-control cells were injected into the
right flank of female BALB/c nude mice (n=20 randomized
mice/group). On day 25, tumors were isolated from experimental
animals and used for further analysis. Tumor diameters were
recorded once every two days and the tumor volume was calculated by
using the following formula: 0.52× smallest diameter × 2× largest
diameter.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Data were analyzed using SPSS 1.0 software (SPSS,
Chicago, IL, USA). Comparisons of data between multiple groups were
analyzed by analysis of variance followed by Tukey's post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
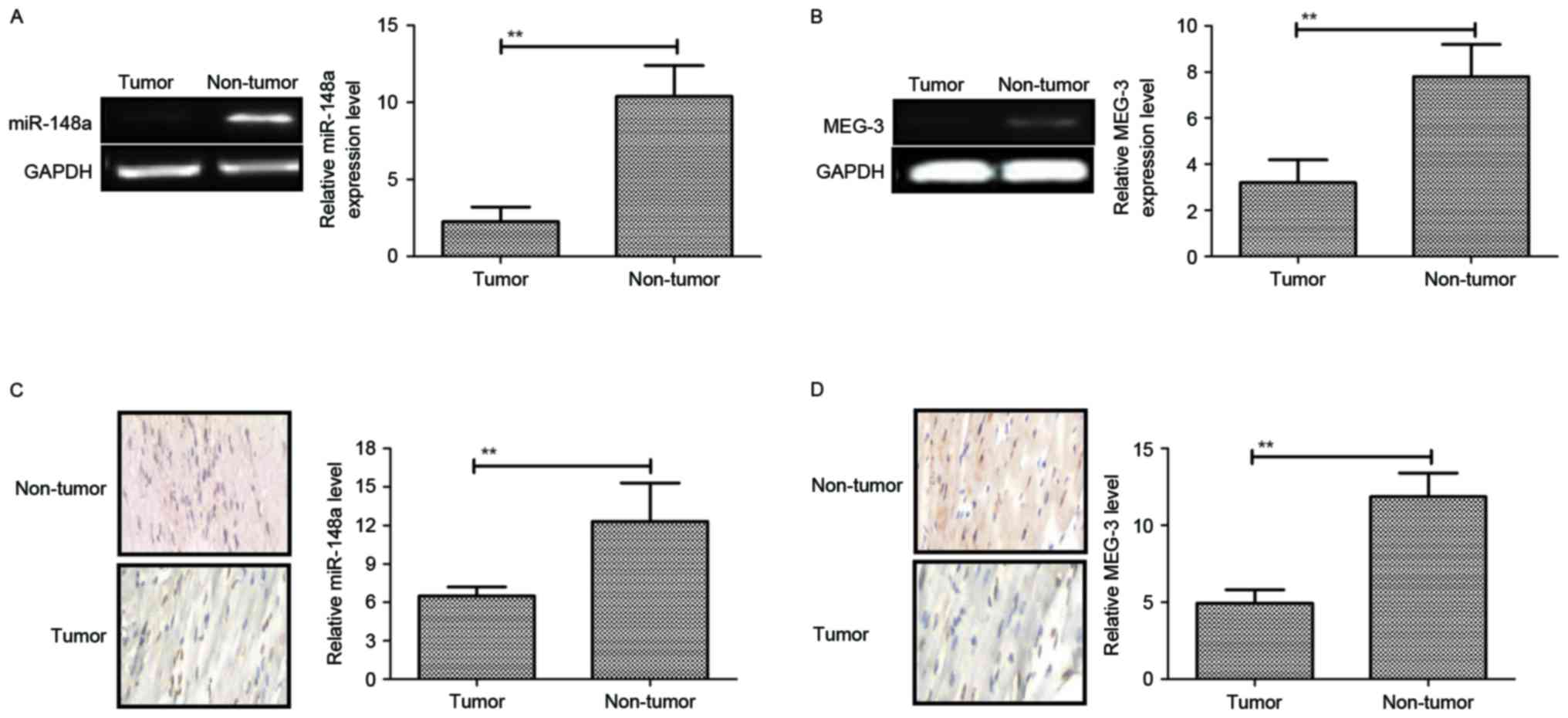
Downregulation of miR-148a and MEG-3
in pancreatic cancer
To explore the role of miR-148a and MEG-3 in
pancreatic cancer, tissue samples from patients and cell lines were
assessed by RT-qPCR and immunohistochemical analysis. Compared with
normal adjacent tissues and a normal pancreatic cell line, miR-148a
and MEG-3 were significantly decreased in pancreatic cancer tissues
and cell lines (Fig. 1A-D). Overall,
these results indicated that miR-148a and MEG-3 were downregulated
in pancreatic cancer cell lines and tumors.
