Introduction
Liver fibrosis is a wound-healing response to a
variety of liver insults, including excessive alcohol intake.
Alcohol abuse and chronic alcohol consumption are major causes of
alcoholic liver disease (ALD) worldwide (1). Alcohol-induced hepatic fibrosis (AHF)
may cause serious hepatic cirrhosis, and is widely accepted as a
milestone event in ALD (2). However,
recent evidence indicates that liver fibrosis is reversible and
that the liver may recover from cirrhosis (3). Therefore, elucidation of the cellular
and molecular mechanisms is urgently required to prevent AHF.
The exact mechanism remains unclear, but certain
changes produced from excessive alcohol consumption have been
clarified. Excessive alcohol damages hepatocytes via the release of
cytosolic proteins and enzymes, such as alanine aminotransferase
(ALT) and aspartate transaminase (AST), into the circulation
(4). Histopathological changes, such
as increased inflammatory cell infiltration, fatty changes,
accumulation of collagenous fibers and necrotic damage, reinforce
toxic liver injury (4). Excessive
alcohol also increases serum hyaluronic acid (HA), type III
precollagen (PCIII) and laminin (LN) levels (1). Indicators of liver fibrogenesis,
including hepatic hydroxyproline collagen I and III levels, are
also over expressed (1). Hepatic
stellate cells (HSCs) may be activated and transformed into
myofibroblast-like cells (5). An
increase in α-smooth muscle actin (α-SMA) stimulates the secretion
of fibrillar collagens and the deposition of a fibrotic matrix
(5). Transforming growth factor-β1
(TGF-β1) and connective tissue growth factor (CTGF) are two
important cytokines involved in the fibrotic and cirrhotic
transformation of the liver and HSCs transformation (6).
Cilostazol is a phosphodiesterase III inhibitor that
increases adenosine 3′,5′-cyclic monophosphate (cAMP) levels, and
is generally used to treat the symptoms of lower extremity
peripheral arterial disease (7).
Regulation of cAMP may inhibit HSCs activation. Recent studies
demonstrated that cilostazol attenuated cholestatic liver injury,
improved hepatic functions and decreased portal hypertension and
hepatic fibrosis (8). Cilostazol
also attenuated HSC activation and protected rats against carbon
tetrachloride-induced liver fibrosis (9). Cilostazol protected rats against
experimental non-alcoholic fatty liver disease via suppression of
mitogen-activated protein kinase activation induced by oxidative
stress and platelet-derived growth factor (10). However, the effects and mechanisms of
cilostazol on AHF are not clear. Therefore, the present study
examined the effects of cilostazol on AHF and investigated its
potential mechanisms of action.
Materials and methods
Animals and reagents
A total of 212 adult male Sprague-Dawley rats (age,
8–9 weeks; weight, 200–220 g) were purchased from the Experimental
Animal Center of Xi'an Central Hospital (Xi'an, China) for use in
the present study. Rats were housed at 24±2°C and 50% relative
humidity with a 12-h light/dark cycle in the Xi'an Central Hospital
Animal Center in accordance with protocols issued by the Xi'an
Central Hospital's Institutional Animal Care and Use Committee
(approval no. XCH-20170923). All rats received standard chow diet
and tap water ad libitum.
Rats were randomly divided into seven groups (20
rats per group) as follows: Control group, Model group, Colchicine
group (positive control), Cilostazol (5 mg/kg) group, Cilostazol
(10 mg/kg) group, Cilostazol (20 mg/kg) group and Cilostazol-only
group (10 mg/kg). Rats in the Model group were exposed to alcohol
as described below. Rats in the Colchicine, Cilostazol (5 mg/kg),
Cilostazol (10 mg/kg), and Cilostazol (20 mg/kg) groups were
exposed to the same dose of alcohol as the Model group and given
colchicine or cilostazol (both Sigma-Aldrich, Merck KGaA,
Darmstadt, Germany). Colchicine (0.1 mg/kg/day) was administered to
rats via gavage for 2 weeks. Cilostazol (5, 10 or 20 mg/kg/day) was
administered to rats via gavage for 2 weeks. Rats received alcohol
for 24 weeks following the completion of colchicine and cilostazol
administration. Rats in the Cilostazol-only group received 10 mg/kg
cilostazol without alcohol for 2 weeks.
Induction of AHF
Rats received alcohol infusions via gavage to induce
liver fibrosis, similar to the procedure used by Zhang et al
(11). Alcohol was administered at
5.0 g/kg/day from 1–4 weeks, 7.0 g/kg/day from 5–8 weeks, 9.0
g/kg/day from 9–12 weeks and 9.5 g/kg/day from 13–24 weeks. Rats
were sacrificed at the end of 24 weeks for the following
assays.
Determination of serum alcohol
dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH)
activities
Blood samples were immediately taken from sacrificed
rats and centrifuged at 3,000 × g for 10 min at 4°C to obtain
serum. ADH and ALDH activities in serum were measured using the
Alcohol Dehydrogenase Activity Assay kit (cat. no. A083-2; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) and Aldehyde
Dehydrogenase Activity Assay kit (cat. no. ALDH-2-G; Suzhou Comin
Biotechnology Co., Ltd., Suzhou, China) via a colorimetric
method.
Determination of liver
hydroxyproline
Liver hydroxyproline was examined as previously
described by Wang et al (12). Briefly, rats were sacrificed and
livers were harvested and cut into slices. Liver slices were
homogenized in 10% (w/v) phosphate buffer (0.5 mol/l potassium
phosphate; cat. no. P3786; Sigma-Aldrich, Merck KGaA) and
hydrolyzed in 12 M HCl at 100°C for 12 h. Following hydrolysis, the
pH was adjusted to pH 7.0 and the samples were centrifuged at 1,000
× g for 10 min at 4°C. The hydroxyproline content in each tissue
sample was examined using the spectrophotometric method, previously
described by Bergman and Loxley (13). Briefly, hydroxyproline was oxidised
by chloramine T (cat no. 402869; Sigma-Aldrich, Merck KGaA) at room
temperature for 5 min. Following oxidation, chloramine T was
removed using perchloric acid (cat no. 311421, Sigma-Aldrich, Merck
KGaA) and Ehrlich's reagent was added to each sample and heated at
60°C for 25 min. Finally, they samples were cooled to room
temperature and the absorbance was measured at a wavelength of 558
nm to calculate the hydroxyproline levels.
Determination of serum levels of
albumin/globulin, enzymes and HA, LN, IV-C and PCIII
Serum levels of albumin, globulin, enzymes [total
protein (TP), total bilirubin (TBIL), ALT, AST, alkaline
phosphatase (AKP) and glutamyltransferase (γ-GT)], HA, LN, type IV
collagen (IV-C) and PCIII were determined using radioimmunoassay
(RIA) kits. Albumin (cat. no. 452106), globulin (cat. no. 325214),
TP (cat. no. 320175), TBIL (cat. no. 235109), ALT (cat. no.
