Introduction
Bronchopulmonary dysplasia (BPD) is a chronic lung
disease commonly observed in premature infants and a major
contributor to long-term morbidity and mortality in premature
infants (1). The pulmonary
inflammatory response induced by mechanical ventilation (MV) and
oxygen toxicity (OT) is crucial to the pathogenesis of BPD, which
is demonstrated by an induction of proinflammatory cytokines and an
enhanced influx of neutrophils and macrophages (2–4).
Upregulation of pro-inflammatory cytokines, which include
interleukin (IL)-1β, IL-8, IL-6 and tumor necrosis factor
(TNF)-α are associated with the development of BPD (5). By contrast, patients with BPD exhibited
a decreased expression of anti-inflammatory cytokines and growth
factors, which include PDGF-A and VEGF-A, which are essential to
angiogenesis and alveolar development (6,7).
IL-6 can activate various signaling cascades and is
involved in several processes, which include bone remodeling, acute
inflammation, cellular proliferation, differentiation and cell
death (8,9). Previous studies investigating the
potential functions of IL-6 demonstrated the importance of IL-6
signaling in the development of the submandibular gland (10,11),
mammary gland remodeling (12),
normal prostate development and prostate malignancy (13), and pulmonary maturation (14). Although the amniotic fluid
concentration of IL-6 was significantly increased in mothers whose
premature infants acquired BPD (5),
the functional role of IL-6 in the development of BPD remains
unknown.
Oxygen-induced lung injury is a known risk factor
associated with the development of BPD (15). High and prolonged oxygen exposure in
newborn rodents is commonly used to study the effect of hyperoxia
in lung development (16). Hyperoxic
lung injury (HLI) is initiated by increased levels of reactive
oxygen species, which is followed by the secretion of
proinflammatory chemokines and cytokines by resident macrophages
and epithelial cells (17). The aim
of the current study was to investigate the effect of genetic
ablation of the IL-6 gene on the inflammatory response of HLI in
newborn mice.
Materials and methods
Animals and neonatal hyperoxic
exposure
Mice homozygous for the IL-6 null mutation (total
number, 30; age, 4–6 weeks) and corresponding wild-type (WT)
littermates (total number, 30; age, 4–6 weeks) were obtained from
The Jackson Laboratory (Bar Harbor, ME, USA; all C57BL/6 mice;
weight, 20 g; sex ratio, 1:1). All mice were housed in separate
cages under controlled temperature (21±1°C) and humidity (35±5%)
condition with a 12-h light/dark cycle and access to food and water
ad libitum. C57BL/6 newborn mice were randomized into either
hyperoxia (85% O2) or normoxia (normal air) groups for
<24 h following birth, followed by normoxia conditions for all
animals for the subsequent 4 days. Nursing mice were changed every
day between hyperoxia and normoxia groups to decrease the toxic
effect of oxygen in maternal mice. All animal experiments were
performed at the Fengcheng Hospital (Shanghai, China) according to
the Animal Use Committee of Fengcheng Hospital.
Lung tissue collection
Mice were anesthetized via intraperitoneal injection
of 50 mg/kg sodium pentobarbital (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). The right lung was removed at day 4 and
snap-frozen in liquid nitrogen for subsequent protein and RNA
analysis. Following PBS perfusion of the pulmonary artery, 10%
neutral buffered formalin (Sigma-Aldrich; Merck KGaA) was used to
dilate the left lung through the trachea (pressure, 25 cm
H2O) and fixed for 1 min. The trachea was tied and the
lungs were removed and fixed in 37% formalin overnight at 4°C. Lung
tissue samples were subsequently embedded in paraffin and cut into
5 µm-thick sections and mounted onto glass slides.
Lung histology and morphometry
Lungs (n=6-8/group; hyperoxia or normoxia) were
intratracheally fixed with 4% buffered paraformaldehyde
(Sigma-Aldrich; Merck KGaA) overnight at 4°C and subsequently
embedded in paraffin. Following paraffin embedding and random
sectioning, hematoxylin and eosin staining at room temperature for
1 h was used to quantitatively examine the alveolar region and
walls (septal density) in two-three randomly selected lung tissue
sections (4 µm) from each mouse. All analyses were performed using
a computer-based system (CAST-Grid 2.1.5; Olympus, Ballerup,
Denmark). Radial alveolar count (RAC) was examined in <30 fields
of view in two-three independent random tissue sections from each
mouse (18).
Terminal deoxynucleotidyl-transferase-mediated dUTP
nick end labeling (TUNEL) analysis of the lung tissue sections was
performed using the In Situ Apoptosis Detection kit (Takara
Bio, Inc., Otsu, Japan), according to the manufacturer's protocol.
The cells were fixed in 37% formalin overnight at 4°C followed by
staining with hematoxylin for 30 min at room temperature. Following
hyperoxia exposure for 4 days, the number of TUNEL-positive cells
in the pulmonary parenchyma from each mouse was examined in six
randomly selected fields under a light microscope. Vectashield
antifade mounting medium (Vector Laboratories, Inc., Burlingame,
CA, USA) was used for mounting. Five mice were used in the control
and experimental groups at each time point. Pulmonary morphology
observation was excluded from fields containing cutting defects,
conducting airways and large arteries or veins.
