Introduction
Cardiac fibrosis is characterized by the excessive
proliferation of interstitial fibroblasts and excessive deposition
of extracellular matrix. It is a common pathophysiologic mechanism
during the development of various cardiovascular diseases such as
atrial fibrillation, hypertensive heart disease, myocardial
infarction and valvular heart diseases. Cardiac fibrosis has a key
role in ventricular remodeling. The main pathological
manifestations of cardiac fibrosis is myocardial stiffness
increase, myocardial systolic and diastolic dysfunction, and
eventually leading to heart failure and sudden death (1,2). The
current treatments available for cardiac fibrosis are not highly
specific and often have many side effects. Therefore, a novel
potential therapeutic agent for cardiac fibrosis is needed.
Acetazolamide is a carbonic anhydrase inhibitor,
which is mainly applied for correct metabolic alkalosis (3) and edematous diseases such as COPD
(4), cerebral edema (5) and chronic heart failure (6). Moreover, Li et al (7) also reported that acetazolamide could
suppress tumor angiogenesis and metastasis in a Lewis lung
carcinoma mouse model. Recently, Lin et al (8) reported that acetazolamide could enhance
the cardioprotective effect of remifentanil in a rat model of
myocardial ischemia/reperfusion injury. However, the effect of
acetazolamide on cardiac fibrosis has not yet been confirmed. We
hypothesized that acetazolamide may have potential usefulness in
attenuating cardiac fibrosis. In this study, we created a mouse
model of pressure overload induced by aortic constriction to
investigate the effect of acetazolamide on cardiac fibrosis and the
potential molecular mechanism.
Materials and methods
Reagents
Acetazolamide was purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). The rabbit anti-α-SMA, collagen
I, TGF-β1 and Smad2 primary antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA).
Ethics statement
Male C57BL/6 mice (8–10 weeks old) were provided by
the Animal Experiment Center of Affiliated Hospital of Jining
Medical University (Jining, China). All aspects of the experimental
protocols were approved by the Animal Care and Use Committee of
Affiliated Hospital of Jining Medical University and conducted in
accordance with the Guide for the Care and Use of Laboratory
Animals, published by the US National Institutes of Health (NIH
Publication no. 85-23, revised 1996). The mice were housed in a
temperature controlled room (21±2°C) with a relative humidity range
of 30 to 40% on a 12:12-h light/dark cycle (lights on at 06:00).
All rats had free access to water and food.
Animal model of pressure overload
The mice were anesthetized with an initial 4%
isoflurane followed by a maintenance dose of 2% isoflurane, then
intubated and ventilated. A midline incision was made at the
sternum. After opening the mediastinal space, the aortic arch was
blunt dissected at the base of the heart. A blunt 27-G injection
needle (OD 0.4 mm) was placed parallel to the aorta between the
left carotid and the right innominate arteries, then the needle and
the aortic arch were tied together using a 7-0 suture. After
removing the needle, a model of aortic constriction was created.
Sham mice underwent the same surgical procedure, the 7-0 suture was
placed in the same position without ligation. After transverse
aortic constriction (TAC) or sham operation, the mice were orally
gavaged with acetazolamide (20 mg/kg/day). There are four groups in
this experiment: i) sham group; ii) sham+acetazolamide group; iii)
TAC group; iv) TAC + ace-tazolamide group, n=10 mice in each group.
After 4 weeks of operation, all mice were sacrificed and the hearts
were harvested. The heart samples were frozen in liquid nitrogen
frozen and then stored at −70°C.
Echocardiography
After 4 weeks of operation, the mice were
anesthetized by isoflurane and the cardiac function was detected
using a rodent animal ultrasonic instrument (Vevo 2100;
VisualSonics, Inc., Toronto, ON, Canada). The interventricular
septum diameter (IVS), left ventricular (LV) posterior wall
thickness (LVPW) and LV ejection fraction (LVEF) were
calculated.