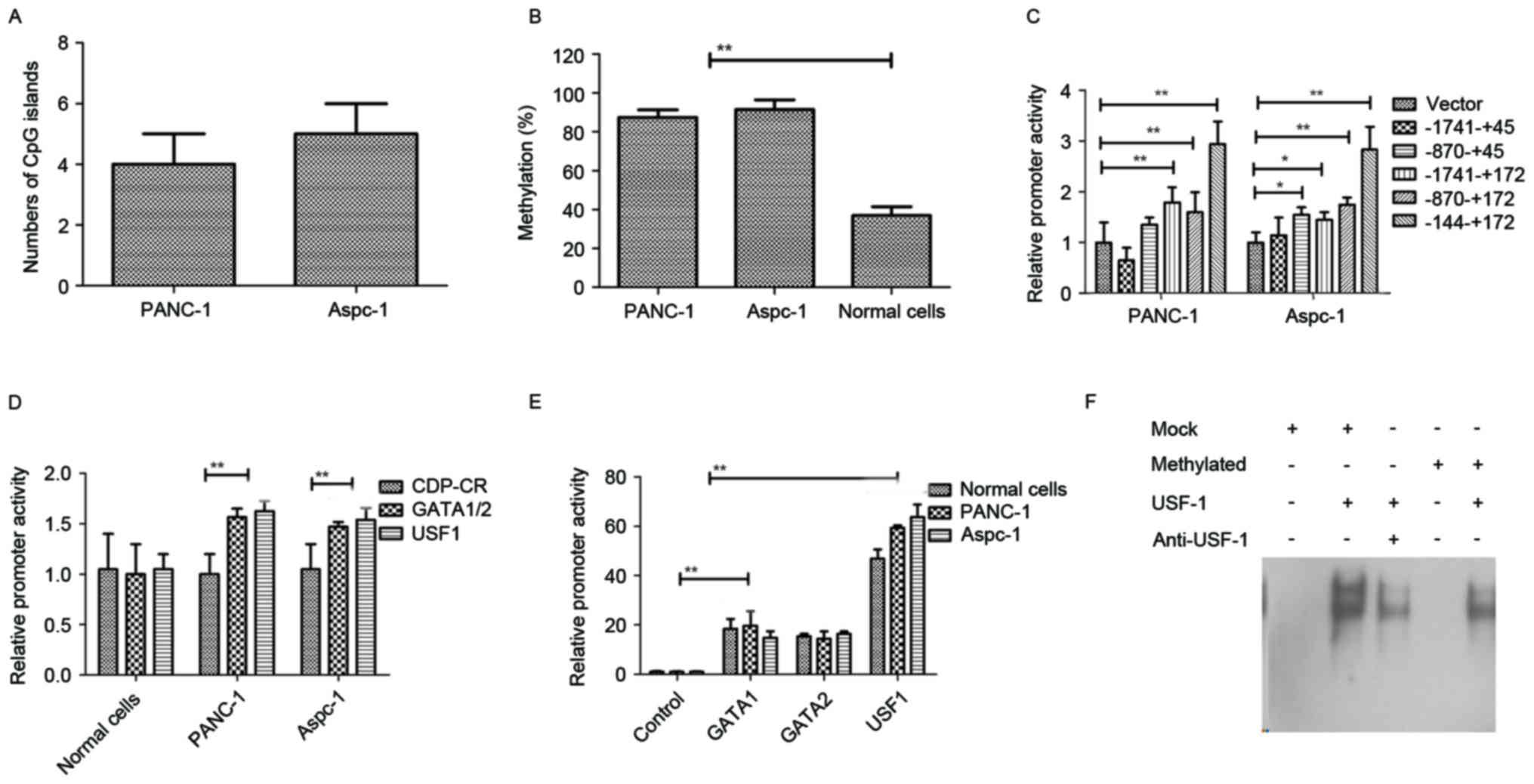
Identification of differential
methylation regions of miR-148a in pancreatic cancer
To investigate of differential methylation regions
of miR-148a in pancreatic cancer, an analysis of the 2,000-bp
upstream DNA sequence of pre-miR-148a and a qMS-PCR were performed
to confirm the extent of methylation and transcription factor
binding sites. As shown in Fig. 2A,
5 putative CpG islands in the promoter of the gene encoding
miR-148a were confirmed in PANC-1 and 4 putative CpG islands in
Aspc-1. As shown in Fig. 2B, these
CpG islands were differentially methylated in
5-aza-2′-deoxycytidine-treated PANC-1 and Aspc-1 cells. As
displayed in Fig. 2C, the CpG sites
within −144 to +172 bp of the promoter of the gene encoding
miR-148a in PANC-1 and Aspc-1 cells were differentially methylated
compared to those in normal pancreatic cells. Luciferase assays
revealed that in pancreatic cancer cell lines, the promoter
activity of a luciferase reporter vector containing clones of
catalytic domain protein-C region (CDP-CR) differential methylation
region mutations was the same as that in normal pancreatic cells,
but those of GATA 1/2 and USF1 mutant reporters were 40–60% higher
than those in normal pancreatic cells (Fig. 2D). In addition, the promoter activity
of a luciferase reporter vector containing differential miR-148a
methylation regions was higher after co-transfection of GATA1,
GATA2 or USF1 compared with control (Fig. 2E). The results in Fig. 2F indicated that the binding affinity
between USF1 and the methylated probe decreased, indicating that
methylcytosines affected the transcription activator USF1. Overall,
these results indicated that the methylation status in pancreatic
tumor cells is high compared to that in normal pancreatic cells and
that GATA1, GATA2 and USF1 are involved in the activation of
differential methylation regions, suggesting that the decreased
ability of USF1 to bind to the methylated DNA may be associated
with the reduction of miR-148a transcription in pancreatic tumor
cells.