Protein extraction and western blot
analysis
Following hyperoxia exposure for 4 days, lungs
(n=4/group) were removed as described above and weighed. Lung
tissue samples were stored at −80°C prior to subsequent analysis.
Total protein was extracted from lung tissue samples using Halt™
Protease Inhibitor Cocktail (100X; cat. no. 1861280; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and high urea buffer (KPO4,
Urea, AppliChem, Darmstadt, Germany). Total protein was quantified
using a bicinchoninic acid assay (cat. no. 23227; Pierce; Thermo
Fisher Scientific, Inc.) and 100 µg protein/lane was separated via
SDS-PAGE on 10% gel. The separated protein was transferred onto
polyvinylidene fluoride membranes using a Bio-Rad trans-blot SD
semi-dry transfer cell (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) and blocked for 2 h at room temperature with blocking buffer
containing 5% skimmed milk. Subsequently, the membranes were
incubated with primary antibodies against β-actin (1:500; cat. no.
SC-8432; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), cleaved
caspase-8 (cat. no. 3259-100; BioVision Inc., Milpitas, CA, USA),
cleaved caspase-6 (cat. no. 9761S) or cleaved caspase-3 (cat. no.
9661; both Cell Signaling Technology, Inc., Danvers, MA, USA)
overnight at 4°C. Following primary incubation, membranes were
incubated for 1 h at room temperature with either goat anti-mouse
or donkey anti-goat horseradish peroxidase-labeled secondary
antibodies (1:1,000; Dako; Agilent Technologies, Inc., Santa Clara,
CA, USA). Protein bands were visualized using the Amersham ECL
Prime Western Blotting Detection reagent (cat. no. RPN2232; GE
Healthcare Life Sciences, Little Chalfont, UK) and protein
expression was quantified using Image Lab software (version 6.0.1;
Bio-Rad Laboratories, Inc.).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from lung tissue samples as
previously described (19). Total
RNA (500 ng) was reverse transcribed into cDNA using the High
Capacity cDNA Reverse Transcription kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.). qPCR was performed using the TaqMan
Universal PCR Master mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and primers for IL-10, IL-12, monocyte
chemoattractant protein (MCP)-1 and TNF-α. IL-10 forward,
5′-GCCTAACATGCTTCGAGATC-3- and reverse, 5′-TGATGTCTGGGTCTTGGTTC-3′;
IL-12 forward, 5′-ATGTACAGCATGCAGCTCGCATC-3′ and reverse,
5′-GGCTTGTTGAGATGATGCTTTGACA-3′; MCP-1 forward,
5′-GTCCCTGTCATGCTTCTGG-3′ and reverse, 5′-GCGTTAACTGCATCTGGCT-3′;
TNF-α forward, 5′-CCCAGGGACCTCTCTCTAATCA-3′ and reverse,
5′-AGCTGCCCCTCAGCTTGAG-3′; β-actin forward,
5′-GTGGGCCGCTCTAGCCACCAA-3′ and reverse,
5′-TCTTTGATGTCACGCACGATTTC-3′. qPCR was performed using the Applied
Biosystems 7500 FAST Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The following thermocycling
conditions were used for the qPCR: Initial denaturation for 2 min
at 94°C; 35 cycles of 30 sec at 94°C, 30 sec at 58°C and 1 min at
72°C; final extension for 10 min at 72°C. The relative mRNA
expression levels were quantified using the comparative critical
threshold method and normalized to β-actin (20).
Cytokine analysis in mouse lung
tissue
Mouse tissue-extracted proinflammatory cytokines
TNF-α, IL-10, IL-12 and MCP-1 were determined in lung tissue
samples using the BD™ Cytometric Bead Array Mouse Inflammation kit
(cat. no. 552364; BD Biosciences, San Jose, CA, USA). The samples
were analyzed using a BD FACSVerse™ flow cytometer with C6 software
(version 1.0.264.21; BD Biosciences).
Statistical analysis
Data were presented as the mean ± standard error.