635921), AST (cat. no. 102307), AKP (cat. no. 471256) and γ-GT
(cat. no. 120523) kits were from Shanghai Institute of Biological
Products Co., Ltd. (Shanghai, China). HA (cat. no. HY-10088), LN
(cat. no. HY-10087), IV-C (cat. no. bs-0806P) and PCIII (cat. no.
HY-E0007) RIA kits were purchased from Beijing Sino-uk Institute of
Biological Technology (Beijing, China). Albumin (A) and globulin
(G) levels were used to calculate the A/G value. Enzyme levels (TP,
TBIL, ALT, AST, AKP, γ-GT) were used to evaluate the degree of
hepatic injury. HA, LN, IV-C and PCIII levels were used to evaluate
the degree of AHF.
Western blot analysis
A liver sample of ~10 g was collected from the left
lobe of the liver and rinsed thoroughly with ice-cold PBS (pH=7.4).
Liver samples were homogenized, and total protein was extracted
using HEPES extraction buffer (Santa Cruz Biotechnology, Inc.).
Total protein was quantified using a bicinchoninic acid assay kit
(Santa Cruz Biotechnology, Inc.) and 40 µg protein/lane were
separated via SDS-PAGE on a 12% gel. The separated proteins were
transferred onto nitrocellulose membranes (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and blocked for 1 h at room temperature
with 5% milk. The membranes were incubated with primary antibodies
against α-SMA (1:1,000: cat. no. 19245), TGF-β1 (1:1,000: cat. no.
3709), CTGF (1:1,000: cat. no. 86641), exchange protein directly
activated by cAMP (Epac)-1 (1:1,000: cat. no. 4155), Epac2
(1:1,000: cat. no. 43239) and β-actin (1:1,000: cat. no. 4970; all
Cell Signaling Technology, Inc., Danvers, MA, USA), and collagen
III (1:1,000; cat. no. sc-271249) and I (1:1,000; cat. no.
sc-376350; Santa Cruz Biotechnology, Inc.) overnight at 4°C.
Membranes were then incubated with a horseradish
peroxidase-conjugated secondary antibody (1:3,000; cat. no.
sc-2354; Santa Cruz Biotechnology, Inc.) for 1 h at 4°C. Protein
bands were visualized using the ECL Plus Western Blotting Detection
Reagents (GE Healthcare Life Sciences, Little Chalfont, UK) and
Bio-Rad ChemiDoc™ Imaging System (Bio-Rad Laboratories, Inc.).
Protein expression was quantified using Image-Quant TL software (GE
Healthcare Life Sciences).
Measurement of hepatic cAMP using
ELISA
Hepatic cAMP was measured using an ELISA kit (cat.
no. RPN225; GE Healthcare, Chicago, IL, USA) and the method
described by Miller et al (14). Hepatic cAMP levels were normalized to
total liver weight, and the results are expressed as pmol/mg
tissue.
Statistical analysis
All data are expressed as the mean ± standard error
of the mean. Statistical significance was determined using one-way
analysis of variance followed by a Dunnett's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of cilostazol on serum ADH and
ALDH activities
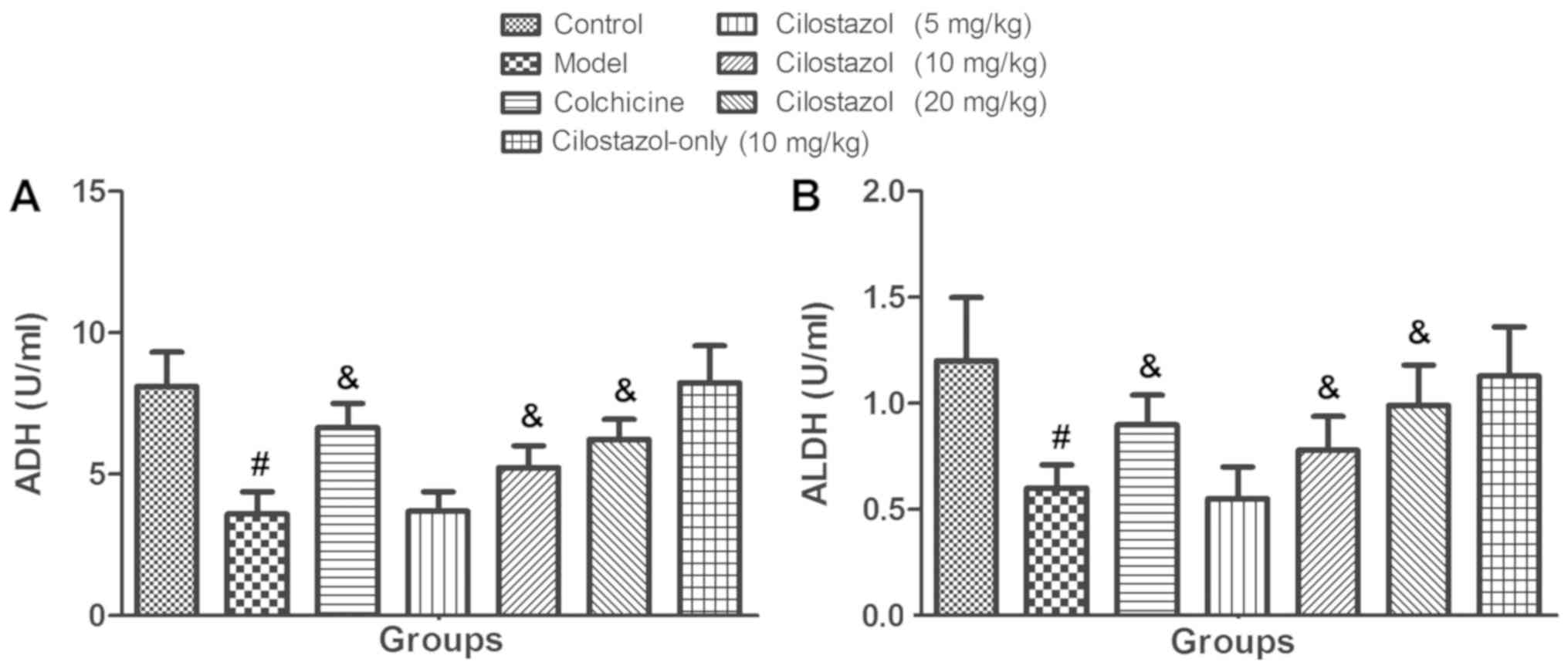
Fig. 1 presents the
serum ADH and ALDH levels. After rats were received treatment with
alcohol for 24 weeks, significantly decreased serum ADH and ALDH
activities were observed in the Model group compared with the
Control group (P<0.05). Rats that received colchicine and
alcohol exhibited a significant recovery of serum ADH and ALDH
activities compared with the Model group (P<0.05). Cilostazol
treatment (5 mg/kg/day) did not significantly alter serum ADH and
ALDH activities, but 10 and 20 mg/kg/day cilostazol significantly
increased these activities compared with the Model group
(P<0.05). Cilostazol treatment (10 mg/kg/day) without alcohol
did not significantly affect serum ADH or ALDH activity compared
with the Control group.