Western blotting
Total proteins were isolated from heart tissues
using a protein extraction kit (Nanjing KeyGen Biotech Co., Ltd.,
Nanjing, China). Total protein concentration was calculated by
bicinchoninic acid (BCA) Protein Assay Kit (Pierce, Rockford, IL,
USA). Gel electrophoresis (10%) was performed to separate the
different molecular weight proteins and then transferred onto
polyvinylidene difluoride membranes. A total of 30 µg proteins were
added into per lane for the electrophoresis. Bull Serum Albumin
(BSA) Blocking buffer (5%) was used as the blocking reagent. The
membrane was incubated with α-SMA, collagen I, TGF-β1,
phospho-Smad2 and Smad2 for overnight at 4°C. After incubation with
the primary antibodies, the membrane was washed in Tris-buffered
saline-tween (TBST) and then incubated with the HRP-conjugated
secondary antibody at room temperature for another 2 h. Rabbit
polyclonal α-SMA antibody (dilution, 1:1,000; cat. no. ab5694);
rabbit monoclonal collagen I antibody (dilution, 1:1,000; cat. no.
ab138492); rabbit monoclonal TGF-β1 antibody (dilution, 1:1,000;
cat. no. ab215715); rabbit monoclonal phospho-Smad2 antibody
(dilution, 1:1,000; cat. no. ab188334); rabbit monoclonal Smad2
antibody (dilution, 1:1,000; cat. no. ab40855); rabbit polyclonal
GAPDH antibody (dilution, 1:1,000; cat. no. ab37168) and secondary
goat anti-rabbit (HRP) IgG antibody (dilution, 1:2,000; cat. no.
ab6721) were all purchased from Abcam (Cambridge, MA, USA).
Immuno-reactive bands were visualized by enhanced chemiluminescence
(ECL) detection kit (Amersham Biosciences, Foster City, CA, USA).
ImageJ software (NIH, Bethesda, MD, USA) was used to measure the
blot signal and density.
Histological assessment of cardiac
fibrosis
The LV tissue samples were fixed in paraformaldehyde
(3.7% in phosphate-buffered saline (PBS), freshly prepared) for 24
h and then embedded in paraffin. LV sections (4–5 µm) were stained
with Masson's trichrome for interstitial fibrosis. The proportion
of the total fibrosis area was observed using a microscope (Nikon,
Tokyo, Japan) and was calculated by ImageJ software (NIH), as the
blue-stained areas divided by the total LV area.
Statistical analysis
SPSS 19.0 software (IBM Corp., Armonk, NY, USA) was
used for statistical analysis. All results were presented as means
± standard deviation (means ± SD). One-way ANOVA followed by post
hoc test (Least Significant Difference) was used to compare the
differences among the different groups. Student's t-test was used
to compare the differences between the two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Acetazolamide attenuates cardiac
dysfunction and interstitial fibrosis induced by TAC
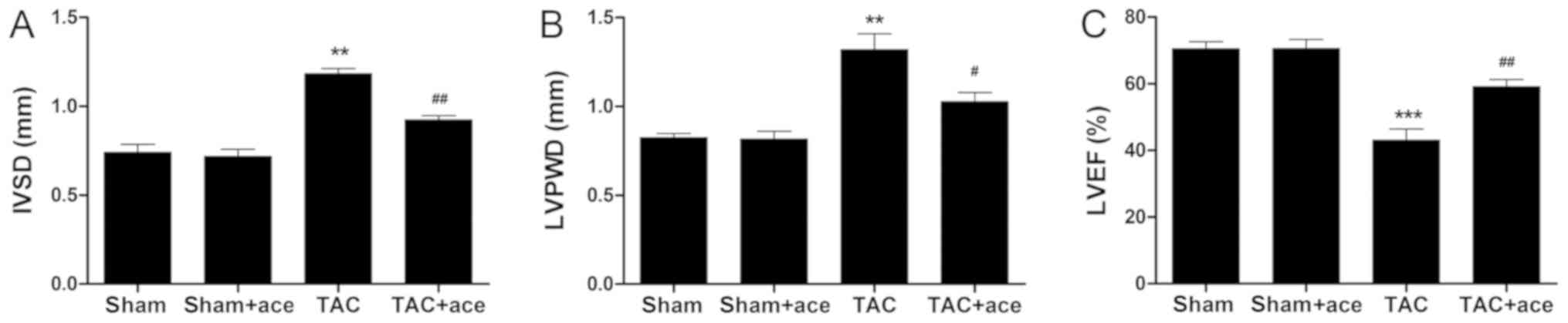
As shown in Fig. 1,
the interventricular septum diastolic dimension (IVSD) and LV
posterior wall thickness diastole (LVPWD) were significantly
thicker in the TAC mice than those in the sham mice (P<0.01,
P<0.01, respectively). Moreover, the LVEF was significantly
decreased in the TAC mice compared with the sham mice (P<0.001).