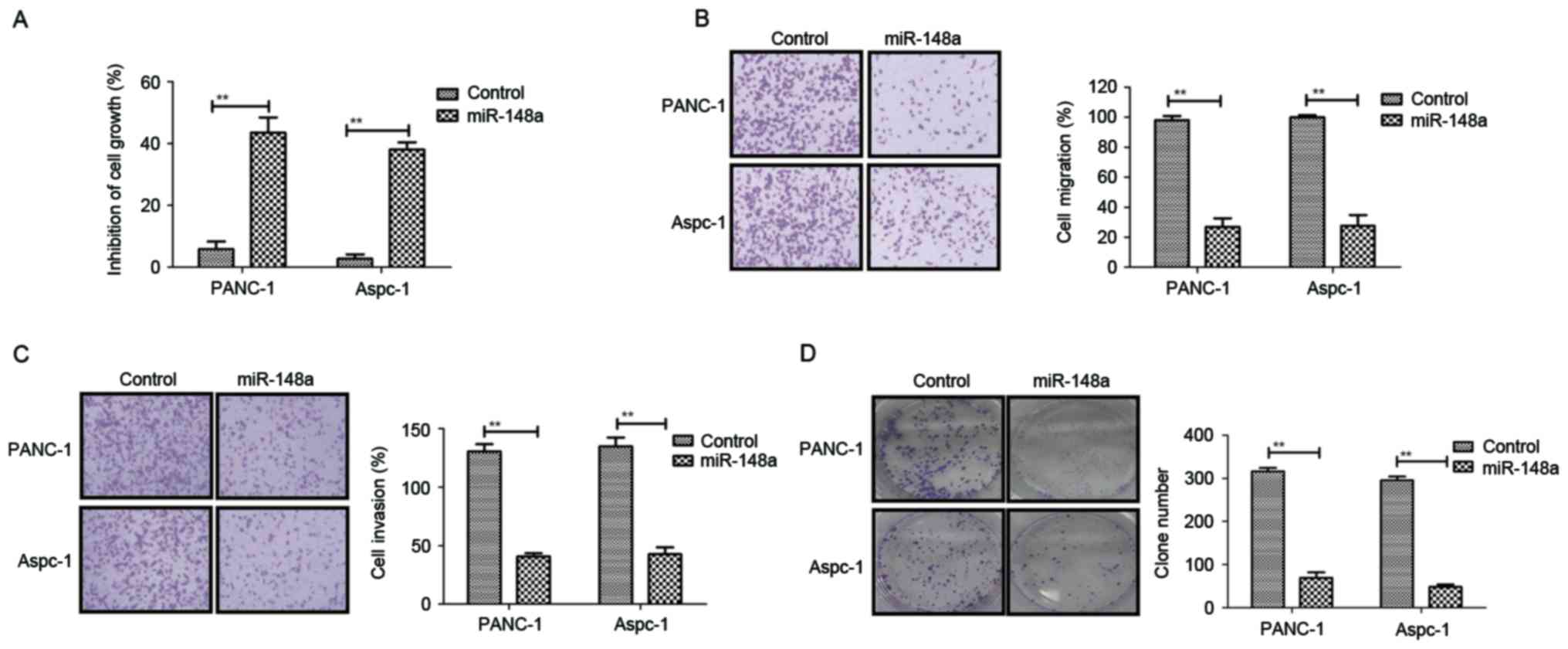
Restoration of miR-148a suppresses
cell growth and metastatic capacity of pancreatic cancer
After confirmation of the downregulation of miR-148a
in pancreatic cancer, the present study further analyzed the
association between miR-148a and pancreatic cancer metastasis.
Subsequently, PANC-1 and Aspc-1 cell lines stably transfected with
miR-148a expression vector were established by lentivirus
infection. It was observed that miR-148a exerted obvious inhibitory
effects on the growth of PANC-1 and Aspc-1 cells (Fig. 3A). In addition, restoration of
miR-148a expression suppressed the migration and invasion of PANC-1
and Aspc-1 cells (Fig. 3B and C).
Furthermore, colony formation assays demonstrated that the number
and size of pancreatic metastasis nodules was markedly inhibited in
PANC-1-miR-148a and Aspc-1-miR-148a cells compared to those in the
miR-148a-vector control groups (Fig.
3D). Conversely, PANC-1 and Aspc-1 cells transfected with
miR-148a showed higher endogenous miR-148a expression, resulting in
increased pancreatic cancer cell apoptosis (Fig. 3E). In addition, miR-148a transfection
in PANC-1 and Aspc-1 cells promoted MEG-3 expression (Fig. 3F). Collectively, the results
suggested that restoration of miR-148a is a negative regulator
inhibiting growth and metastatic potential in pancreatic cancer
cells.