Effects of cilostazol on liver
hydroxyproline levels
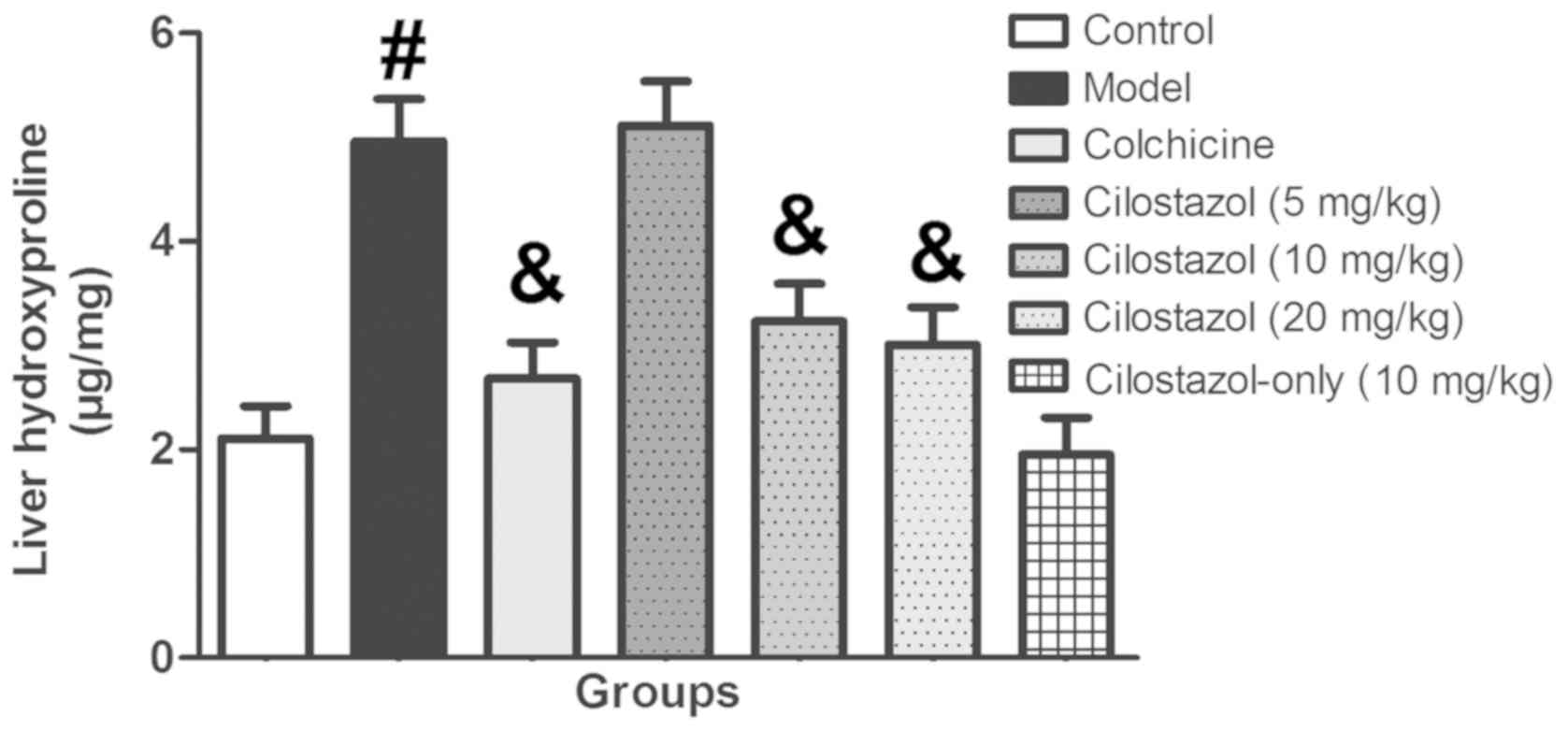
Fig. 2 presents the
liver hydroxyproline levels. Liver hydroxyproline levels in the
Model group were significantly increased compared with the Control
group (P<0.05). Colchicine treatment significantly recovered
liver hydroxyproline levels compared with the Model group
(P<0.05). Cilostazol treatment (5 mg/kg/day) did not
significantly alter liver hydroxyproline levels, but 10 and 20
mg/kg/day cilostazol significantly decreased these levels compared
with the Model group (P<0.05). Cilostazol treatment (10
mg/kg/day) without alcohol did not significantly alter liver
hydroxyproline levels compared with the control group.
Effects of cilostazol on serum A/G
ratio, protein, enzyme, HA, LN, IV-C and PCIII levels
Table I presents the
effects of cilostazol on the serum A/G ratio. Alcohol treatment
reduced the serum A/G ratio from 1.08±0.11 to 0.63±0.08
(P<0.05). Colchicine and 10 and 20 mg/kg/day cilostazol
significantly increased the serum A/G ratio to 0.94±0.12, 0.86±0.08
and 0.91±0.07, respectively (P<0.05 vs. Model group). Cilostazol
treatment (10 mg/kg/day) without alcohol did not significantly
alter serum A, G or A/G levels. Table
II presents the levels of serum protein and enzymes (TP, TBIL,
ALT, AST, AKP and γ-GT). Alcohol treatment significantly increased
serum levels of TP, TBIL, ALT, AST, AKP and γ-GT compared with the
Control group (P<0.05). Colchicine and 10 and 20 mg/kg/day
cilostazol significantly inhibited the increase in these proteins
and enzymes compared with the Model group (all P<0.05).
Treatment with 10 mg/kg/day cilostazol without alcohol did not
significantly alter serum protein or enzymes compared with the
Control group. Table III presents
the serum levels of HA, LN, IV-C and PCIII in rats. Alcohol
treatment significantly increased these indicators of AHF compared
with the Control group (all P<0.05). However, co-treatment with
colchicine or 10 mg/kg/day and 20 mg/kg/day cilostazol
significantly decreased these indicators compared with the Model
group (all P<0.05). Treatment with cilostazol (10 mg/kg/day)
without alcohol did not significantly alter serum levels of HA, LN,
IV-C or PC III compared with the Control group.
 | Table I.Effects of Cilostazol on serum
albumin, globulin and A/G in rats. |
Table I.