By contrast, acetazolamide administration significantly decreased
the IVSD and LVPWD, and inhibited the reduction in LVEF induced by
TAC (P<0.01, P<0.05, P<0.01, respectively).
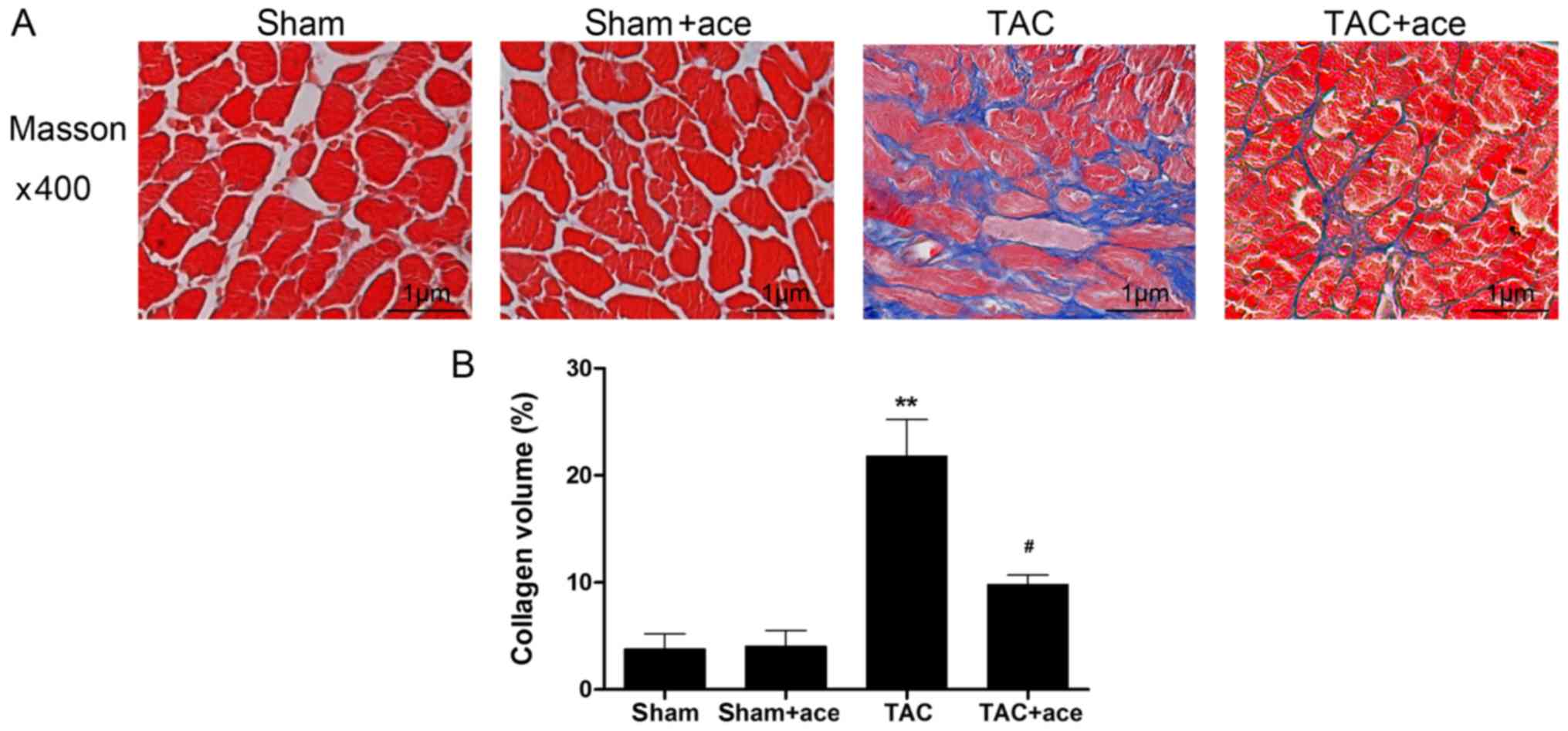
As shown in Fig. 2,
the interstitial collagen volume was substantially increased in the
TAC group compared with the sham group (P<0.01). By contrast,
acetazolamide administration significantly inhibited TAC-induced
interstitial fibrosis (P<0.05).