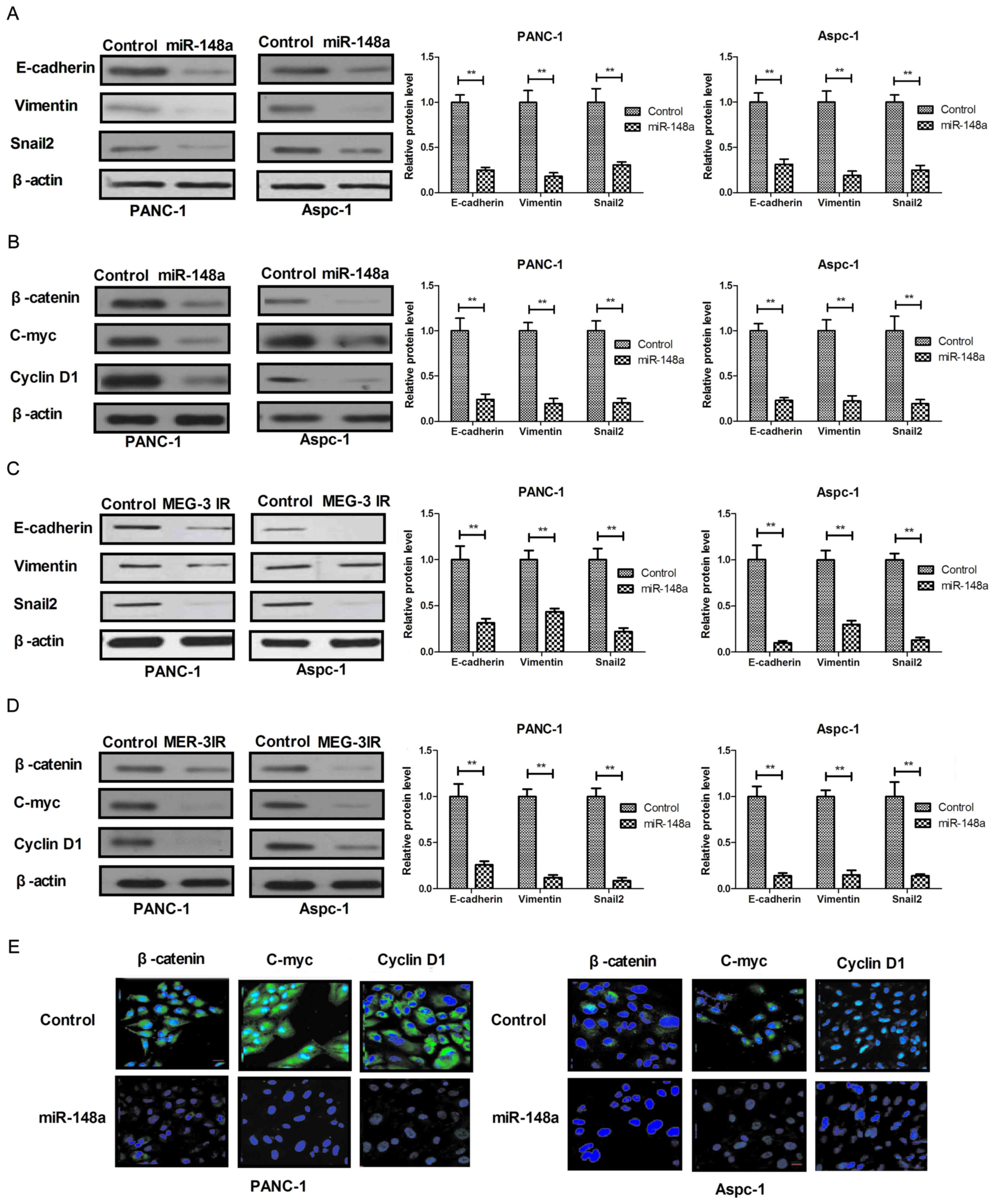
Restoration of miR-148a inhibits
mesenchymal-epithelial transition (MET) through Wnt/β-catenin
signaling pathway mediated via MEG-3 in pancreatic cancer
cells
Restoration of miR-148a led to inhibition of
pancreatic cancer cells. In order to investigate the underlying
molecular mechanisms and associated signal pathways, the present
study assessed the effect of miR-148a on MET through the
MEG-3-imediated Wnt/β-catenin signaling pathway in pancreatic
cancer cells. The results in Fig. 4A
demonstrated that restoration of miR-148a markedly suppressed the
expression of epithelial cell markers (E-cadherin, Vimentin and
Snail2) in PANC-1 and Aspc-1 cells. In addition, restoration of
miR-148a inhibited the expression of β-catenin, C-myc and Cyclin D1
in PANC-1 and Aspc-1 cells (Fig.
4B). A further experiment revealed that recombinant MEG-3
treatment also suppressed EMT and Wnt/β-catenin signaling pathways
in PANC-1 and Aspc-1 cells (Fig. 4C and
D). In addition, restoration of miR-148a decreased endogenous
β-catenin levels and suppressed the activity of nuclear
translocation (Fig. 4E). Taken
together, the results suggested that miR-148a regulates MEG-3
expression to activate the Wnt/β-catenin signaling pathway to drive
EMT in pancreatic cancer cells, which explains why the restoration
of miR-148a inhibits cell growth and metastatic capacity of
pancreatic cancer.