Effects of Cilostazol on serum
albumin, globulin and A/G in rats.
| Measurement | Control | Model | Colchicine | Cilostazol (5
mg/kg) | Cilostazol (10
mg/kg) | Cilostazol (20
mg/kg) | Cilostazol-only (10
mg/kg) |
|---|
| Albumin (g/l) | 41.2±6.5 | 40.3±5.5 | 40.6±5.4 | 41.6±4.3 | 40.7±4.7 | 41.1±3.9 | 40.3±5.4 |
| Globulin (g/l) | 38.2±4.2 | 63.5±4.8 | 43.1±4.9 | 60.3±5.1 | 47.6±3.9 | 45.1±4.3 | 39.9±4.9 |
| A/G | 1.08±0.11 |
0.63±0.08a |
0.94±0.12b | 0.69±0.07 |
0.86±0.08b |
0.91±0.07b | 1.12±0.15 |
| Table II.Serum levels of protein and enzymes
in rats. |
Table II.
Serum levels of protein and enzymes
in rats.
| Measurement | Control | Model | Colchicine | Cilostazol (5
mg/kg) | Cilostazol (10
mg/kg) | Cilostazol (20
mg/kg) | Cilostazol-only (10
mg/kg) |
|---|
| TP (g/l) | 70.6±6.9 |
88.3±7.1a |
72.1±6.8b | 85.4±7.5 |
75.2±5.9b |
71.0±6.6b | 66.7±5.7 |
| TBIL (µmol/l) | 1.7±0.4 |
7.8±1.3a |
3.2±0.5b | 7.6±1.1 |
3.8±0.3b |
3.5±0.6b | 1.6±0.5 |
| ALT (U/l) | 50.3±10.5 |
154.5±20.6a |
80.9±21.3b | 139.6±22.6 |
92.3±23.4b |
87.8±21.8b | 55.2±8.9 |
| AST (U/l) | 132.5±23.3 |
254.6±36.6a |
166.8±35.9b | 241.6±30.8 |
195.7±34.2b |
184.2±31.3b | 125.5±21.4 |
| AKP (U/l) | 135.4±36.5 |
251.2±35.2a |
157.4±26.5b | 239.2±23.5 |
188.1±25.5b |
167.4±34.2b | 132.9±33.7 |
| γ-GT (U/l) | 3.5±1.2 |
21.2±2.4a |
6.5±2.1b | 19.9±2.7 |
10.3±3.1b |
8.2±2.8b | 3.4±1.3 |
| Table III.Serum levels of HA, LN, IV-C and
PCIII in rats. |
Table III.
Serum levels of HA, LN, IV-C and
PCIII in rats.
| Measurement | Control | Model | Colchicine | Cilostazol (5
mg/kg) | Cilostazol (10
mg/kg) | Cilostazol (20
mg/kg) | Cilostazol-only (10
mg/kg) |
|---|
| HA | 201.6±32.3 |
451.3±34.5a |
233.4±36.1b | 457.8±39.1 |
286.4±32.2b |
259.3±28.7b | 195.6±23.4 |
| LN | 33.1±2.8 |
56.9±3.8a |
38.9±4.1b | 58.7±4.5 |
44.6±5.1b |
40.5±3.4b | 32.6±3.1 |
| IV-C | 18.4±3.1 |
39.1±3.5a |
21.3±2.7b | 38.7±3.4 |
25.2±4.4b |
23.3±3.7b | 17.9±2.6 |
| PCIII | 47.6±10.3 |
210.7±32.4a |
103.8±10.9b | 203.9±24.5 |
169.8±13.6b |
150.6±16.8b | 45.8±9.5 |
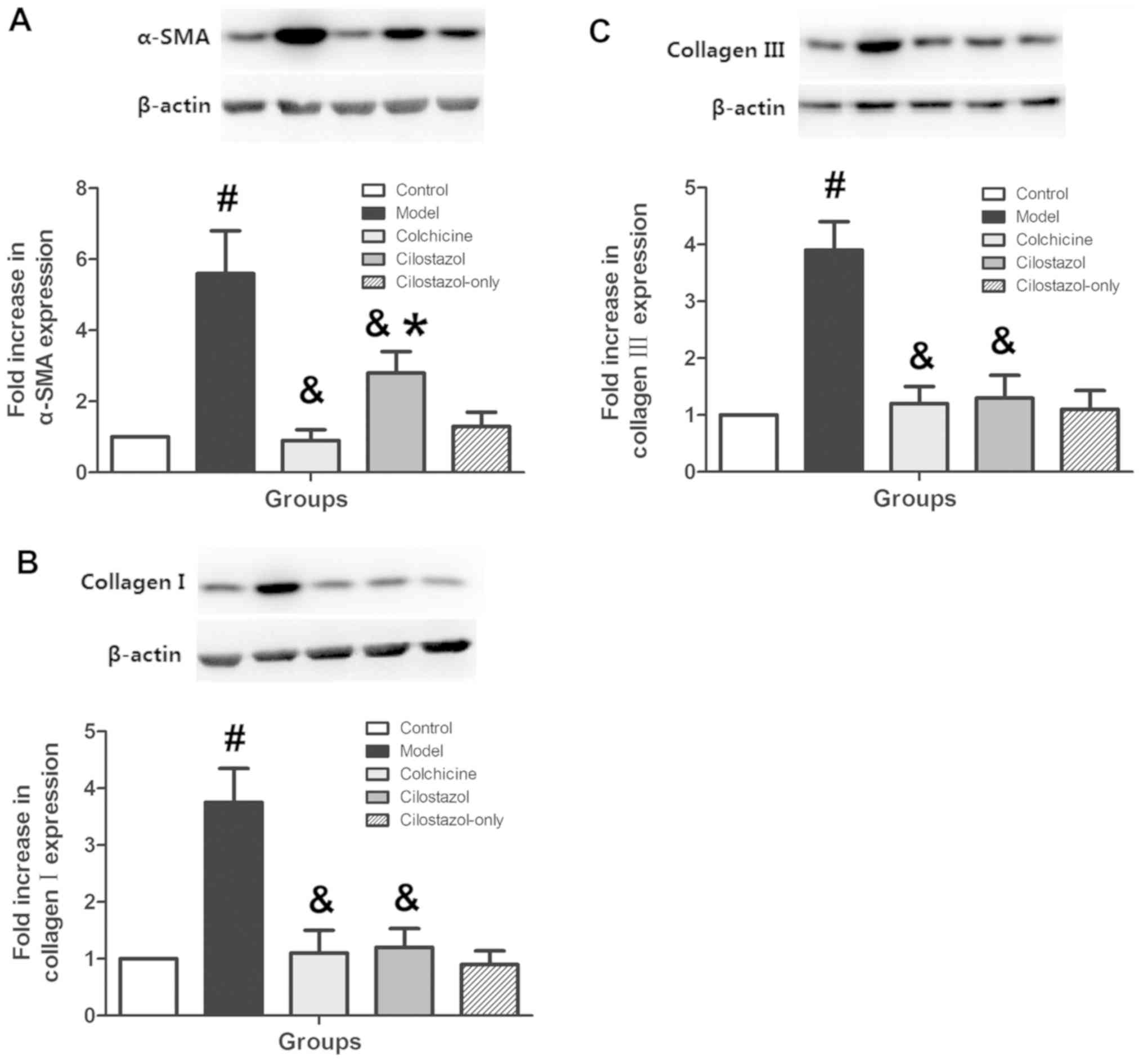
Effects of cilostazol on liver α-SMA
and collagen III and I expression
α-SMA and collagen III and I expression were
measured in liver tissue using western blotting to confirm the
effects of cilostazol on AHF. Protein levels of α-SMA and collagen
III and I increased significantly in the Model group compared with
the Control group (P<0.05; Fig.