Acetazolamide inhibits the TAC-induced
increase in the expression of α-SMA and collagen I proteins
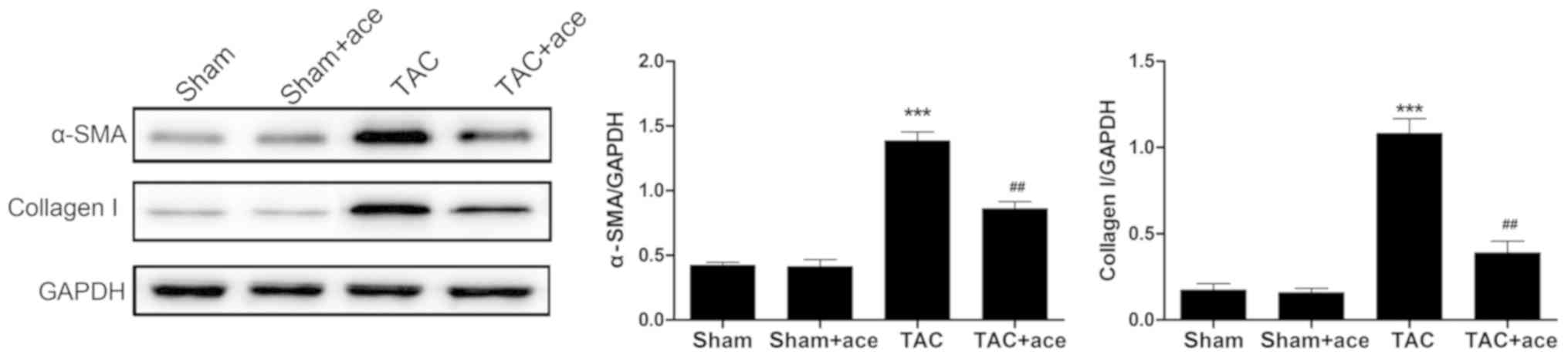
As shown in Fig. 3,
the expression of α-SMA and collagen I proteins were significantly
increased in TAC group compared with the sham group (P<0.001,
P<0.001, respectively). Acetazolamide administration reduced the
expression of α-SMA and collagen I proteins in contrast to the TAC
group (P<0.01, P<0.01, respectively).
