Introduction
Osteoporosis refers to a systemic skeletal disease
characterized by a low bone mass and deterioration of the
microstructure, followed by an increase in bone fragility and
fracture (1). As one main type of
osteoporosis, postmenopausal osteoporosis (PO) results from a
scarcity of endogenous oestrogen in postmenopausal women (2). Clinically, the two major manifestations
for PO patients are low bone mass density and fracture, and even
the occurrence of vertebral compression fractures during routine
activities without a specific fall or injury (3). Consequently, PO imposes a great burden
on life and work both for the individual and society. Thus, an
effective prevention, diagnosis and treatment before the occurrence
of fractures for this disease is urgently needed (4).
Microarray has become a powerful technology that
simultaneously monitors the expression levels of thousands of
genes, and provides many basic applications, such as functional
genomics, molecular pathway modeling and tumor classification
(5). The most commonly in use is the
comparison of gene expression discrepancy between two different
statuses (normal controls versus disease and treated versus
untreated samples). Furthermore, under such an experimental setup,
how to investigate a gene whose expressed value is remarkably
different between the two status is a dominate challenge. A variety
of methods have been proposed to detect significant genes across
controls and cases in the past few years (6,7).
However, it has been revealed that the most significant genes
obtained from distinct research are typically inconsistent for a
specific disease because of batch effects and method difference
(8).
Therefore, to avoid inconsistent results and enhance
the feasibility and confidence of the results, an integrated method
was proposed in the present study. The integrated method combined
three commonly used methods, including the convergent evidence (CE)
method, rank product (RP) algorithm and combing P-values, to
identify informative genes for PO. Subsequently, hub genes were
identified by accessing topological analysis of mutual information
network (MIN) which were constructed based on the informative
genes. Moreover, hub gene functions were determined by Gene
Ontology (GO) enrichment analysis for informative genes.
Materials and methods
Preparing gene data
A gene expression dataset [E-MEXP-1618 (9,10)] was
downloaded for PO from the online ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/). In brief,
E-MEXP-1618 consisted of 39 normal controls and 45 PO samples, and
deposited on the A-AFFY-44-Affymetrix GeneChip Human Genome U133
Plus 2.0 [HG-U133_Plus_2] Platform. Subsequently, duplicated or
invalid probes in the data were removed, and the retained probes
were converted into gene symbols. Ultimately, 20,544 genes were
obtained for subsequent exploitation.
Ranking genes by multiple
algorithms
Currently, an effective method to investigate and
prioritize candidate genes in complex diseases is integration of
gene data from multiple evidence layers (11). Thus, to recognize informative genes
generated from multiple independent lines of investigation for PO
patients, we integrated 3 commonly used approaches, CE method, RP
algorithm and combining P-values.
CE method
CE method, a variant of the famous PageRank
algorithm, is implemented in the GenRank Bioconductor package
(http://bioconductor.org/packages/GenRank). This method
aggregates ranks of genes using a weighted vote counting method
that the genes were ranked according to the self-importance and the
number of each evidence layer was detected (12). In addition, the algorithm permits us
to integrate evidence from studies where P-values and effect sizes
are unavailable or the gene sets varies across research. During
this process, the weighted arithmetic mean of each gene, a CE
score, was calculated as the given formula (13):
CE(g)=CE(L1)n(L1)+CE(L2)n(L2)+⋯CE(Ln)n(Ln)
of which CE(Li) referred to the
self-importance of gene g in evidence layer i, while
n(Li) represented the number of genes within evidence
layer i.
RP algorithm
To the best of our knowledge, RP method has earlier
been widely used to carry out differential expression and
meta-analysis in microarray-based gene expression datasets
(5,14). Furthermore, it is also a simple, but
powerful to rank genes that are consistently ranked high in
replicated microarray tests (5).
Thus, we used the RP algorithm to identify consistently highly
ranked genes across various evidence layers. Supposing that RPi, g
was the position of gene g in the list of genes in the ith
replicate or layer among k replicates sorted by decreasing
fold-change (FC), the RP value for a gene g was computed as
following:
RP(g)=(∏i=1kRPi,g)1/k
Consequently, all genes in the gene lists were
sorted according to their RP values. Note that genes possessing the
smallest RP values are the most fascinating candidates and
researchers could choose some of them for further
investigation.