All statistical analyses were preformed using SPSS software
(version 20; IBM Corporation, Armonk, NY, USA). One-way analysis of
variance followed by Tukey's post hoc test (for parametric data) or
Kruskal-Wallis test (for non-parametric data) was used to analyze
differences among multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
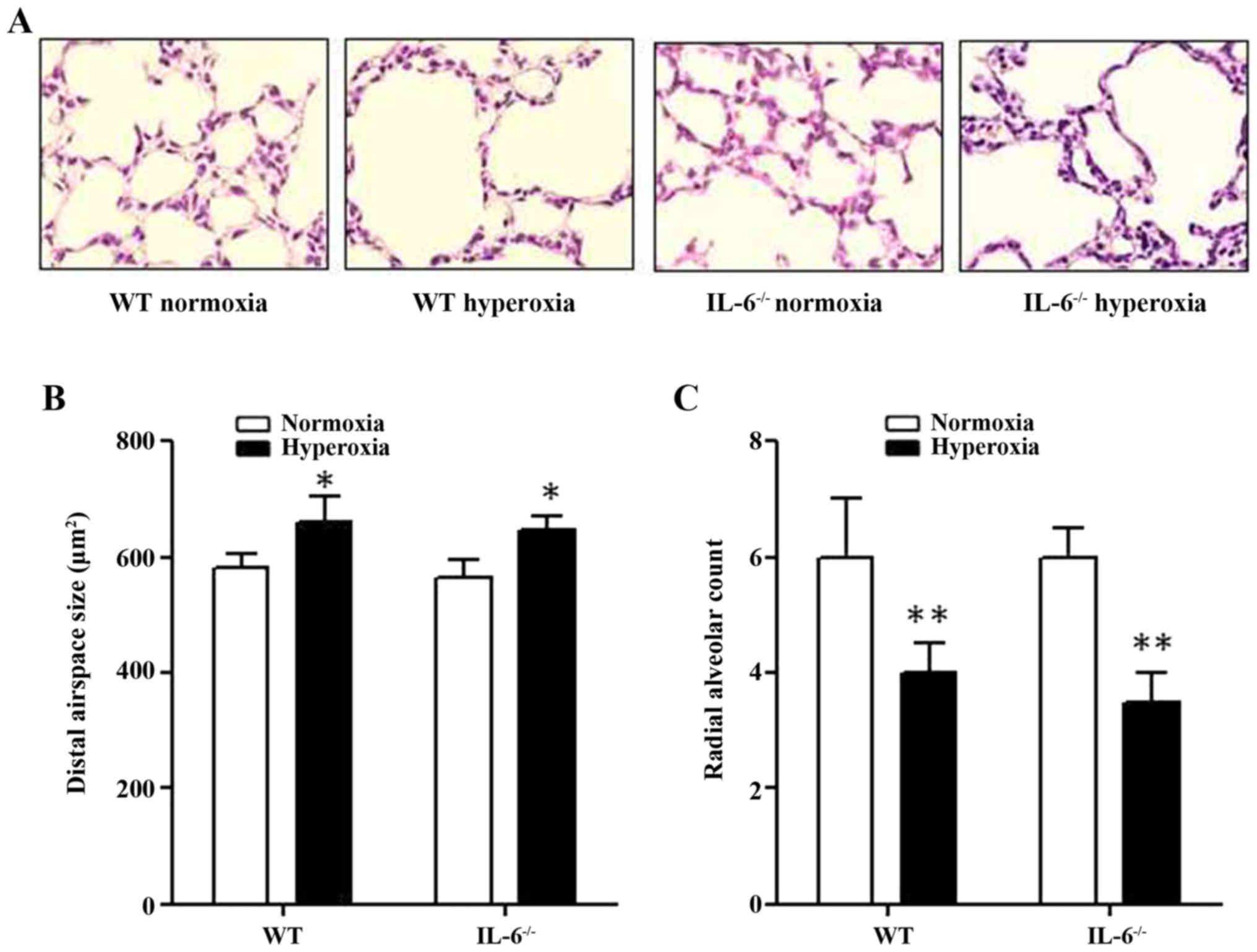
Hyperoxia suppresses alveoli
development in IL-6 null and WT mice
Histological assessment of lung tissue samples
confirmed that IL-6 null and WT mice in the normoxia group
developed terminal airways with normal organization. By contrast,
the lung structure of IL-6 null and WT mice in the hyperoxia group
appeared to be damaged, leading to simplification and dilation of
the alveoli (Fig. 1A). The average
alveolar airspace size was significantly increased in 4-day old WT
and IL-6 null mice in the hyperoxia group compared with the
normoxia group (Fig. 1B). In
addition, the RAC, which reflects the quantity of alveoli, was
significantly decreased in 4-day old WT and IL-6 null mice in the
hyperoxia group compared with the normoxia group (Fig. 1C). Furthermore, no significant
difference was observed in the alveolar airspace size or the RAC
between IL-6 null and WT mice in either the normoxia or hyperoxia
group.