Acetazolamide inhibits the activation
of TGF-β1/Smad2 signaling pathway
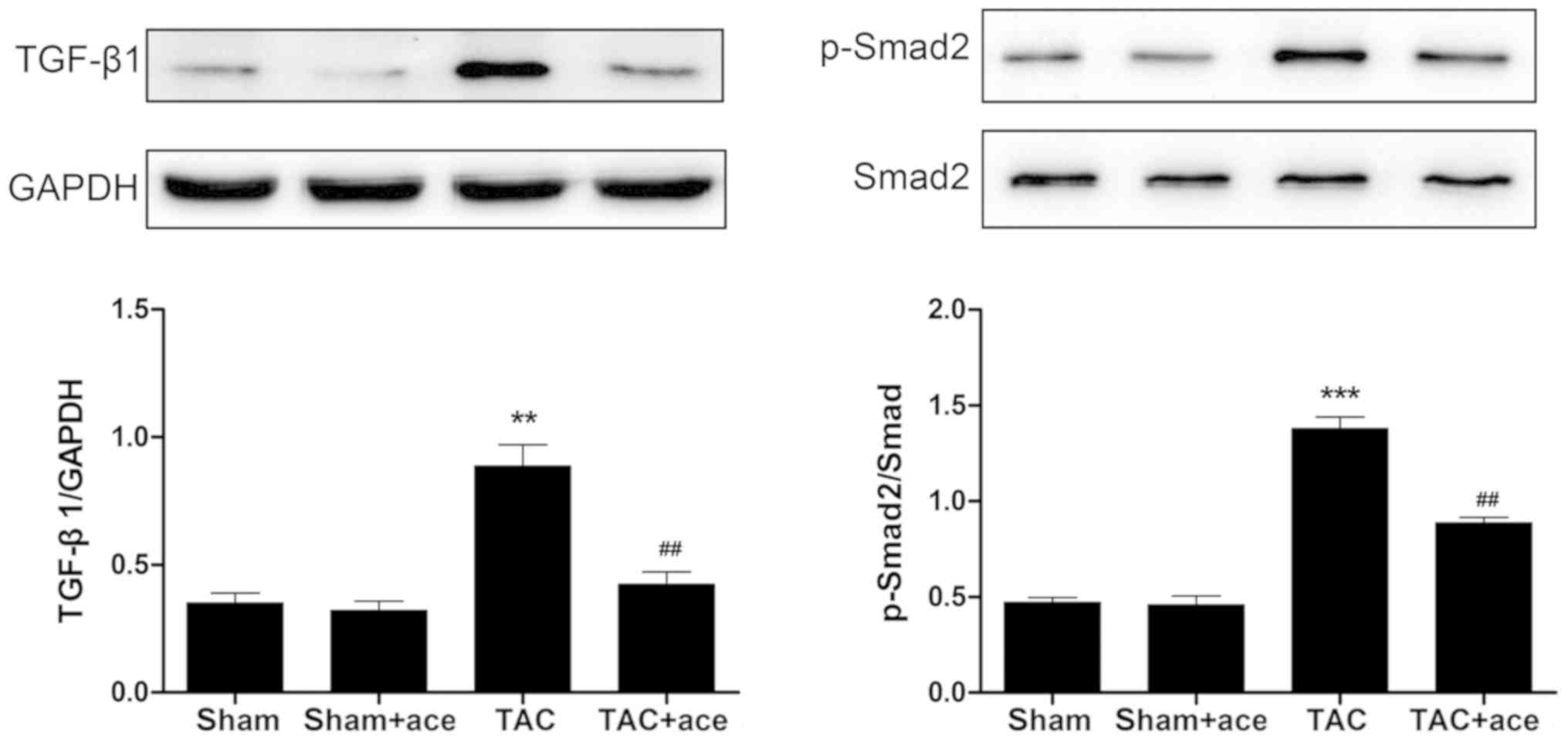
As shown in Fig. 4,
the expression of TGF-β1 and phosphorylation level of Smad2 were
significantly increased in TAC group compared with the sham group
(P<0.01, P<0.001, respectively). Acetazolamide administration
reduced the expression of TGF-β1 and phosphorylation level of Smad2
in contrast to the TAC group (P<0.01, P<0.01,
respectively).
Discussion
To the best of our knowledge, the present study
provides the first report that acetazolamide is able to inhibit
cardiac fibrosis and dysfunction induced by pressure overload in
mice. The anti-fibrotic effect of acetazolamide was confirmed by
the reduction of collagen volume in myocardial interstitium and the
inhibition of α-SMA and collagen I protein expression.