Combining P-values
Estimation of P-values from the same null nypothesis
was the precondition of this method. When a null assumption was
examined on distinct sets of data, the individual P-values may
effectively be integrated to supply a more powerful test than any
individual one (15). Each P-value
abides by a uniform distribution, in case the null hypothesis is
true, and the statistical model is correct. The Fisher's exact test
was utilized to combine the P-values, which is a statistical test
to compare binary outcomes between two groups (16). In consequence, we obtained P-value
for each gene and corrected them by false discovery rate (FDR)
implemented in Benjamini-Hochberg (BH) method (17).
Extracting informative genes
As mentioned above, based on 20,544 genes in the
gene data, we obtained three different gene lists ranked by CE, RP
and combining P-values, respectively. Subsequently, we selected the
top 100 genes from the three gene lists separately. Taking
intersections between any two of them was a good way to overcome
the inconsistent results from the top 100 genes. These common genes
might be more significant than the others, and thus we defined them
as the informative genes for PO patients. Analyses were conducted
based on the informative genes to investigate hub genes and gene
functions in the progression of PO.
Constructing network and topological
analysis
Constructing network
To investigate the co-operations and interactions
between any two of informative genes, a MIN was constructed based
on the context likelihood of relatedness (CLR) algorithm. CLR
algorithm is an information-theoretic approach that applies a
generalization of the pairwise correlation coefficient called
mutual information for each pair of genes (18). For a pair of genes × and y, an edge
score, Sxy, was derived (19):
Sxy2=Sx2+Sy2
of which
Sx=max(0,Mxy-μMxσMx)
where μMx represents the sample
mean of the empirical distribution of the values
Mxy, and σMx refers to
the standard deviation Mxy. Similarly,
Sy was calculated. Finally, Cytoscape software