3). Colchicine (0.1 mg/kg/day) and cilostazol (10 mg/kg/day)
significantly decreased the levels of these proteins compared with
the Model group (P<0.05). Treatment with cilostazol (10
mg/kg/day) without alcohol did not significantly alter liver α-SMA
or collagen III and I expression compared with controls.
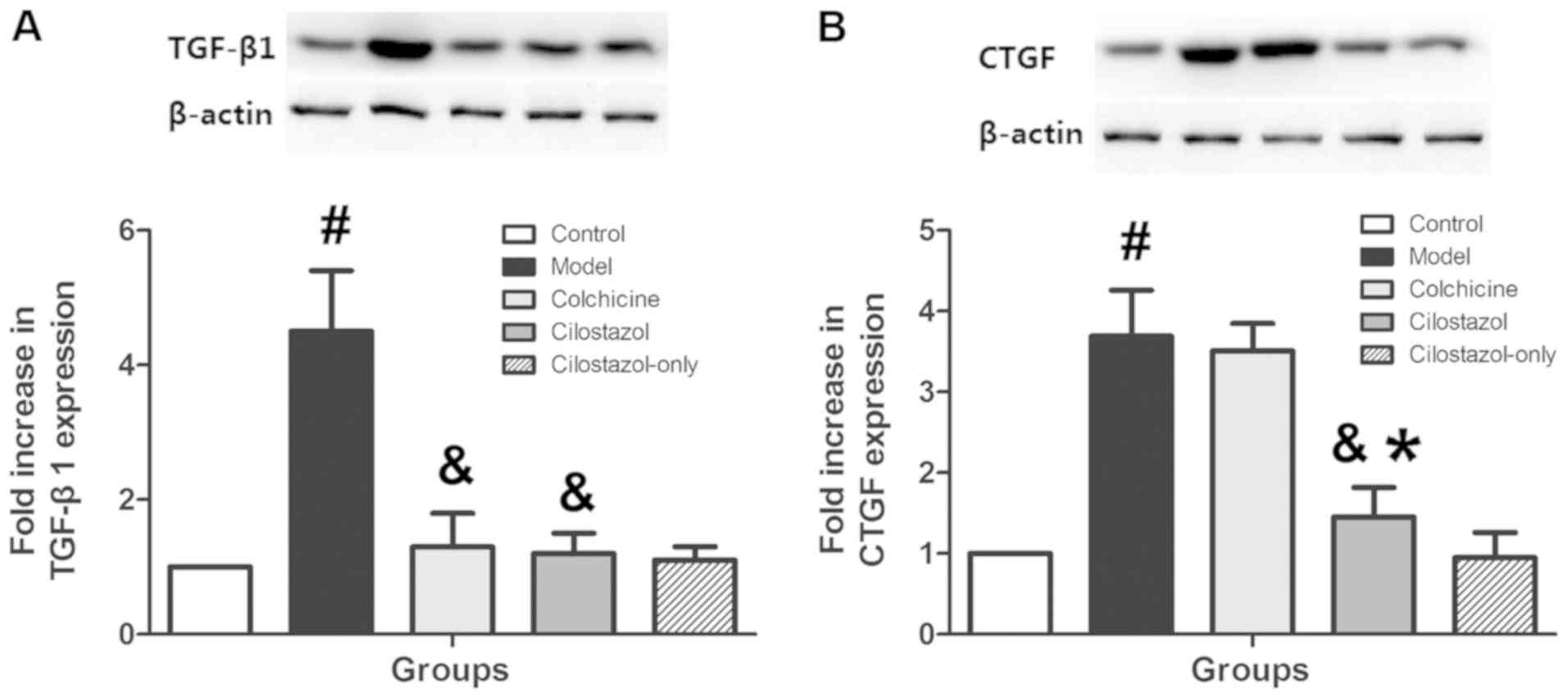
Effects of cilostazol on liver TGF-β1,
CTGF, Epac1/2 and cAMP levels
TGF-β1, CTGF and Epac1/2 expression were measured in
liver tissue using western blotting, and cAMP levels were measured
using ELISA to investigate the mechanisms of cilostazol on AHF.
TGF-β1 expression was significantly increased in the Model group
compared with the Control group (P<0.05), and colchicine (0.1
mg/kg/day) and cilostazol (10 mg/kg/day) significantly inhibited
this increase compared with the Model group (P<0.05; Fig. 4A). CTGF expression was significantly
increased in the Model group compared with the Control group
(P<0.05), and 10 mg/kg/day cilostazol significantly inhibited
this increase (P<0.05 vs. Model group; Fig. 4B). Colchicine (0.1 mg/kg/day) did not
significantly alter CTGF expression compared with the Model group.