Restoration of miR-148a in pancreatic
cancer cells leads to reduced tumor growth and invasion
To confirm the therapeutic effects of miR-148a
restoration on cellular responses, the tumor growth in mice
inoculated with PANC-1-miR-148a or PANC-1-miR-mimic (control) was
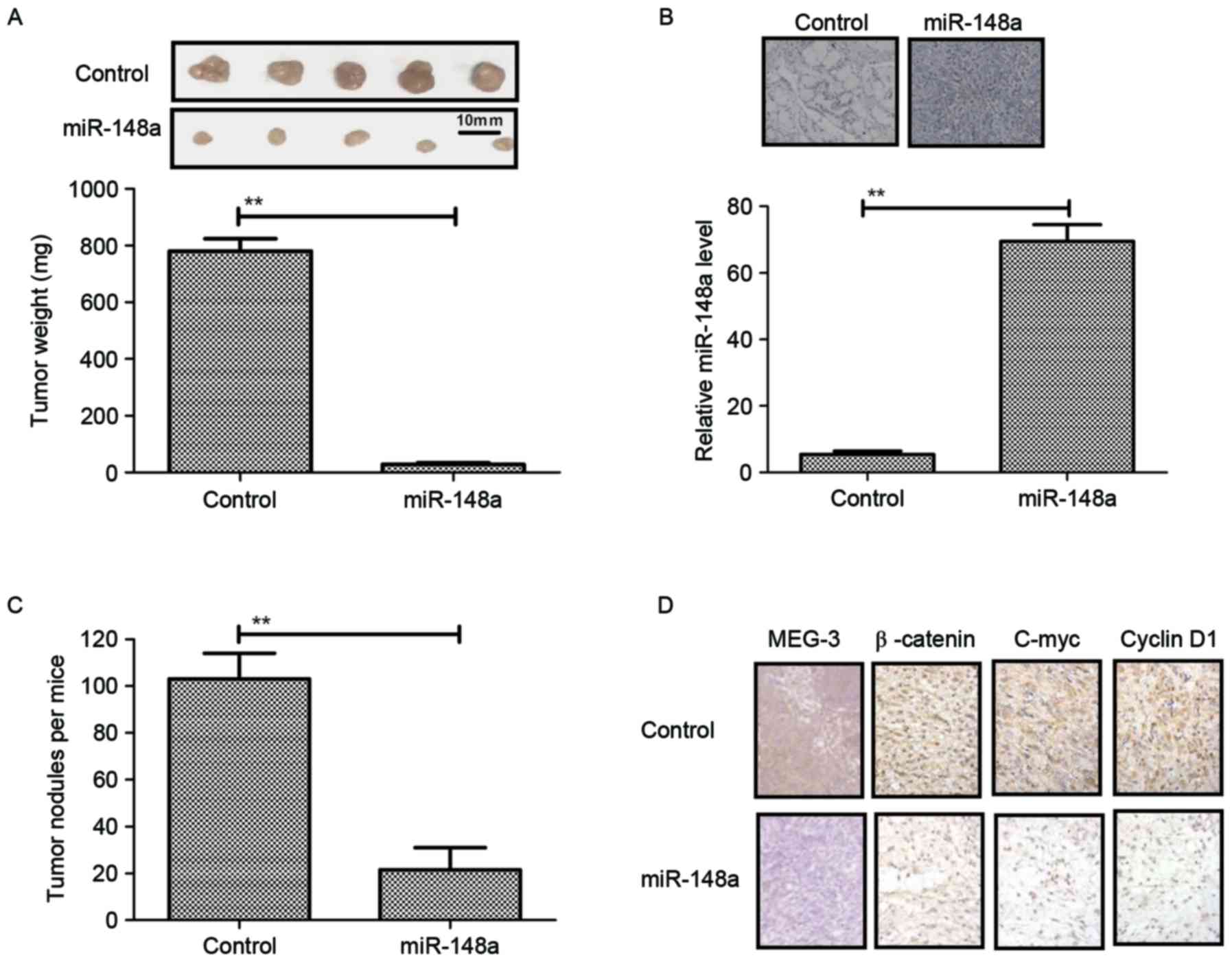
assessed. The results in Fig. 5A
showed that mice implanted with PANC-1-miR-148a hardly formed any
tumor lesions, while those inoculated with PANC-1-vector formed
obvious tumor tissue masses. In addition, histochemical analysis
and RT-qPCR confirmed the overexpression of miR-148a in the tumors
(Fig. 5B). To investigate the
effects of miR-148a expression on the metastatic potential of
PANC-1 cells, the total amount of pancreatic tumor nodules in each
animal was evaluated. It was observed that PANC-1-miR-148a
transfection decreased the amount of pancreatic tumor nodules in
the mice harboring PANC-1 cells (Fig.
5C). Furthermore, the expression levels of the mediator MEG-3
and Wnt/β-catenin signaling pathway proteins were assessed in the
pancreatic cancer tissues in vivo. As shown in Fig. 5D, it was found that MEG-3 expression
was upregulated, while β-catenin, C-myc and Cyclin D1 expression
levels were downregulated in tumors from mice implanted with
PANC-1-miR-148a compared to those in the vector group.
Collectively, these results indicated that miR-148a restoration in
PANC-1 cells not only impaired Wnt/β-catenin-induced cell growth
and invasion via MEG-3 in vitro but also in vivo.
Discussion
A large number of studies have indicated that miRNAs
constitute complex and efficient regulatory networks via
post-transcriptional regulation in cellular biochemistry (26,31).
Recently, post-transcriptional regulation of miRNAs in various
cancer cells has drawn the attention of scientists in the field of
cancer therapy (32,33). Aberrant miRNA expression in various
tumor cell types has been evidenced to have important roles in
carcinogenesis, tumorigenesis, tumor suppression and tumor
progression (34–36). The present study investigated the
function of miR-148a in pancreatic cancer. The results indicated
that miR-148a is frequently downregulated in human pancreatic
cancer cells and tissues and that its downregulation is
significantly associated with growth, invasion and metastasis
through regulating the MEG-3-induced Wnt/β-catenin signaling
pathway in pancreatic cancer. Further analysis revealed that
restoration of miR-148a suppressed pancreatic carcinoma cell
growth, colony formation, migration and invasion in vitro as
well as tumor growth and metastasis in vivo. The functional
target molecule of miR-148a was identified as MEG-3 in pancreatic
cancer cells and tumors. The results of the present study suggested
that miR-148a is a potential tumor suppressor and inhibits
metastasis of pancreatic cancer, and that its restoration may
contribute to tumor regression in patients with pancreatic
cancer.