(http://www.cytoscape.org/) was used to
visualize MIN for PO (20).
Topological analysis
For purpose of deeply investigating biological
significance of nodes in the MIN, topological centrality analysis
of degree index was conducted. Degree quantifies the local topology
of each gene by summing up the number of its adjacent genes (j) and
gives a simple count of the number of interplays of a given node
(21). Besides, the nodes on the top
of degree distribution (≥10% quantile) in the MIN were described as
hub genes for PO.
Gene functional enrichment
analysis
The Biological Networks Gene Ontology tool (BiNGO),
is a tool to identify which GO terms are significantly
overrepresented in a set of gene lists, and utilizes Cytoscape's
versatile visualization environment to generate an intuitional and
customizable visual representation of the results (22). Thus, we applied the BiNGO to perform
the GO enrichment analysis for informative genes in this study. The
GO terms with P<0.001 were considered to be predominant
functional themes, and screened for mapping to the GO hierarchy.
Most importantly, the term at the top of P distribution (≥10%
quantile) among the significant gene sets were defined as hub gene
functions of PO.
Results
Informative genes
In the present study, three commonly used methods of
ranking genes were implemented to prioritize significant genes for
PO patients. As a result, we obtained three gene lists dependent
upon their CE scores, RP scores and P-values, respectively. The top
100 genes which might be more important than others were selected
from each gene list for further analysis. Although there were
differences among the three kinds of top 100 genes, we found that a
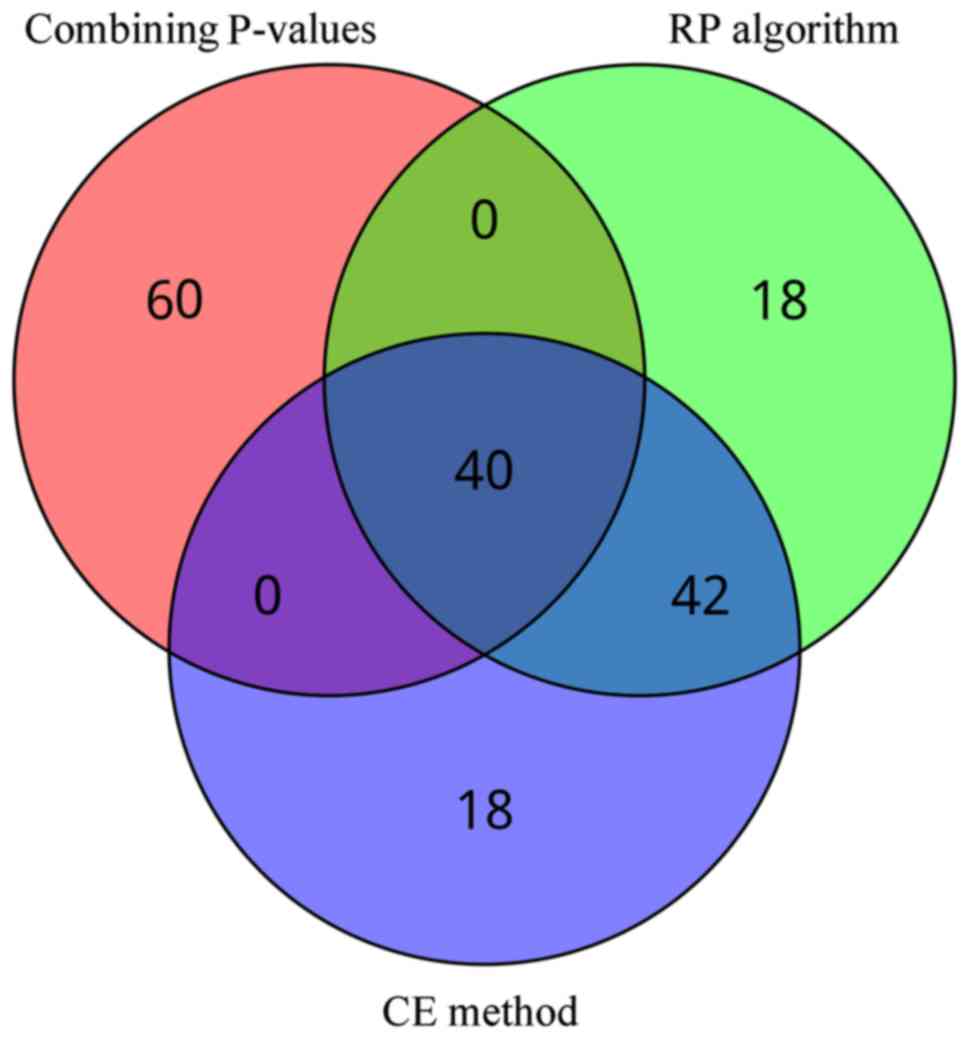
certain number of genes were common. As shown in Fig. 1, 82 common genes from two or even all
three methods were identified, of which 40 genes were common to the
three methods. All 82 common genes were defined as informative
genes of PO patients, where 40 genes were attributed to the numbers