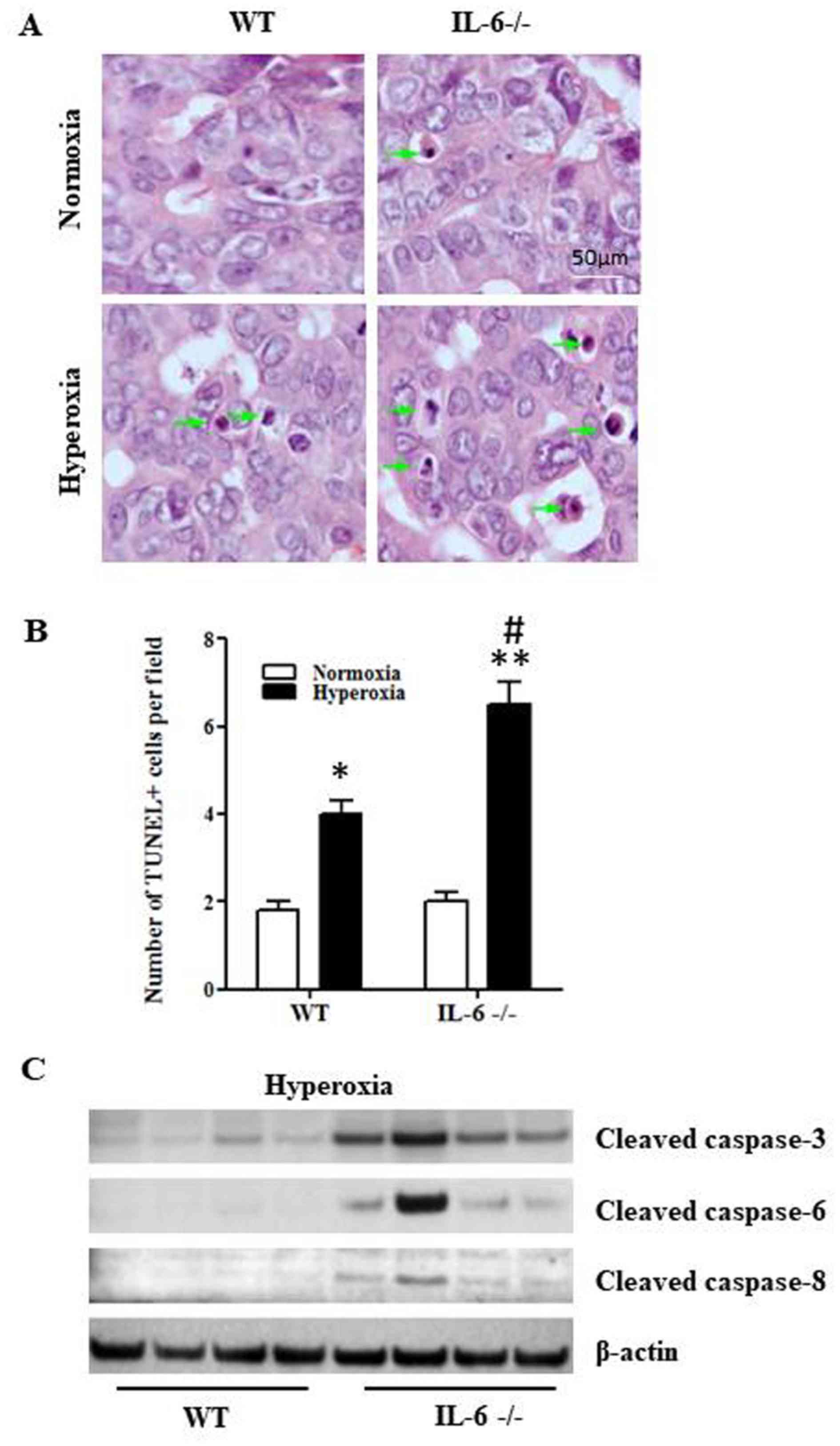
Hyperoxia promotes cell apoptosis and
pulmonary inflammation in IL-6 null mice
To determine whether IL-6 deficiency augments
hyperoxia-induced apoptosis, the TUNEL assay was performed on lung
tissue sections from 4-day old IL-6 null and WT mice exposed to
normoxia or hyperoxia. The number of TUNEL-positive cells in lung
tissue samples was significantly increased in 4-day old WT and IL-6
null mice in the hyperoxia group compared with the normoxia group
(Fig. 2A and B). In addition, the
number of TUNEL-positive cells was significantly increased in IL-6
null mice compared with WT mice in the hyperoxia group (Fig. 2B). Furthermore, the relative protein
expression levels of cleaved caspase-8, caspase-6 and caspase-3
were increased in IL-6 null mice compared with WT mice in the
hyperoxia group (Fig. 2C).
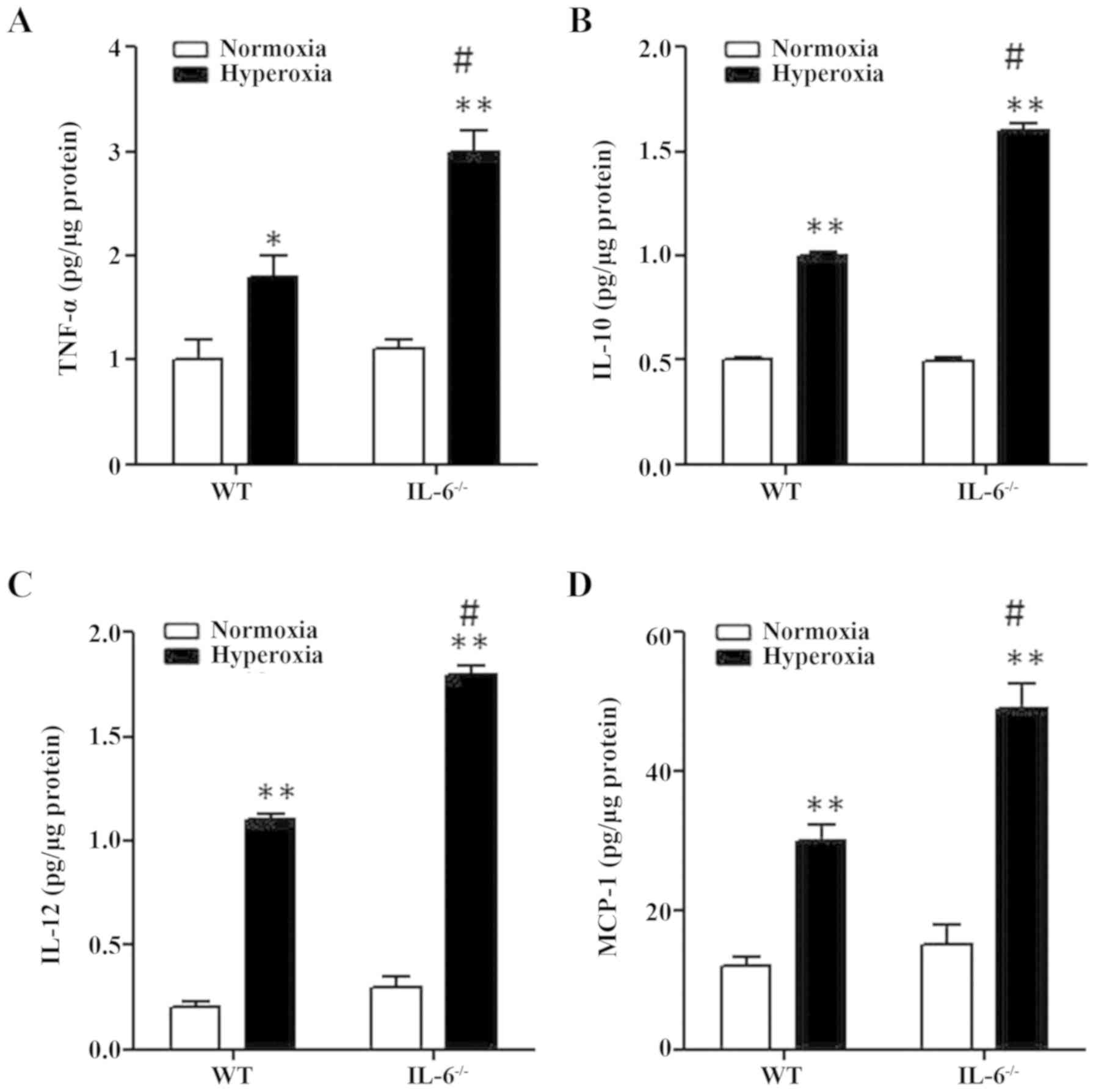
Effects of IL-6 on the production of
pro-inflammatory cytokines in hyperoxia-exposed mice
To investigate the role of IL-6 on pulmonary
inflammation, mouse tissue-extracted pro-inflammatory cytokines
were examined in lung tissue samples from 4-day old IL-6 null and
WT mice exposed to normoxia or hyperoxia. The protein expression
levels of MCP-1, TNF-α, IL-12 and IL-10 were significantly
increased in WT and IL-6 null mice in the hyperoxia group compared
with the normoxia group (Fig. 3). In
addition, the protein levels of pro-inflammatory cytokines were
significantly increased in IL-6 null mice compared with WT mice in
the hyperoxia group (Fig. 3).
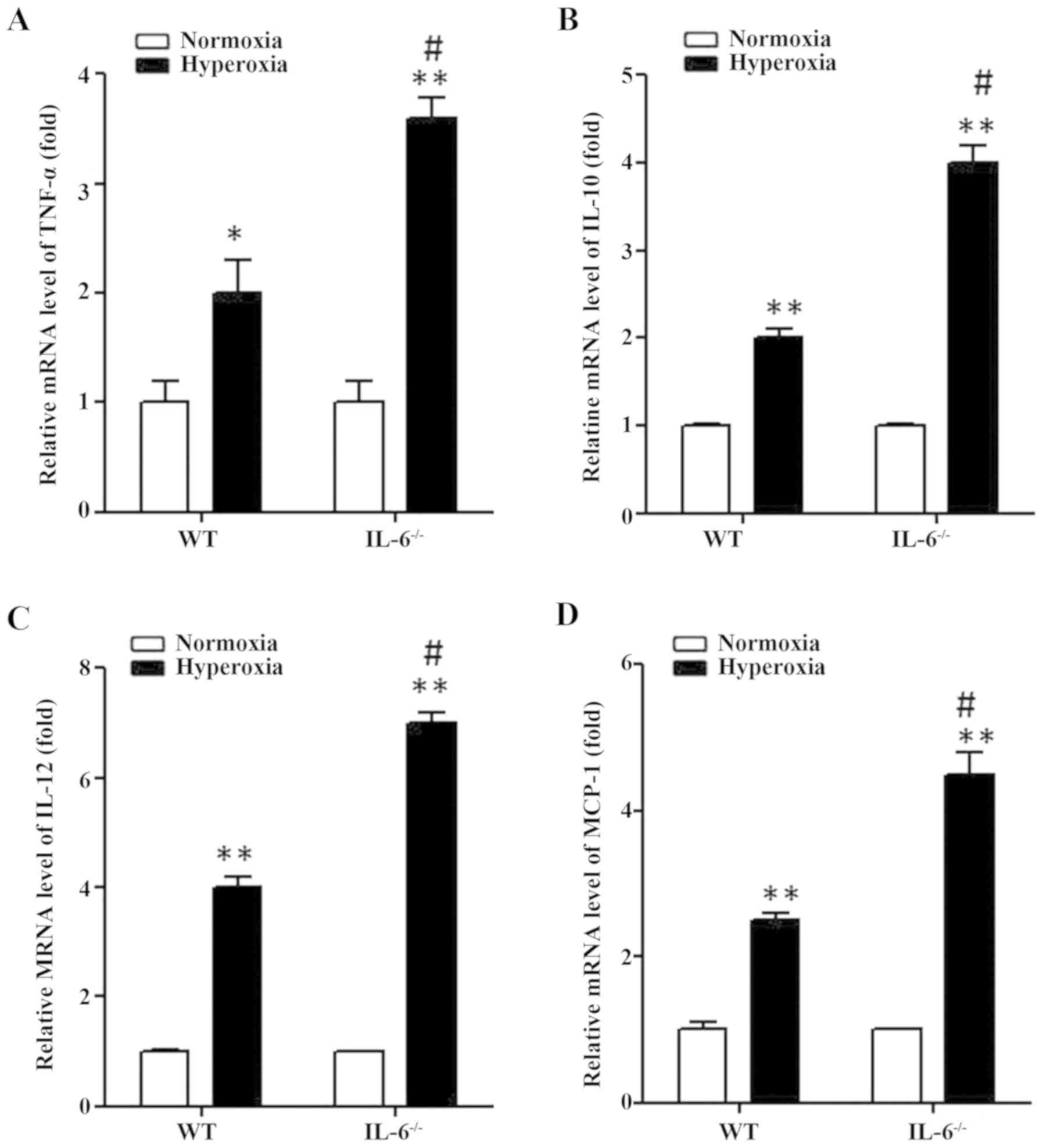
Expression levels of MCP-1, TNF-α,
IL-12 and IL-10 are enhanced in hyperoxia-exposed IL-6 null
mice
As the protein expression levels of MCP-1,
TNF-α, IL-12 and IL-10 were significantly increased in IL-6