Treatment with cilostazol (10 mg/kg/day) without alcohol did not
significantly affect liver TGF-β1 or CTGF expression compared with
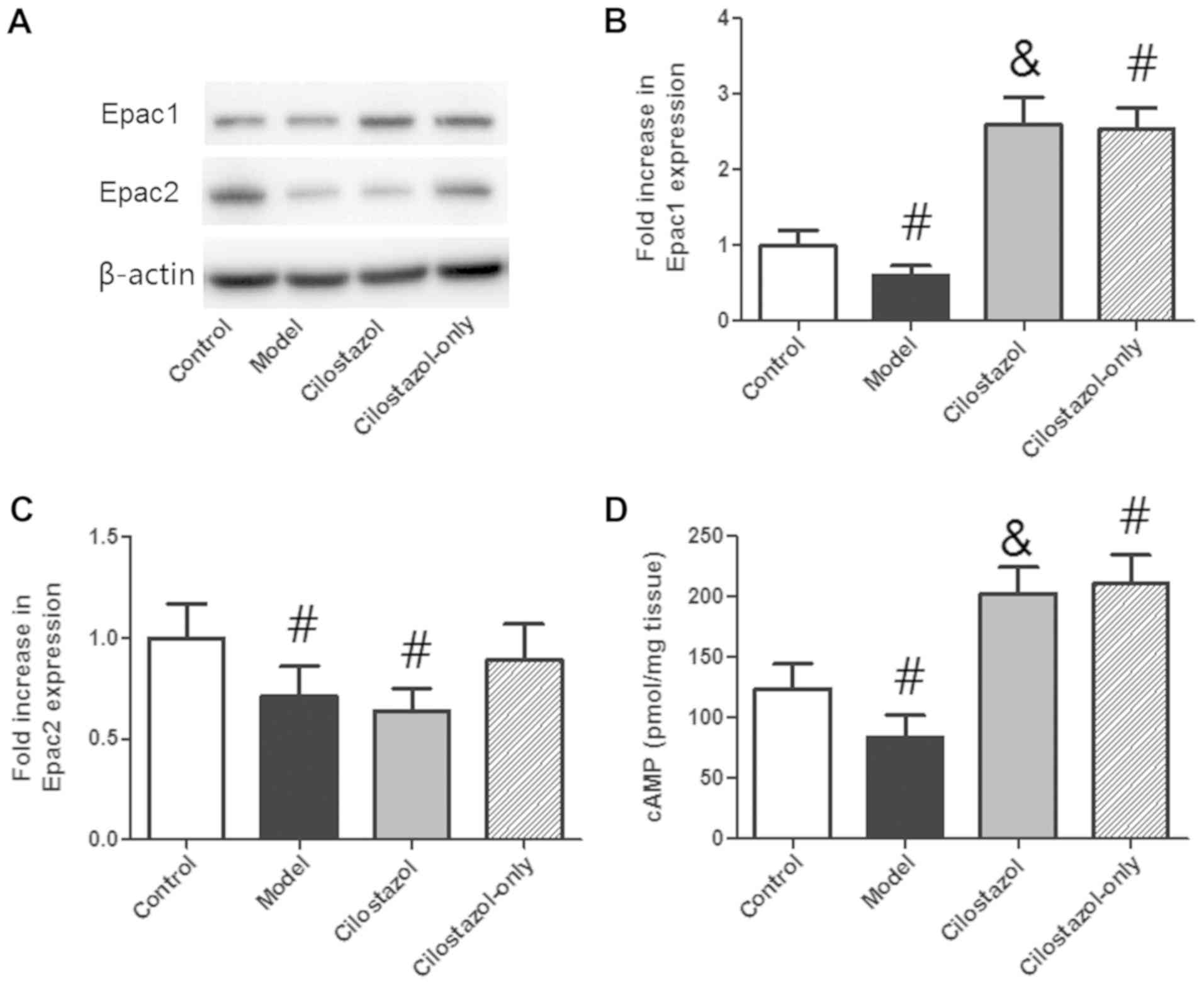
the Control group. Epac1 expression was decreased significantly in
the Model group compared with the Control group (P<0.05), and
cilostazol greatly enhanced Epac1 expression (P<0.05 compared
with the Model group; Fig. 5A and
B). Cilostazol treatment without alcohol also significantly
increased Epac1 expression compared with the Control group
(P<0.05). Epac2 expression was significantly decreased in the
Model and Cilostazol groups compared with the Control group
(P<0.05; Fig. 5A and C). There
was no significant difference between the Model and Cilostazol
groups. Cilostazol treatment without alcohol did not significantly
alter Epac2 expression compared with the Control group. cAMP levels
were significantly decreased in the Model group compared with the
Control group (P<0.05; Fig. 5D),
and cilostazol significantly enhanced these levels (P<0.05 vs.
Model group). Cilostazol treatment without alcohol also
significantly increased cAMP levels compared with the Control group
(P<0.05; Fig. 5D).
Discussion
Chronic alcohol consumption produces many harmful
consequences, and liver failure is one of the most serious effects.
Chronic alcohol consumption may lead to fatty liver disease,
alcoholic hepatitis and alcoholic liver fibrosis (15). Liver fibrosis is an important public
health concern because of its high morbidity and mortality
(16). The present study examined
the effects and mechanisms of the phosphodiesterase III inhibitor
cilostazol, which is clinically used to treat lower extremity
peripheral arterial disease, on alcohol-induced liver fibrosis. The
results demonstrated that in alcohol-treated rats, the levels of
liver hydroxyproline, α-SMA, collagen III and collagen I, and serum
levels of HA, LN, IV-C and PCIII in rats were all significantly
increased. These data indicated that the model of AHF was
successful. Cilostazol significantly increased serum ADH and ALDH
activities and decreased liver hydroxyproline levels. Cilostazol
increased the serum A/G ratio and inhibited serum TP, TBIL, ALT,
AST, AKP and γ-GT, HA, LN, IV-C and PCIII levels. Western blotting
revealed that cilostazol effectively decreased α-SMA, collagen III
and I, TGF-β1 and CTGF expression in the liver. Cilostazol
significantly increased Epac1 expression and cAMP level in liver
tissue.
The development of AHF is associated with the
oxidation of alcohol to acetaldehyde, which stimulates the
production of extracellular matrix (ECM) components, such as type I
collagen, via HSC activation (17).
The activation of HSCs is a milestone event in the development of
AHF as these cells are the primary source of ECM in the response of
the liver to alcohol consumption (18). HSCs transform to myofibroblast-like
cells, proliferate and eventually become fibrogenic (19). Alcohol is metabolized via various
catabolic metabolic pathways. ADH is the primary enzyme that
oxidizes alcohol to acetaldehyde (20), which is converted to acetate via ALDH
(21). The present study treated
rats with alcohol for 24 weeks and demonstrated a significant
decrease in serum ADH and ALDH activities in the Model group. This
decrease suggests that alcohol was deposited in the liver tissue
and induced hepatotoxicity over time. Colchicine or cilostazol
administration with alcohol significantly recovered serum ADH and
ALDH activities. These results suggest that cilostazol enhances ADH
and ALDH activities to accelerate alcohol metabolism and protect
the liver from alcohol assault.