from 1 to 40, such as LYZ, LCN2 and EEF2 (Table I).
 | Table I.Informative genes. |
Table I.
Informative genes.
| No. | Gene | No. | Gene | No. | Gene | No. | Gene |
|---|
| 1 | LYZ | 22 | RPL23A | 43 | CAMP | 64 | FTH1P5 |
| 2 | LCN2 | 23 | RPL37 | 44 | UBC | 65 | MMP9 |
| 3 | EEF2 | 24 | PRG2 | 45 | RPL11 | 66 | HLA-A |
| 4 | COX1 | 25 | RPL32 | 46 | AHSP | 67 | RPS5 |
| 5 | HBB | 26 | RNASE3 | 47 | NCOA4 | 68 | TALDO1 |
| 6 | UBB | 27 | ELANE | 48 | AZU1 | 69 | RPL3 |
| 7 | S100A9 | 28 | RNASE2 | 49 | TUBA1C | 70 | CA2 |
| 8 | TMSB4X | 29 | RPS2 | 50 | RPL8 | 71 | CEACAM8 |
| 9 | LTF | 30 | GAPDH | 51 | RPL12 | 72 | PFN1 |
| 10 | RPL39 | 31 | RPS17 | 52 | RPL10A | 73 | HLA-B |
| 11 | S100A12 | 32 | RPL27 | 53 | RPS25 | 74 | PF4 |
| 12 | RPS4X | 33 | HMGN2 | 54 | HBD | 75 | RPL35 |
| 13 | ND2 | 34 | RPLP0 | 55 | RPL34 | 76 | COX2 |
| 14 | DEFA4 | 35 | RPS3 | 56 | RPS7 | 77 | IGLL3P |
| 15 | TPT1 | 36 | CLC | 57 | MPO | 78 | BLVRB |
| 16 | RPS18 | 37 | CTSG | 58 | UBA52 | 79 | ANP32B |
| 17 | ACTB | 38 | RPL19 | 59 | TMSB10 | 80 | HLA-C |
| 18 | RPL9 | 39 | OAZ1 | 60 | RPL6 | 81 | RPL41 |
| 19 | FTL | 40 | ND4 | 61 | RPL29 | 82 | PRG3 |
| 20 | HBM | 41 | RPL24 | 62 | MNDA |
|
|
| 21 | RPL30 | 42 | PPBP | 63 | FAU |
|
|
Hub genes
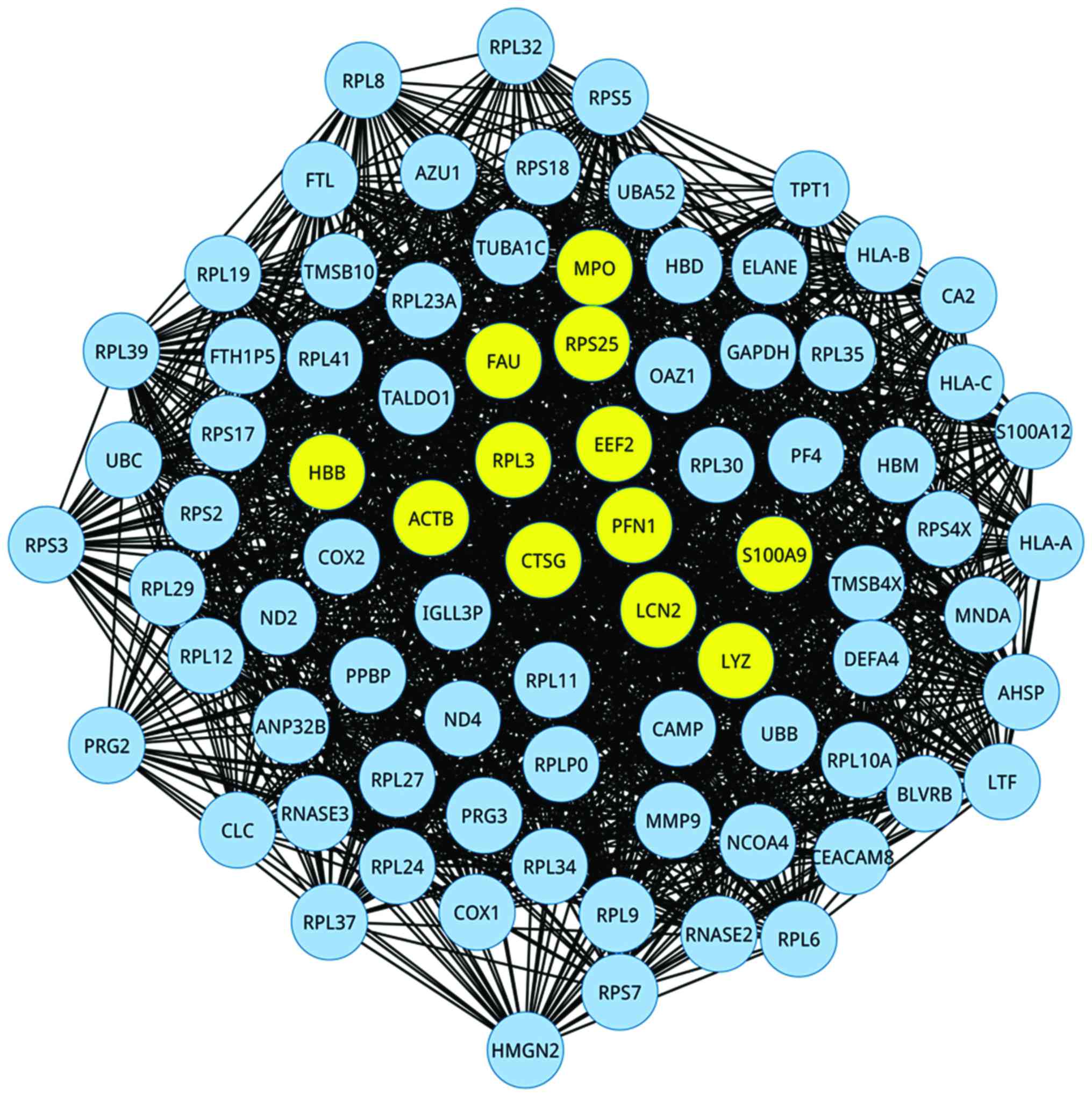
Since genes often work together in complex disease
rather than individually, a MIN based on the informative genes of
PO was constructed using the CLR algorithm, as illustrated in
Fig. 2. There were 82 nodes and
1,741 edges in the MIN, which suggested that all of the informative
genes were mapped to the PO-associated network. Furthermore,
topological centrality of degree analysis was conducted to screen
crucial genes in the MIN, and we denoted the nodes at the top 10%
of degree distribution as hub genes for PO. In consequence, a total
of 8 hub genes were identified, including PFN1 (Degree = 63), EEF2
(Degree = 54), S100A9 (Degree = 53), LCN2 (Degree = 52), ACTB
(Degree = 51), FAU (Degree = 50), RPS25 (Degree = 50) and CTSG
(Degree = 50).