null mice compared with WT mice in the hyperoxia group,
transcriptional activation of these inflammatory genes was examined
in lung tissue samples by RT-qPCR. Similarly, the mRNA expression
levels of MCP-1, TNF-α, IL-12 and IL-10 were significantly
increased in WT and IL-6 null mice in the hyperoxia group compared
with the normoxia group (Fig. 4). In
addition, MCP-1, TNF-α, IL-12 and IL-10 transcription was
significantly enhanced in IL-6 null mice compared with WT mice in
the hyperoxia group (Fig. 4).
Discussion
Despite advances in neonatal medicine, BPD remains a
major cause of mortality and long-term morbidity in premature
infants with a birth weight <1,000 g (21–23). As
a form of chronic lung disease, BPD arises following OT and MV
required for the treatment of acute respiratory distress syndrome
(ARDS) in premature infants (24).
Pulmonary injury following birth at the terminal canalicular stage
can disrupt the normal progression of pulmonary generation, leading
to alveolar simplification with larger alveoli, fewer septa and
decreased vessel density (25),
which can be associated with pulmonary hypertension (26).
IL-6 is a highly versatile cytokine involved in
several processes, including cellular proliferation,
differentiation and cell death (8,9).
Previous studies have demonstrated the importance of IL-6 in
several inflammatory pulmonary diseases including ARDS (27), chronic obstructive pulmonary disease
(28), non-small cell lung cancer
(29) and asthma (30). The effect of IL-6 pre- and post-birth
has been investigated following the identification that IL-6
elevation decreases the incidence of respiratory distress syndrome
in premature infants (31). In
addition, animal models of inflammation inside the amniotic