Pancreatic cancer is a common malignancy worldwide
and with the increasing rate of newly diagnosed cases, is bound to
become the second leading cause of cancer-associated mortality in
the future (8,37). Based on the findings of previous
studies, a better understanding of cancer pathology, earlier
diagnosis and systemic therapies and the development of more
efficacious drugs may improve the five-year survival rate of
patients with pancreatic cancer (37–39).
However, >85% of patients are newly diagnosed with pancreatic
cancer at the advanced stage, which contributes to the difficulty
of their treatment and dismal prognosis (40). At present, treatment options for
pancreatic cancer are based on chemotherapy and radiotherapy
regimens. Although numerous studies, which have proposed different
schedules to improve the overall survival and outcomes for patients
with pancreatic cancer, are encouraging additional studies are
still required to corroborate the molecular mechanisms (1,41,42). The
present study provided novel insight and potential therapeutic
approaches for the treatment of pancreatic cancer, suggesting the
potential application of miR-148a for the treatment of human
cancers.
Certain miRNAs are involved in tumorigenesis,
progression and metastasis in the majority of cancer cells, and are
potentially involved in the regulation of proliferation-associated
cellular processes, including mitosis and chromosome replication
(43). The present study indicated
that miR-148a acts as a tumor suppressor in pancreatic cancer and
it is downregulated through epigenetic silencing by promoter
methylation. Although it has been reported that miR-148a silencing
by hypermethylation activates the integrin-mediated signaling
pathway in nasopharyngeal carcinoma, the detailed mechanisms of the
biological roles of miR-148a have largely remained elusive
(44). The present study identified
that ectopic expression of the MEG-3 gene is controlled by miR-148a
expression in human pancreatic cancer cells and tumors, and that
MEG-3 overexpression significantly inhibited the proliferation and
invasion for pancreatic cancer cells.
MEG-3 encodes a long non-coding RNA that has been
recently shown to regulate tumorigenesis through its interaction
with miRNA (13). Liu et al
(45) assessed the expression levels
and molecular mechanisms of the lncRNA-encoding gene MEG-3 in
gallbladder cancer and the results indicated that
pcDNA-MEG3-transfected cells reduced the activity and clone counts
of gallbladder cancer cells and increased p53 and apoptosis. In
addition, Luo et al (46)
showed that overexpression of lncRNA MEG-3 inhibits cell
proliferation and invasion, and induces apoptosis in prostate
cancer in vitro as well as in vivo via reducing the
protein expression of B-cell lymphoma 2 (Bcl-2), enhancing
Bcl-2-associated X protein and activating caspase 3. Furthermore,
Chunharojrith et al (17)
indicated that pituitary tumor growth is suppressed by MEG-3
lncRNA, leading to cell cycle arrest at the G1 phase. The results
of the present study revealed that overexpression of MEG-3 resulted
in downregulation of the Wnt/β-catenin signaling pathway in
pancreatic cancer. Furthermore, inhibition of MEG-3 expression
abrogated inhibitory effects from miR-148a restoration in
pancreatic cancer cells.
In conclusion, the present study investigated the
inhibitory effects of miRNA-148a in pancreatic cancer and
demonstrated its anti-proliferative and anti-metastatic potential,
which may be utilized for cancer treatment. Lower expression levels
of miRNA-148a caused by DNA hypermethylation were associated with
pancreatic tumor growth and aggressive behavior. The present study
provided a potential rationale for developing epigenetic therapies
through restoration of tumor-suppressor miRNA-148a to inhibit
invasion and metastasis by downregulating the Wnt/β-catenin
signaling pathway through upregulating MEG-3 in pancreatic cancer.