Hub gene functions
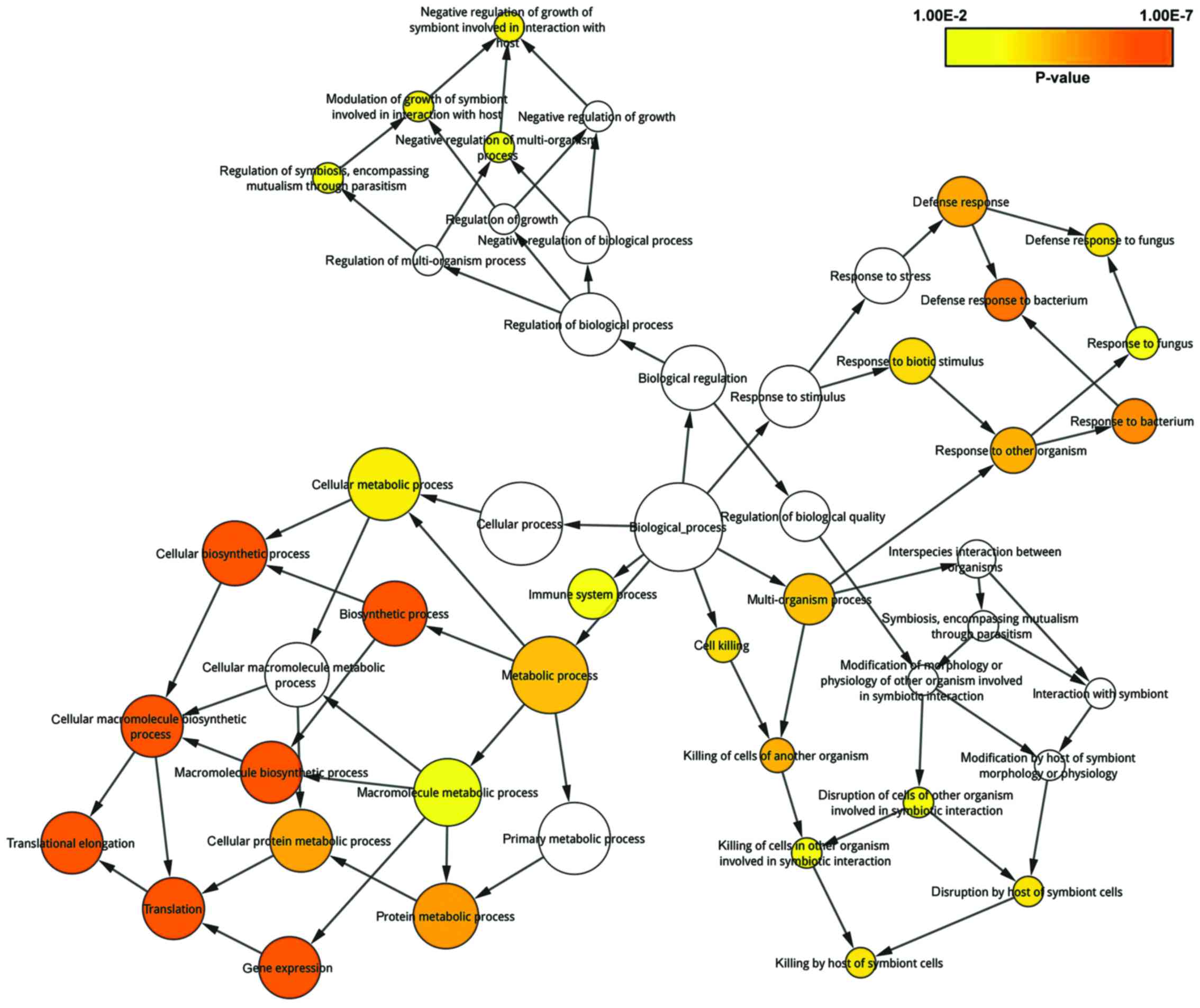
GO enrichment analysis was conducted on the
informative genes by the BiNGO plugin in order to investigate
significant gene functions in the progression of PO. The result
displayed that 49 GO BP terms were determined when setting the
threshold as P<0.001 (Fig. 3).