Acetazolamide also inhibited the activation of TGF-β1/Smad2
signaling pathway. These findings support the conclusion that
acetazolamide possibly is a potential therapeutic agent for the
prevention of cardiac fibrosis.
Cardiac fibrosis is an important hallmark during the
development of ventricular remodeling, and is a key pathological
foundation of cardiac dysfunction and malignant cardiovascular
events (2). The development of
cardiac fibrosis is associated with the activation of renin
angiotensin-aldosterone system, oxidative stress and a variety of
cytokines. TGF-β1 is a cytokine that performed a variety of
biological functions including promoting cell proliferation and
differentiation, promoting collagen synthesis, and inhibiting
collagen degradation. It has been demonstrated that TGF-β1 is one
of the most important factors for inducing cardiac fibrosis
(9). During the development of
cardiac fibrosis, activated TGF-β1 can promote cardiac fibroblast
differentiation into myofibroblasts, as reflected by the expression
of α-SMA (10). Rosenkranz et
al (11) reported that the
overexpression of TGF-β1 could induce cardiac fibrosis and
hypertrophy in transgenic mice. Furthermore, in a rat model of
pressure-overload, Kuwahara et al (12) found that TGF-β1 function blocking
could inhibit cardiac fibrosis and dysfunction. Collagen I is
secreted by myofibroblasts and is the most abundant collagen type
in the myocardium, constituting ~80% of the extracellular matrix
(1). The overexpression of collagen
I in transgenic mice displayed significant cardiac fibrosis and
dysfunction (13). Similarly, in
this study, we created a pressure overload model to induce cardiac
fibrosis. Our results showed that the mice displayed significant
cardiac fibrosis after 4 weeks of TAC, as confirmed by Masson
staining and increased collagen volume. The expression of TGF-β1,
α-SMA and collagen I proteins was also markedly increased in the
pressure-overloaded myocardium. Acetazolamide administration
significantly attenuated cardiac fibrosis and inhibited the
expression of TGF-β1, α-SMA and collagen I in the
pressure-overloaded myocardium.
TGF-β1/Smad signaling is the main pathway during the
development of cardiac fibrosis (9,14). The
protein Smads are the key downstream signaling molecules triggered
by TGF-β1 and then induce the expression of pro-fibrotic target
genes (9). Lei et al
(15) reported that Smad2 siRNA
could significantly inhibit TGF-β1-induced fibrotic changes in rat
cardiac fibroblasts. Huang et al (16) also found that Smad3 activation could
induce cardiac fibrosis in a myocardial remodeling model.
Conversely, Smad7 activation could inhibit cardiac fibrosis and
dysfunction induced by angiotensin II (17). Similarly, in this study, we found
that the phosphorylation level of Smad2 was significantly increased
in the TAC mice. Acetazolamide administration reduced the
phosphorylation level of Smad2 in the pressure-overloaded
myocardium. Our results demonstrated that acetazolamide
significantly attenuated cardiac fibrosis and dysfunction induced
by pressure overload through inhibiting the TGF-β1/Smad2 signaling
pathway.
In conclusion, this is the first study to identify
that acetazolamide inhibit the development of cardiac fibrosis. The
molecular mechanism involved in the anti-fibrotic effect of
acetazolamide was possibly through inhibiting TGF-β1/Smad2
signaling pathway. Our results suggest that acetazolamide may be
used as a therapeutic agent for the prevention of cardiac fibrosis.