membrane confirmed these results (24). Elevated IL-6 concentrations were
associated with BPD progression, which suggests that upregulated
IL-6 expression may be used as a reliable predictor of BPD
progression (32,33). In the current study, IL-6 gene
deficiency in pulmonary development treated with hyperoxia enhanced
the production of several pro-inflammatory cytokines and cell
death. However, treatment with hyperoxia did not promote pulmonary
development in neonatal mice.
IL-10 is considered to be an anti-inflammatory
cytokine (34). In the current
study, the expression level of IL-10 was significantly increased in
WT and IL-6 null mice in the hyperoxia group compared with the
normoxia group. Protection against hyperoxia may be involved,
however, once it fails, apoptosis is automatically triggered and
TNF-α is induced to perform its pro-apoptotic function
(35). The current study
demonstrated that IL-6 deficiency enhanced the expression of
TNF-α following exposure to hyperoxia. As a member of the
TNF superfamily, TNF-α is one of the most potent inducers of
apoptosis (36).
TNF-α-induced apoptosis is mediated primarily through the
activation of type I receptors, the death domain of which recruits
multiple signaling proteins, which together form part of an
apoptotic cascade. However, further studies are required to
determine the mechanisms underlying TNF-α-induced
apoptosis.
Apoptosis is a form of programmed cell death, which
is an essential mechanism to regulate embryonic development and
cell differentiation (37).
Following treatment with MV in premature babies, apoptosis occurs
as a result of oxygen toxicity, septic shock, oxygen deficiency, a
decrease in enzymes against oxidants and proteases as well as
volutrauma/barotrauma (38).