The darker the node, the more significant the GO term for PO was.
In addition, we ordered the 49 terms in descending order of their
P-values, and defined the top 10% as hub terms enriched by
informative genes. Specifically, the hub gene functions were
comprised of translational elongation (P=7.317E-49), translation
(P=9.21E-33), cellular macromolecule biosynthetic process
(P=3.37E-16), macromolecule biosynthetic process (P=5.92E-16), and
gene expression (P=8.44E-14).
Discussion
A better understanding of pathological mechanism
underlying PO may give crucial insights for prevention and
treatment in the progression of this disease. Hence, in this study,
we predicted hub genes and gene functions for PO by integrating CE
method, RP algorithm and combing P-values approach. In brief, the
CE method integrates single-case assessment data by converging data
gathered across multiple settings, or measures, thereby supplying
an overall criterion-referenced outcome (23). Importantly, a conceptually similar
gene-level integration of CE method had been smoothly applied to
prioritize candidate genes in neuropsychiatric diseases (24). In addition, the RP algorithm provides
a powerful statistics for defining significant genes in microarray
tests, and needs only a few well-justified hypothesis on the data
because of its non-parametric nature (5). Besides, the combing P-values approach
is a typical and efficient method to rank genes by their P-values.
Above all, we merged the three methods to enhance the feasibility
and precision of our research. Consequently, we obtained 82
informative genes for PO patients.
In general, a gene is inclined to interact with
other genes, and the co-operated genes often actively participate
in the same biological processes. Moreover, network-based approach
is able to extract informative and significant genes dependent upon
bio-molecular networks and provides a quantifiable characterization
of the molecular networks that characterize the complicated
interplays and the complex interwoven associations that administer
cellular functions (25,26). Thus, a network was constructed on the
informative genes, termed with MIN. The hub genes were extracted
from the MIN by accessing the degree centrality analysis. As a
result, a total of 8 hub genes were identified, PFN1, EEF2, S100A9,
LCN2, ACTB, FAU, RPS25 and CTSG. Taking S100A9 as an example,
S100A9 (S100 calcium binding protein A9) is a member of the S100
family of proteins containing 2 EF-hand calcium-binding motifs.
S100 proteins involve in many biological processes, including
proliferation, apoptosis, differentiation, inflammation,
Ca2+ homeostasis, as well as migration/invasion by
interplays with various target proteins such as receptors, enzymes,
transcription factors, cytoskeletal subunits and nucleic acids
(27). Specifically, calcium
supplementation in PO markedly changed bone mineral and organic
matrix quality (28). In addition,
S100A9 expressed earlier than the appearance of neutrophilics in
the airway of neutrophilic inflammation, and directly activated
inflammasome in the airway in asthma (29,30). Due
to its expression in inflammation, S100A9 had been indicated to
play a significant role in cell proliferation (31). Similarly, the significant role of
S100A9 in PO may also attribute to its activities in cell
proliferation. Most importantly, this is the first clue of the
correlation between S100A9 and PO patients.