Chronic alcohol assault also promotes the release of
cytosolic proteins and enzymes, such as TBIL, ALT, AST, AKP and
γ-GT, into the circulation (4). The
present study demonstrated that alcohol treatment significantly
increased serum levels of TP, TBIL, ALT, AST, AKP and γ-GT.
However, colchicine and 10 and 20 mg/kg/day cilostazol
significantly inhibited the release of these protein and enzymes.
Liver hydroxyproline level increased significantly in the Model
group, and 10 and 20 mg/kg/day cilostazol treatment significantly
decreased these levels. Cilostazol significantly increased the
serum A/G ratio. Cilostazol also significantly inhibited the
increased levels of serum HA, LN, IV-C and PCIII, which are
indicators of liver fibrogenesis. These results suggest that
cilostazol effectively inhibited biomarkers of liver fibrogenesis
and collagen deposition. This mechanism of action may delay the
progression of hepatic fibrosis and alleviate hepatic injury.
α-SMA is a marker of HSC transformation into
myofibroblast-like cells and the secretion of fibrillar collagens
(collagen III and I) (5). α-SMA and
collagen III and I expression were measured in liver tissue using
western blotting to confirm the effect of cilostazol on AHF. The
protein levels of α-SMA and collagen III and I expression increased
significantly in the Model group. Rats treated with 0.1 mg/kg/day
colchicine or 10 mg/kg/day cilostazol exhibited significantly
decreased levels. These results suggest that cilostazol prevents
initiation of the fibrotic process and synthesis of excessive
connective tissue components.
CTGF is a cysteine-rich peptide that is synthesized
and secreted by fibroblastic cells following TGF-β activation. CTGF
is a downstream mediator of TGF-β-induced fibroblast proliferation
(22). Previous studies demonstrated
an upregulation of CTGF expression in numerous fibrotic diseases,
such as atherosclerosis, pancreas, kidney, and liver fibrosis
(23,24). Duncan et al (22) demonstrated that CTGF mediated
TGF-β-induced fibroblast collagen synthesis, and the inhibition of
CTGF synthesis prevented granulation tissue formation via the
inhibition of collagen synthesis and fibroblast accumulation.
TGF-β1 and CTGF expression were measured in liver tissue to further
investigate the mechanism of cilostazol on AHF. TGF-β1 expression
increased significantly following 24 weeks of alcohol
administration, and 0.1 mg/kg/day colchicine and 10 mg/kg/day
cilostazol significantly inhibited this increase. CTGF expression
was also significantly increased in the Model group, and 10
mg/kg/day cilostazol significantly inhibited this increase.
Colchicine (0.1 mg/kg/day) did not alter CTGF expression. These
results reveal that the TGF-β1/CTGF pathway is involved in the
protective effects of cilostazol in AHF. Cilostazol inhibited CTGF
expression, and the positive control treatment, colchicine, did not
affect expression.
The effects of cilostazol were measured on cAMP
level and Epac1/2 expression in liver tissues to further examine
the association between cilostazol and TGF-β1/CTGF signaling in the
antifibrotic action of cilostazol. Previous studies have reported
that the cAMP/Epac1/2 pathway served a key role in the effect of
cilostazol in other tissues and cells including, bone, aortic
endothelial cells and progenitor cells (25–27). A
number of previous studies revealed the importance of Epac1 and
Epac2 as mediators of the antifibrotic effects of cAMP (28–30).
Epac1 and Epac2 exhibit different cAMP-binding sites. Increased
cAMP restrains fibroblast function to exert its anti-fibrotic
effects. The mechanisms of these effects include fibroblast
proliferation inhibition, fibroblast death and ECM protein
synthesis inhibition (31).
Activation of Epac1 or Epac2 in different tissues inhibits
profibrotic actions in the body, such as collagen and DNA synthesis
(31). The present results
demonstrated that Epac1 expression and cAMP level decreased
significantly in the Model group, and cilostazol greatly enhanced
this reduction. However, cilostazol did not significantly alter
Epac2 expression. These results indicate that cilostazol
effectively activates the cAMP/Epac1 signaling pathway in liver
tissue. Multiple studies previously established the association
between cAMP and TGF-β1/CTGF. Huang et al (32) demonstrated that liraglutide improved
myocardial fibrosis following myocardial infarction via inhibition
of CTGF and activation of cAMP in mice. Weng et al (33) revealed that high expression of TGF-β1
and its downstream pathways in MDCK cells produced a significant
and negative effect of cAMP-PKA on TGF-β1-induced p-ERK1/2 and FN
expression. Satish et al (34) also demonstrated that increasing cAMP
levels potentially inhibited myofibroblast formation and the
accumulation of ECM components via inhibition of TGF-β1 stimulation
of α-SMA, CTGF, and collagen I and III. Therefore, it is reasonable
to hypothesize that cilostazol inhibits TGF-β1/CTGF expression via
activation of the cAMP/Epac1 signaling pathway in liver tissue to
suppress hepatic fibrosis development.
The present study demonstrated that cilostazol
protected rats against AHF via suppression of TGF-β1/CTGF
activation. Further studies are required to confirm the exact
mechanisms, but these results provide a novel potential strategy to
prevent AHF and associated liver injury. The present study focused
on the preventive effects of cilostazol on AHF. However, whether
cilostazol reverses AHF is not clear. The therapeutic effect of
cilostazol on AHF requires further study.
Acknowledgements
Not applicable.
Funding
The present study was funded by Dr Kun Han
(Department of Gastroenterology, Xi'an Central Hospital (Shaanxi,
China).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KH designed the study and prepared the manuscript.
YZ performed the experiments. ZY collected and analyzed the
data.
Ethics approval and consent to
participate
The present study was approved by the Xi'an Central
Hospital's Institutional Animal Care and Use Committee (Xi'an,
China; approval number: XCH-20170923).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lin X, Zhang S, Huang R, Wei L, Tan S,
Liang S, Tian Y, Wu X, Lu Z and Huang Q: Helenalin attenuates
alcohol-induced hepatic fibrosis by enhancing ethanol metabolism,
inhibiting oxidative stress and suppressing HSC activation.
Fitoterapia. 95:203–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Duddempudi AT: Immunology in alcoholic
liver disease. Clin Liver Dis. 16:687–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kumar M and Sarin SK: Is cirrhosis of the
liver reversible? Indian J Pediatr. 74:393–399. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jayakumar T, Ramesh E and Geraldine P:
Antioxidant activity of the oyster mushroom, Pleurotus ostreatus,
on CCl(4)-induced liver injury in rats. Food Chem Toxicol.