Further research is needed to investigate the effect and mechanism
of acetazolamide in cardiac fibroblasts in vitro.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QH and RZ designed the study and performed the
experiments. QH, TiW and TaW established the animal models. QH and
TiW collected the data. TiW and TaW analyzed the data. QH and RZ
prepared the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Animal Care and Use
Committee of Affiliated Hospital of Jining Medical University
(Jining, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Creemers EE and Pinto YM: Molecular
mechanisms that control interstitial fibrosis in the
pressure-overloaded heart. Cardiovasc Res. 89:265–272. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bar A, Cies J, Stapleton K, Tauber D,
Chopra A and Shore PM: Acetazolamide therapy for metabolic
alkalosis in critically ill pediatric patients. Pediatr Crit Care
Med. 16:e34–e40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fontana V, Santinelli S, Internullo M,
Marinelli P, Sardo L, Alessandrini G, Borgognoni L, Ferrazza AM,
Bonini M and Palange P: Effect of acetazolamide on post-NIV
metabolic alkalosis in acute exacerbated COPD patients. Eur Rev Med
Pharmacol Sci. 20:37–43. 2016.PubMed/NCBI
|
|
5
|
Bremer AM, Yamada K and West CR: Ischemic
cerebral edema in primates: Effects of acetazolamide, phenytoin,
sorbitol, dexamethasone, and methylprednisolone on brain water and
electrolytes. Neurosurgery. 6:149–154. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Apostolo A, Agostoni P, Contini M,
Antonioli L and Swenson ER: Acetazolamide and inhaled carbon
dioxide reduce periodic breathing during exercise in patients with
chronic heart failure. J Card Fail. 20:278–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li XJ, Xiang Y, Ma B and Qi XQ: Effects of
acetazolamide combined with or without NaHCO3 on
suppressing neoplasm growth, metastasis and aquaporin-1 (AQP1)
protein expression. Int J Mol Sci. 8:229–240. 2007. View Article : Google Scholar
|
|
8
|
Lin PT, Chen WH, Zheng H, Lai ZM and Zhang
LC: Involvement of AQP 1 in the cardio-protective effect of
remifentanil post-conditioning in ischemia/reperfusion rats. Int J
Clin Exp Med. 8:12736–12745. 2015.PubMed/NCBI
|
|
9
|
Dobaczewski M, Chen W and Frangogiannis
NG: Transforming growth factor (TGF)-β signaling in cardiac
remodeling. J Mol Cell Cardiol. 51:600–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu M, Han M, Li J, Xu X, Li T, Que L, Ha
T, Li C, Chen Q and Li Y: 17beta-estradiol inhibits angiotensin
II-induced cardiac myofibroblast differentiation. Eur J Pharmacol.
616:155–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosenkranz S, Flesch M, Amann K, Haeuseler
C, Kilter H, Seeland U, Schlüter KD and Böhm M: Alterations of
beta-adrenergic signaling and cardiac hypertrophy in transgenic
mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol.
283:H1253–H1262. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuwahara F, Kai H, Tokuda K, Kai M,
Takeshita A, Egashira K and Imaizumi T: Transforming growth
factor-beta function blocking prevents myocardial fibrosis and
diastolic dysfunction in pressure-overloaded rats. Circulation.
106:130–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miller AD and Tyagi SC: Mutation in
collagen gene induces cardiomyopathy in transgenic mice. J Cell
Biochem. 85:259–267. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lei B, Hitomi H, Mori T, Nagai Y, Deguchi
K, Mori H, Masaki T, Nakano D, Kobori H, Kitaura Y, et al: Effect
of efonidipine on TGF-β1-induced cardiac fibrosis through
Smad2-dependent pathway in rat cardiac fibroblasts. J Pharmacol
Sci. 117:98–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang XR, Chung AC, Yang F, Yue W, Deng C,
Lau CP, Tse HF and Lan HY: Smad3 mediates cardiac inflammation and
fibrosis in angiotensin II-induced hypertensive cardiac remodeling.
Hypertension. 55:1165–1171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei LH, Huang XR, Zhang Y, Li YQ, Chen HY,
Yan BP, Yu CM and Lan HY: Smad7 inhibits angiotensin II-induced
hypertensive cardiac remodelling. Cardiovasc Res. 99:665–673. 2013.
View Article : Google Scholar : PubMed/NCBI
|