Moreover, functional gene sets enriched by
informative genes were identified based on the GO enrichment
analysis. Consequently, a total of 5 hub gene functions were
gained, including cellular macromolecule biosynthetic process,
translational elongation, translation, gene expression and
macromolecule biosynthetic process. Interestingly, we found that 2
of 5 hub gene functions belonged to the biosynthetic process.
Macromolecule biosynthetic process refers to the chemical reactions
and pathways resulting in formation of a macromolecule, any
molecule of high relative molecular mass, the structure of which
essentially comprises the multiple repetitions of units derived,
actually or conceptually, from molecules of low relative molecular
mass. Mackiewicz et al revealed that macromolecule
biosynthesis was a key function of sleep (32). Furthermore, it had been demonstrated
that the most obviously involved processes for breast cancer were
translation, cellular biosynthesis, and macromolecular biosynthesis
(33). The change of hormonal
readiness was the common point across breast cancer and PO. Thus we
might infer it was the medium for the gene function macromolecular
biosynthesis process and PO patients.
In conclusion, we identified 8 hub genes and 5 hub
gene functions for PO by an integrated method. Our results provide
potential biomarkers for prevention and therapy of PO patients, and
further help uncover the pathological mechanism underlying this
progression. Whereas the validations of these genes have not
finished, and future study should focus on this aspect.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HoC and PT designed the study. LZ collected the
data. HuC performed the statistical analysis. WZ preparated the
figures. HoC, QZ and XL drafted the manuscript and interpreted the
data. YG is accountable for the accuracy or integrity of the work.
YG and PT revised the manuscrript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kanis JA, Burlet N, Cooper C, Delmas PD,
Reginster JY, Borgstrom F and Rizzoli R: European Society for
Clinical and Economic Aspects of Osteoporosis and Osteoarthritis
(ESCEO): European guidance for the diagnosis and management of
osteoporosis in postmenopausal women. Osteoporos Int. 19:399–428.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marcus R: Post-menopausal osteoporosis.
Best Pract Res Clin Obstet Gynaecol. 16:309–327. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Unni S, Yao Y, Milne N, Gunning K, Curtis
JR and LaFleur J: An evaluation of clinical risk factors for
estimating fracture risk in postmenopausal osteoporosis using an
electronic medical record database. Osteoporos Int. 26:581–587.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cosman F, de Beur SJ, LeBoff MS, Lewiecki
EM, Tanner B, Randall S and Lindsay R: National Osteoporosis
Foundation: Clinician's guide to prevention and treatment of
osteoporosis. Osteoporos Int. 25:2359–2381. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Breitling R, Armengaud P, Amtmann A and
Herzyk P: Rank products: A simple, yet powerful, new method to
detect differentially regulated genes in replicated microarray
experiments. FEBS Lett. 573:83–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu Y, Koyutürk M, Barnholtz-Sloan JS and
Chance MR: Gene interaction enrichment and network analysis to
identify dysregulated pathways and their interactions in complex
diseases. BMC Syst Biol. 6:652012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li RH, Zhang AM, Li S, Li TY, Wang LJ,
Zhang HR, Li P, Jia XJ, Zhang T, Peng XY, et al: Multiple
differential expression networks identify key genes in rectal
cancer. Cancer Biomark. 16:435–444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ein-Dor L, Kela I, Getz G, Givol D and
Domany E: Outcome signature genes in breast cancer: Is there a
unique set? Bioinformatics. 21:171–178. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tharmaratnam K, Sperrin M, Jaki T, Reppe S
and Frigessi A: Tilting the lasso by knowledge-based
post-processing. BMC Bioinformatics. 17:3442016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reppe S, Refvem H, Gautvik VT, Olstad OK,
Høvring PI, Reinholt FP, Holden M, Frigessi A, Jemtland R and
Gautvik KM: Eight genes are highly associated with BMD variation in
postmenopausal Caucasian women. Bone. 46:604–612. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanduri C and Jarvela I: GenRank: An
R/Bioconductor package for prioritization of candidate genes.
bioRxiv 048264. doi: https://doi.org/10.1101/048264.
|
|
12
|
Morrison JL, Breitling R, Higham DJ and
Gilbert DR: GeneRank: Using search engine technology for the
analysis of microarray experiments. BMC Bioinformatics. 6:2332005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oikkonen J, Onkamo P, Järvelä I and
Kanduri C: Convergent evidence for the molecular basis of musical
traits. Sci Rep. 6:397072016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang K, Cheng Y, Liao WK and Choudhary A:
Mining millions of reviews: a technique to rank products based on
importance of reviews. Proceedings of the 13th International
Conference on Electronic Commerce. ICEC'11 [2378116] doi:
10.1145/2378104.2378116.
|
|
15
|
Westfall PH: Combining P Values.