Apoptosis serves an essential role in the degeneration of type II
alveolar epithelial cells in adults with ARDS (39). However, type II alveolar epithelial
cells exhibit decreased cell death in chronic interstitial
pneumonitis (40). Although
apoptosis is a prominent event in the lungs of animals injured by
inhaling 100% O2, hyperoxia-induced epithelial cell
death occurs as a result of necrosis, and not apoptosis (41).
In the current study, apoptotic cell death occurred
in the lungs of neonatal mice exposed to hyperoxia as demonstrated
by caspase stimulation and TUNEL staining. Furthermore, loss of
IL-6 function through gene ablation enhanced caspase activation and
increased the number of TUNEL-positive cells. These results suggest
that the crosstalk between inflammation and cell death may be
involved in hyperoxia-induced lung injury in BPD. However, further
studies are required to determine whether increased cell death and
pulmonary development is linked with poor long-term prognosis.
In conclusion, the current study demonstrated the
effect of IL-6 gene deficiency in hyperoxia-induced lung injury.
The findings revealed the versatility of IL-6 as a potentially
novel therapeutic approach to attenuate the injury caused by OT and
MV.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL contributed to the conception and design of the
study and analysis of the results. GW contributed to the design of
the study and analysis of the results. SL, CW and JZ performed the
experiments, analyzed data, prepared figures and drafted the
manuscript. CW approved the final version of the manuscript. JZ
edited and revised the manuscript.
Ethics approval and consent to
participate
All procedures and protocols were approved by the
Animal Use Committee of Fengcheng Hospital (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bhandari A and Panitch HB: Pulmonary
outcomes in bronchopulmonary dysplasia. Semin Perinatol.
30:219–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bhandari V: Hyperoxia-derived lung damage
in preterm infants. Semin Fetal Neonatal Med. 15:223–229. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hillman NH, Moss TJ, Kallapur SG,
Bachurski C, Pillow JJ, Polglase GR, Nitsos I, Kramer BW and Jobe
AH: Brief, large tidal volume ventilation initiates lung injury and
a systemic response in fetal sheep. Am J Respir Crit Care Med.
176:575–581. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Woods SJ, Waite AA, O'Dea KP, Halford P,
Takata M and Wilson M R: Kinetic profiling of in vivo lung cellular
inflammatory responses to mechanical ventilation. Am J Physiol Lung
Cell Mol Physiol. 308:L912–L921. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoon BH, Romero R, Jun JK, Park KH, Park
JD, Ghezzi F and Kim BI: Amniotic fluid cytokines (interleukin-6,
tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8)
and the risk for the development of bronchopulmonary dysplasia. Am
J Obstet Gynecol. 177:825–830. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bry K and Lappalainen U: Pathogenesis of
bronchopulmonary dysplasia: The role of interleukin 1beta in the
regulation of inflammation-mediated pulmonary retinoic acid
pathways in transgenic mice. Semin Perinatol. 30:121–128. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Speer CP: Pulmonary inflammation and
bronchopulmonary dysplasia. J Perinatol. 26 (Suppl 1):S57–S64.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dame JB and Juul SE: The distribution of
receptors for the pro-inflammatory cytokines interleukin (IL)-6 and
IL-8 in the developing human fetus. Early Hum Dev. 58:25–39. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xing Z, Gauldie J, Cox G, Baumann H,
Jordana M, Lei XF and Achong MK: IL-6 is an antiinflammatory
cytokine required for controlling local or systemic acute
inflammatory responses. J Clin Invest. 101:311–320. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jaskoll T and Melnick M: Submandibular
gland morphogenesis: Stage-specific expression of TGF-alpha/EGF,
IGF, TGF-beta, TNF, and IL-6 signal transduction in normal
embryonic mice and the phenotypic effects of TGF-beta2, TGF-beta3,
and EGF-r null mutations. Anat Rec. 256:252–268. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Melnick M, Chen H, Zhou YM and Jaskoll T:
Interleukin-6 signaling and embryonic mouse submandibular salivary
gland morphogenesis. Cells Tissues Organs. 168:233–245. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jobe AH and Ikegami M: Antenatal
infection/inflammation and postnatal lung maturation and injury.
Respir Res. 2:27–32. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao L, Hart S, Cheng J, Melenhorst JJ,
Bierie B, Ernst M, Stewart C, Schaper F, Heinrich PC, Ullrich A, et
al: Mammary gland remodeling depends on gp130 signaling through
Stat3 and MAPK. J Biol Chem. 279:44093–44100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Culig Z, Steiner H, Bartsch G and Hobisch
A: Interleukin-6 regulation of prostate cancer cell growth. J Cell
Biochem. 95:497–505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thebaud B and Abman SH: Bronchopulmonary
dysplasia: Where have all the vessels gone? Roles of angiogenic
growth factors in chronic lung disease. Am J Respir Crit Care Med.
175:978–985. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berger J and Bhandari V: Animal models of
bronchopulmonary dysplasia. The term mouse models. Am J Physiol
Lung Cell Mol Physiol. 307:L936–L947. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukumoto J, Fukumoto I, Parthasarathy PT,
Cox R, Huynh B, Ramanathan GK, Venugopal RB, Allen-Gipson DS,
Lockey RF and Kolliputi N: NLRP3 deletion protects from
hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol.