44:1989–1996. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shyu MH, Kao TC and Yen GC: Hsian-tsao
(Mesona procumbens Heml.) prevents against rat liver fibrosis
induced by CCl(4) via inhibition of hepatic stellate cells
activation. Food Chem Toxicol. 46:3707–3713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ihn H: Pathogenesis of fibrosis: Role of
TGF-beta and CTGF. Curr Opin Rheumatol. 14:681–685. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marcial JM, Pérez R, Vargas P and
Franqui-Rivera H: Non-invasive therapy of peripheral arterial
disease. Bol Asoc Med P R. 107:52–57. 2015.PubMed/NCBI
|
|
8
|
Abdel Kawy HS: Cilostazol attenuates
cholestatic liver injury and its complications in common bile duct
ligated rats. Eur J Pharmacol. 752:8–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saito S, Hata K, Iwaisako K, Yanagida A,
Takeiri M, Tanaka H, Kageyama S, Hirao H, Ikeda K, Asagiri M and
Uemoto S: Cilostazol attenuates hepatic stellate cell activation
and protects mice against carbon tetrachloride-induced liver
fibrosis. Hepatol Res. 44:460–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujita K, Nozaki Y, Wada K, Yoneda M, Endo
H, Takahashi H, Iwasaki T, Inamori M, Abe Y, Kobayashi N, et al:
Effectiveness of antiplatelet drugs against experimental
non-alcoholic fatty liver disease. Gut. 57:1583–1591. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang XH, Yan M, Liu L, Wu TJ, Ma LL and
Wang LX: Expression of discoidin domain receptors (DDR2) in
alcoholic liver fibrosis in rats. Arch Med Res. 41:586–592. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang L, Potter JJ, Rennie-Tankersley L,
Novitskiy G, Sipes J and Mezey E: Effects of retinoic acid on the
development of liver fibrosis produced by carbon tetrachloride in
mice. Biochim Biophys Acta. 1772:66–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bergman I and Loxley R: New
spectrophotometric method for the determination of proline in
tissue hydrolyzates. Anal Chem. 42:702–706. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller RA, Chu Q, Xie J, Foretz M, Viollet
B and Birnbaum MJ: Biguanides suppress hepatic glucagon signalling
by decreasing production of cyclic AMP. Nature. 494:256–260. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Purohit V, Gao B and Song BJ: Molecular
mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res.
33:191–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Magdaleno F, Blajszczak CC and Nieto N:
Key events participating in the pathogenesis of alcoholic liver
disease. Biomolecules. 7(pii): E92017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mello T, Ceni E, Surrenti C and Galli A:
Alcohol induced hepatic fibrosis: Role of acetaldehyde. Mol Aspects
Med. 29:17–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo Z, Liu H, Sun X, Guo R, Cui R, Ma X
and Yan M: RNA interference against discoidin domain receptor 2
ameliorates alcoholic liver disease in rats. PLoS One.
8:e558602013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jelski W, Orywal K and Szmitkowski M:
Effects of H2-blockers on alcohol dehydrogenase (ADH) activity. Pol
Merkur Lekarski. 25:531–533. 2008.(In Polish). PubMed/NCBI
|
|
21
|
Sung CK, Kim SM, Oh CJ, Yang SA, Han BH
and Mo EK: Taraxerone enhances alcohol oxidation via increases of
alcohol dehyderogenase (ADH) and acetaldehyde dehydrogenase (ALDH)
activities and gene expressions. Food Chem Toxicol. 50:2508–2514.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Duncan MR, Frazier KS, Abramson S,
Williams S, Klapper H, Huang X and Grotendorst GR: Connective
tissue growth factor mediates transforming growth factor
beta-induced collagen synthesis: Down-regulation by cAMP. FASEB J.
13:1774–1786. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y
and Leask A: Tumor necrosis factor alpha suppresses the induction
of connective tissue growth factor by transforming growth
factor-beta in normal and scleroderma fibroblasts. J Biol Chem.
275:15220–15225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Blom IE, Sa S, Goldschmeding R,
Abraham DJ and Leask A: CTGF expression in mesangial cells:
Involvement of SMADs, MAP kinase, and PKC. Kidney Int.
62:1149–1159. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hashimoto A, Tanaka M, Takeda S, Ito H and
Nagano K: Cilostazol induces PGI2 production via activation of the
downstream Epac-1/Rap1 signaling cascade to increase intracellular
calcium by PLCε and to activate p44/42 MAPK in human aortic
endothelial cells. PLoS One. 10:e01328352015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee DH, Lee HR, Shin HK, Park SY, Hong KW,
Kim EK, Bae SS, Lee WS, Rhim BY and Kim CD: Cilostazol enhances
integrin-dependent homing of progenitor cells by activation of
cAMP-dependent protein kinase in synergy with Epac1. J Neurosci
Res. 89:650–660. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ke K, Safder AM, Sul OJ, Suh JH, Joe Y,
Chung HT and Choi HS: Cilostazol attenuates ovariectomy-induced
bone loss by inhibiting osteoclastogenesis. PLoS One.
10:e01248692015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gloerich M and Bos JL: Epac: Defining a
new mechanism for cAMP action. Annu Rev Pharmacol Toxicol.
50:355–375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grandoch M, Roscioni SS and Schmidt M: The
role of Epac proteins, novel cAMP mediators, in the regulation of
immune, lung and neuronal function. Br J Pharmacol. 159:265–284.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Breckler M, Berthouze M, Laurent AC,
Crozatier B, Morel E and Lezoualc'h F: Rap-linked cAMP signaling
Epac proteins: Compartmentation, functioning and disease
implications. Cell Signal. 23:1257–1266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Insel PA, Murray F, Yokoyama U, Romano S,
Yun H, Brown L, Snead A, Lu D and Aroonsakool N: cAMP and Epac in
the regulation of tissue fibrosis. Br J Pharmacol. 166:447–456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang DD, Huang HF, Yang Q and Chen XQ:
Liraglutide improves myocardial fibrosis after myocardial
infarction through inhibition of CTGF by activating cAMP in mice.
Eur Rev Med Pharmacol Sci. 22:4648–4656. 2018.PubMed/NCBI
|
|
33
|
Weng L, Wang W, Su X, Huang Y, Su L, Liu
M, Sun Y, Yang B and Zhou H: The effect of cAMP-PKA activation on
TGF-β1-induced profibrotic signaling. Cell Physiol Biochem.
36:1911–1927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Satish L, Gallo PH, Baratz ME, Johnson S
and Kathju S: Reversal of TGF-β1 stimulation of α-smooth muscle
actin and extracellular matrix components by cyclic AMP in
Dupuytren's-derived fibroblasts. BMC Musculoskelet Disord.
12:1132011. View Article : Google Scholar : PubMed/NCBI
|