Encyclopedia of Biostatistics. Armitage P and Colton T: Jonh Wiley
& Sons, Ltd.; Chichester, UK: pp. 987–991. 2005
|
|
16
|
Fay MP: Confidence intervals that match
Fisher's exact or Blaker's exact tests. Biostatistics. 11:373–374.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc B. 57:289–300. 1995.
|
|
18
|
Cover TM and Thomas JA: Elements of
information theory. Jonh Wiley & Sons Ltd.; Chichester, UK:
2012
|
|
19
|
Meyer PE, Lafitte F and Bontempi G: minet:
A R/Bioconductor package for inferring large transcriptional
networks using mutual information. BMC Bioinformatics. 9:4612008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Methods Mol Biol. 696:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haythornthwaite C: Social network
analysis: An approach and technique for the study of information
exchange. Libr Inf Sci Res. 18:323–342. 1996. View Article : Google Scholar
|
|
22
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Busse RT, Elliott SN and Kratochwill TR:
Convergent evidence scaling for multiple assessment indicators:
Conceptual issues, applications, and technical challenges. J Appl
Sch Psychol. 26:149–161. 2010. View Article : Google Scholar
|
|
24
|
Ayalew M, Le-Niculescu H, Levey DF, Jain
N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R,
et al: Convergent functional genomics of schizophrenia: From
comprehensive understanding to genetic risk prediction. Mol
Psychiatry. 17:887–905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu ZP, Wang Y, Zhang XS and Chen L:
Network-based analysis of complex diseases. IET Systems Biology.
6:22–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen L, Wang RS and Zhang XS:
Reconstruction of gene regulatory networks. Biomolecular Networks.
John Wiley & Sons, Inc.; pp. 47–87. 2009, View Article : Google Scholar
|
|
27
|
Donato R, Cannon B, Sorci G, Riuzzi F, Hsu
K, Weber D and Geczy C: Functions of S100 Proteins. Curr Mol Med.
13:242013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paschalis EP, Gamsjaeger S, Hassler N,
Fahrleitner-Pammer A, Dobnig H, Stepan JJ, Pavo I, Eriksen EF and
Klaushofer K: Vitamin D and calcium supplementation for three years
in postmenopausal osteoporosis significantly alters bone mineral
and organic matrix quality. Bone. 95:41–46. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pawar H, Srikanth SM, Kashyap MK, Sathe G,
Chavan S, Singal M, Manju HC, Kumar KV, Vijayakumar M, Sirdeshmukh
R, et al: Downregulation of S100 calcium binding protein A9 in
esophageal squamous cell carcinoma. Sci World J. 2015:3257212015.
View Article : Google Scholar
|
|
30
|
Lee TH, Jang AS, Park JS, Kim TH, Choi YS,
Shin HR, Park SW, Uh ST, Choi JS, Kim YH, et al: Elevation of S100
calcium binding protein A9 in sputum of neutrophilic inflammation
in severe uncontrolled asthma. Ann Allergy Asthma Immunol.
111:268–275.e1. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gebhardt C, Németh J, Angel P and Hess J:
S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol.
72:1622–1631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mackiewicz M, Shockley KR, Romer MA,
Galante RJ, Zimmerman JE, Naidoo N, Baldwin DA, Jensen ST,
Churchill GA and Pack AI: Macromolecule biosynthesis: A key
function of sleep. Physiol Genomics. 31:441–457. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miller WR, Larionov A, Renshaw L, Anderson
TJ, Walker JR, Krause A, Sing T, Evans DB and Dixon JM: Gene
expression profiles differentiating between breast cancers
clinically responsive or resistant to letrozole. J Clin Oncol.
27:1382–1387. 2009. View Article : Google Scholar : PubMed/NCBI
|