The results suggested that miRNA-148a may be a potential
anti-cancer agent for the treatment of patients with pancreatic
carcinoma to complement clinical cancer management strategies.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Educational
Department of Zhejiang Province (grant no. Y201534189).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
YSu performed the experiments. QZhu, MZ, WY, HS, YSh
and QZha prepared and analyzed experimental data. FY designed the
experiment. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was performed in strict accordance
with the recommendations in the Guide for the Care and Use of
Laboratory Animals of the National Institutes of Health. This study
was approved by the Institutional Review Board and Ethics Committee
of Wenzhou Medical University (approval no. 0812-10421C4).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dumstrei K, Chen H and Brenner H: A
systematic review of serum autoantibodies as biomarkers for
pancreatic cancer detection. Oncotarget. 7:11151–11164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klompmaker S, de Rooij T, Korteweg JJ, van
Dieren S, van Lienden KP, van Gulik TM, Busch OR and Besselink MG:
Systematic review of outcomes after distal pancreatectomy with
coeliac axis resection for locally advanced pancreatic cancer. Br J
Surg. 103:941–949. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paiella S, Sandini M, Gianotti L,
Butturini G, Salvia R and Bassi C: The prognostic impact of
para-aortic lymph node metastasis in pancreatic cancer: A
systematic review and meta-analysis. Eur J Surg Oncol. 42:616–624.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Subramani R, Gangwani L, Nandy SB,
Arumugam A, Chattopadhyay M and Lakshmanaswamy R: Emerging roles of
microRNAs in pancreatic cancer diagnosis, therapy and prognosis
(Review). Int J Oncol. 47:1203–1210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang J, Li J, Zhu R, Zhang H, Zheng Y, Dai
W, Wang F, Shen M, Chen K, Cheng P, et al: K-ras mutational status
in cytohistological tissue as a molecular marker for the diagnosis
of pancreatic cancer: A systematic review and meta-analysis. Dis
Markers. 2014:5737832014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spadavecchia J, Movia D, Moore C, Maguire
CM, Moustaoui H, Casale S, Volkov Y and Prina-Mello A: Targeted
polyethylene glycol gold nanoparticles for the treatment of
pancreatic cancer: From synthesis to proof-of-concept in vitro
studies. Int J Nanomedicine. 11:791–822. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldsmith C, Price P, Cross T, Loughlin S,
Cowley I and Plowman N: Dose-volume histogram analysis of
stereotactic body radiotherapy treatment of pancreatic cancer: A
focus on duodenal dose constraints. Semin Radiat Oncol. 26:149–156.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spadi R, Brusa F, Ponzetti A, Chiappino I,
Birocco N, Ciuffreda L and Satolli MA: Current therapeutic
strategies for advanced pancreatic cancer: A review for clinicians.
World J Clin Oncol. 7:27–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang K, Lu W, Qin W and Wu Y: Neoadjuvant
therapy for patients with borderline resectable pancreatic cancer:
A systematic review and meta-analysis of response and resection
percentages. Pancreatology. 16:28–37. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blogowski W, Bodnarczuk T and Starzynska
T: Concise review: Pancreatic cancer and bone marrow-derived stem
cells. Stem Cells Transl Med. 5:938–945. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyoshi N, Wagatsuma H, Wakana S,
Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA, Kaneko-Ishino
T and Ishino F: Identification of an imprinted gene, Meg3/Gtl2 and
its human homologue MEG3, first mapped on mouse distal chromosome
12 and human chromosome 14q. Genes Cells. 5:211–220. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oczkowicz M, Ropka-Molik K, Piorkowska K,
Rozycki M and Rejduch B: Frequency of DLK1 c.639C>T polymorphism
and the analysis of MEG3/DLK1/PEG11 cluster expression in muscle of
swine raised in Poland. Meat Sci. 88:627–630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Benetatos L, Vartholomatos G and
Hatzimichael E: MEG3 imprinted gene contribution in tumorigenesis.
Int J Cancer. 129:773–779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kagami M, O'Sullivan MJ, Green AJ, Watabe
Y, Arisaka O, Masawa N, Matsuoka K, Fukami M, Matsubara K, Kato F,
et al: The IG-DMR and the MEG3-DMR at human chromosome 14q32.2:
Hierarchical interaction and distinct functional properties as
imprinting control centers. PLoS Genet. 6:e10009922010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Rice K, Wang Y, Chen W, Zhong Y,
Nakayama Y, Zhou Y and Klibanski A: Maternally expressed gene 3
(MEG3) noncoding ribonucleic acid: Isoform structure, expression,
and functions. Endocrinology. 151:939–947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang X, Zhou Y, Mehta KR, Danila DC,
Scolavino S, Johnson SR and Klibanski A: A pituitary-derived MEG3
isoform functions as a growth suppressor in tumor cells. J Clin
Endocrinol Metab. 88:5119–5126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chunharojrith P, Nakayama Y, Jiang X, Kery
RE, Ma J, De La Hoz, Ulloa CS, Zhang X, Zhou Y and Klibanski A:
Tumor suppression by MEG3 lncRNA in a human pituitary tumor derived
cell line. Mol Cell Endocrinol. 416:27–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lv J, Qiu M, Xia W, Liu C, Xu Y, Wang J,
Leng X, Huang S, Zhu R, Zhao M, et al: High expression of long
non-coding RNA SBF2-AS1 promotes proliferation in non-small cell
lung cancer. J Exp Clin Cancer Res. 35:752016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Costales MG, Rzuczek SG and Disney MD:
Comparison of small molecules and oligonucleotides that target a
toxic, non-coding RNA. Bioorg Med Chem Lett. 26:2605–2609. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Victoria Martinez B, Dhahbi JM, Nunez
Lopez YO, Lamperska K, Golusinski P, Luczewski L, Kolenda T, Atamna
H, Spindler SR, Golusinski W and Masternak MM: Circulating small
non-coding RNA signature in head and neck squamous cell carcinoma.
Oncotarget. 6:19246–19263. 2015.PubMed/NCBI
|
|
21
|
Nowacki FC, Swain MT, Klychnikov OI, Niazi
U, Ivens A, Quintana JF, Hensbergen PJ, Hokke CH, Buck AH and
Hoffmann KF: Protein and small non-coding RNA-enriched
extracellular vesicles are released by the pathogenic blood fluke
Schistosoma mansoni. J Extracell Vesicles. 4:286652015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marchand V and Branlant C: Quantification
and quality control of a small non-coding RNA preparation. Methods
Mol Biol. 1296:17–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marti E and Estivill X: Small non-coding
RNAs add complexity to the RNA pathogenic mechanisms in
trinucleotide repeat expansion diseases. Front Mol Neurosci.