305:C182–C189. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Emery JL and Mithal A: The number of
alveoli in the terminal respiratory unit of man during late
intrauterine life and childhood. Arch Dis Child. 35:544–547. 1960.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ito M, Nagano N, Arai Y, Ogawa R,
Kobayashi S, Motojima Y, Go H, Tamura M, Igarashi K, Dennery PA and
Namba F: Genetic ablation of Bach1 gene enhances recovery from
hyperoxic lung injury in newborn mice via transient upregulation of
inflammatory genes. Pediatr Res. 81:926–931. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vohr BR, Wright LL, Dusick AM, Mele L,
Verter J, Steichen JJ, Simon NP, Wilson DC, Broyles S, Bauer CR, et
al: Neurodevelopmental and functional outcomes of extremely low
birth weight infants in the National Institute of Child Health and
Human Development Neonatal Research Network, 1993–1994. Pediatrics.
105:1216–1226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lemons JA, Bauer CR, Oh W, Korones SB,
Papile LA, Stoll BJ, Verter J, Temprosa M, Wright LL, Ehrenkranz
RA, et al: Very low birth weight outcomes of the National Institute
of Child health and human development neonatal research network,
January 1995 through December 1996. NICHD Neonatal Research
Network. Pediatrics. 107:E12001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allen J, Zwerdling R, Ehrenkranz R,
Gaultier C, Geggel R, Greenough A, Kleinman R, Klijanowicz A,
Martinez F, Ozdemir A, et al: Statement on the care of the child
with chronic lung disease of infancy and childhood. Am J Respir
Crit Care Med. 168:356–396. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jobe AH and Bancalari E: Bronchopulmonary
dysplasia. Am J Respir Crit Care Med. 163:1723–1729. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beers MF and Mulugeta S: The biology of
the ABCA3 lipid transporter in lung health and disease. Cell Tissue
Res. 367:481–493. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mourani PM, Ivy DD, Gao D and Abman SH:
Pulmonary vascular effects of inhaled nitric oxide and oxygen
tension in bronchopulmonary dysplasia. Am J Respir Crit Care Med.
170:1006–1013. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Doganci A, Sauer K, Karwot R and Finotto
S: Pathological role of IL-6 in the experimental allergic bronchial
asthma in mice. Clin Rev Allergy Immunol. 28:257–270. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chung KF: Cytokines in chronic obstructive
pulmonary disease. Eur Respir J. (Suppl 34):S50–S59. 2001.
View Article : Google Scholar
|
|
30
|
Bihl M, Tamm M, Nauck M, Wieland H,
Perruchoud AP and Roth M: Proliferation of human non-small-cell
lung cancer cell lines: Role of interleukin-6. Am J Respir Cell Mol
Biol. 19:606–612. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimoya K, Taniguchi T, Matsuzaki N,
Moriyama A, Murata Y, Kitajima H, Fujimura M and Nakayama M:
Chorioamnionitis decreased incidence of respiratory distress
syndrome by elevating fetal interleukin-6 serum concentration. Hum
Reprod. 15:2234–2240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kramer BW, Kramer S, Ikegami M and Jobe
AH: Injury, inflammation, and remodeling in fetal sheep lung after
intra-amniotic endotoxin. Am J Physiol Lung Cell Mol Physiol.
283:L452–L459. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moss TJ, Newnham JP, Willett KE, Kramer
BW, Jobe AH and Ikegami M: Early gestational intra-amniotic
endotoxin: Lung function, surfactant, and morphometry. Am J Respir
Crit Care Med. 165:805–811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saraiva M and O'Garra A: The regulation of
IL-10 production by immune cells. Nat Rev Immunol. 10:170–181.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu Y, Jiang W, Liu S, Su X and Zhou S:
Combined effect of tnf-alpha polymorphisms and hypoxia on
steroid-induced osteonecrosis of femoral head. Int J Clin Exp
Pathol. 8:3215–3219. 2015.PubMed/NCBI
|
|
36
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng
P, Hogan RN, Gilpin C and Levine B: Autophagy gene-dependent
clearance of apoptotic cells during embryonic development. Cell.
128:931–946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hargitai B, Szaboó V, Hajdú J, Harmath A,
Pataki M, Farid P, Papp Z and Szende B: Apoptosis in various organs
of preterm infants: Histopathologic study of lung, kidney, liver,
and brain of ventilated infants. Pediatr Res. 50:110–114. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Korfei M, Ruppert C, Mahavadi P, Henneke
I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al:
Epithelial endoplasmic reticulum stress and apoptosis in sporadic
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
178:838–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bardales RH, Xie SS, Schaefer RF and Hsu
SM: Apoptosis is a major pathway responsible for the resolution of
type II pneumocytes in acute lung injury. Am J Pathol. 149:845–852.
1996.PubMed/NCBI
|
|
41
|
Kazzaz JA, Xu J, Palaia TA, Mantell L,
Fein AM and Horowitz S: Cellular oxygen toxicity. Oxidant injury
without apoptosis. J Biol Chem. 271:15182–15186. 1996. View Article : Google Scholar : PubMed/NCBI
|