6:452013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martens-Uzunova ES, Olvedy M and Jenster
G: Beyond microRNA-novel RNAs derived from small non-coding RNA and
their implication in cancer. Cancer Lett. 340:201–211. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prensner JR and Chinnaiyan AM: The
emergence of lncRNAs in cancer biology. Cancer Discov. 1:391–407.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Munagala R, Aqil F and Gupta RC: Exosomal
miRNAs as biomarkers of recurrent lung cancer. Tumour Biol.
37:10703–10714. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dimitrova DI, Yang X, Reichenbach NL,
Karakasidis S, Sutton RE, Henderson EE, Rogers TJ and Suhadolnik
RJ: Lentivirus-mediated transduction of PKR into CD34(+)
hematopoietic stem cells inhibits HIV-1 replication in
differentiated T cell progeny. J Interferon Cytokine Res.
25:345–360. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yallop CA and Svendsen I: The effects of
G418 on the growth and metabolism of recombinant mammalian cell
lines. Cytotechnology. 35:101–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wai-Hoe L, Wing-Seng L, Ismail Z and
Lay-Harn G: SDS-PAGE-based quantitative assay for screening of
kidney stone disease. Biol Proced Online. 11:145–160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seviour EG, Sehgal V, Lu Y, Luo Z, Moss T,
Zhang F, Hill SM, Liu W, Maiti SN, Cooper L, et al: Functional
proteomics identifies miRNAs to target a p27/Myc/phospho-Rb
signature in breast and ovarian cancer. Oncogene. 35:691–701. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Madhavan D, Peng C, Wallwiener M, Zucknick
M, Nees J, Schott S, Rudolph A, Riethdorf S, Trumpp A, Pantel K, et
al: Circulating miRNAs with prognostic value in metastatic breast
cancer and for early detection of metastasis. Carcinogenesis.
37:461–470. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian W, Liu J, Pei B, Wang X, Guo Y and
Yuan L: Identification of miRNAs and differentially expressed genes
in early phase non-small cell lung cancer. Oncol Rep. 35:2171–2176.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Endzelins E, Melne V, Kalnina Z,
Lietuvietis V, Riekstiņa U, Llorente A and Linē A: Diagnostic,
prognostic and predictive value of cell-free miRNAs in prostate
cancer: A systematic review. Mol Cancer. 15:412016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masliah-Planchon J, Garinet S and Pasmant
E: RAS-MAPK pathway epigenetic activation in cancer: miRNAs in
action. Oncotarget. 7:38892–38907. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mathe A, Scott RJ and Avery-Kiejda KA:
MiRNAs and other epigenetic changes as biomarkers in triple
negative breast cancer. Int J Mol Sci. 16:28347–28376. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee JC, Ahn S, Paik KH, Kim HW, Kang J,
Kim J and Hwang JH: Clinical impact of neoadjuvant treatment in
resectable pancreatic cancer: A systematic review and meta-analysis
protocol. BMJ Open. 6:e0104912016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kunk PR, Bauer TW, Slingluff CL and Rahma
OE: From bench to bedside a comprehensive review of pancreatic
cancer immunotherapy. J Immunother Cancer. 4:142016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kristensen A, Vagnildhaug OM, Gronberg BH,
Kaasa S, Laird B and Solheim TS: Does chemotherapy improve
health-related quality of life in advanced pancreatic cancer? A
systematic review. Crit Rev Oncol Hematol. 99:286–298. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Paiella S, Salvia R, Ramera M, Girelli R,
Frigerio I, Giardino A, Allegrini V and Bassi C: Local ablative
strategies for ductal pancreatic cancer (Radiofrequency Ablation,
Irreversible Electroporation): A review. Gastroenterol Res Pract.
2016:45083762016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gagliardi AR, Soong D and Gallinger S:
Identifying factors influencing pancreatic cancer management to
inform quality improvement efforts and future research: A scoping
systematic review. Pancreas. 45:161–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fan Y, Hu J, Feng B, Wang W, Yao G, Zhai J
and Li X: Increased risk of pancreatic cancer related to gallstones
and cholecystectomy: A systematic review and Meta-analysis.
Pancreas. 45:503–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stuopelyte K, Daniunaite K, Jankevicius F
and Jarmalaite S: Detection of miRNAs in urine of prostate cancer
patients. Medicina (Kaunas). 52:116–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li HP, Huang HY, Lai YR, Huang JX, Chang
KP, Hsueh C and Chang YS: Silencing of miRNA-148a by
hypermethylation activates the integrin-mediated signaling pathway
in nasopharyngeal carcinoma. Oncotarget. 5:7610–7624. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu B, Shen ED, Liao MM, Hu YB, Wu K, Yang
P, Zhou L and Chen WD: Expression and mechanisms of long non-coding
RNA genes MEG3 and ANRIL in gallbladder cancer. Tumour Biol.
37:9875–9786. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Luo G, Wang M, Wu X, Tao D, Xiao X, Wang
L, Min F, Zeng F and Jiang G: Long Non-coding RNA MEG3 inhibits
cell proliferation and induces apoptosis in prostate cancer. Cell
Physiol Biochem. 37:2209–2220. 2015. View Article : Google Scholar : PubMed/NCBI
|