Introduction
Diabetic retinopathy (DR) is the most common and
serious complication of diabetes mellitus (DM) and is a major
vision-threatening eye disease in the working-age population
(1). Despite a new generation of
medications and modern vitreoretinal microsurgery in clinical
treatments, the prevalence of DR has still risen dramatically over
the last two decades (2). Therefore,
further understanding of the pathogenesis of DR is required in
order to improve the presently available clinical therapies.
Proliferative DR (PDR) is the advanced,
sight-threatening stage of DR characterized by ischemia-induced
pathological preretinal neovascularization and uncontrolled
production of extracellular matrix (ECM) proteins associated with
the outgrowth of epiretinal fibrovascular membranes (FVMs) at the
vitreoretinal interface (3). An
increasing amount of evidence has suggested that endothelial cells
may undergo endothelial-mesenchymal transition (EndMT) under high
glucose (HG) stimulation, which contributes to pathological
fibrosis in PDR (4–6). EndMT is a complex biological process in
which endothelial cells lose their specific markers, including
cluster of differentiation 31 (CD31) and vascular endothelial
(VE)-cadherin, and express higher levels of mesenchymal markers
including α-smooth muscle actin (α-SMA), fibroblast-specific
protein 1 (FSP1) and fibronectin (FN) (7,8). A more
comprehensive understanding of the molecular mechanisms involved in
the EndMT process may be used to provide a novel therapeutic
approach to attenuate the formation of FVMs associated with
PDR.
MicroRNAs (miRNAs/miRs) are small noncoding
regulatory RNA molecules, ~18–22 nucleotides in length, which
post-transcriptionally regulate gene expression by binding the
3′-untranslated regions (3′-UTRs) of target mRNAs to repress their
translation or decrease their stability (9). Previous studies have reported that a
number of miRNAs, including miR-21 (10), miR-23b-3p (11), miR-195 (12) and miR-126 (13), may be involved in the development of
DR. Our recent studies revealed that the expression level of the
miR-29a/b cluster decreased in the retina tissue of diabetic rats,
which may serve as an important biomarker in DR (14,15). An
increasing amount of evidence has suggested that the miR-29 family
could serve a role in the development of fibrosis of multiple
organs, including renal, pulmonary and cardiac fibrosis (16–18).
Therefore, the current study hypothesized that modulating the
miR-29a/b cluster may provide a novel therapeutic strategy for
fibrosis associated with PDR.
In the present study, human retinal microvascular
endothelial cells (HRMECs) were used to elucidate the potential
involvement of the miR-29a/b cluster in HG-induced EndMT. It was
demonstrated that the expression level of miR-29a/b was decreased
in HG-treated HRMECs, and overexpression of miR-29a/b inhibited the
EndMT of HRMECs induced by HG. Further mechanistic studies
indicated that neurogenic locus notch homolog protein 2 (Notch2)
was a direct target of miR-29a/b and was involved in the
progression of EndMT.
Materials and methods
Cells culture
Primary HRMECs isolated from a single donor eye were
purchased from Cell Systems (Kirkland, WA, USA) and cultured in
endothelial basal medium (Lonza Group, Ltd., Basel, Switzerland)
supplemented with 10% fetal bovine serum, endothelial cell growth
supplements (EGM SingleQuots; Lonza Group, Ltd.) and 1%
penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). All cultures were incubated at 37°C in a
5% CO2 humidified atmosphere. Cells at passage 3–6 were
used for subsequent experiments.
Cell stimulation
HRMECs were cultured in conditioned medium with
various concentrations of D-glucose (Sigma-Aldrich; Merck KGaA,
Darmstadt Germany) and were treated in 6 groups with the following
for 7 days: 0 mM glucose, 5 mM glucose [serving as the normal
glucose (NG) group], 15 mM glucose, 30 mM glucose (HG group), 50 mM
glucose and 5 mM glucose + 25 mM mannitol (serving as the osmotic
control). Cells were subsequently stimulated with HG or osmotic
control for either 1, 3, 5 or 7 days. In certain experiments, a
selective inhibitor of endogenous transforming growth factor-β
(TGF-β) signaling (SB431542; 10 µM; Sigma-Aldrich; Merck KGaA) was
added to cultures 1 h prior to treatment with NG or HG.
Cell transfection
HRMECs were seeded in six-well plates at a density
of 1×105 cells/well for 24 h. A total of 100 nM miR-29a
and miR-29b mimics or inhibitors, and non-targeting control (NC)
were subsequently transfected into cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). All miRNA mimics, inhibitors and miR-NC
oligonucleotides were purchased from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China), the sequences of which are listed in Table I. The pcDNA-enhanced green
fluorescent protein (EGFP) vector with Notch2 cDNA (pcDNA-Notch2)
and the blank pcDNA-EGFP plasmids (pcDNA) were purchased from
Shanghai GeneChem Co., Ltd. (Shanghai, China). 1 mg pcDNA-Notch2
expression plasmid or blank pcDNA plasmid were transfected into
HRMECs (5×106 cells/well) and the mock group was
untransfected. At 24 h following transfection, HRMECs were washed
with PBS and recorded using a laser-scanning confocal microscope
(LSM710; Carl Zeiss AG, Oberkochen, Germany) to evaluate the
transfection efficiency. Four images were obtained per well in two
channels: HRMECs body and GFP. Transfection efficacy was calculated
manually using the following formula: % transfection efficacy =
green fluorescent cell number/cell number ×100.
 | Table I.Sequences of microRNAs and primers
used in the current study. |
Table I.
Sequences of microRNAs and primers
used in the current study.
| microRNA/primer
name | Sequence
(5′→3′) |
|---|
| miR-29a mimic |
UAGCACCAUCUGAAAUCGGUUA |
| miR-29b mimic |
UAGCACCAUUUGAAAUCAGUGUU |
| miR-29a
inhibitor |
UAACCGAUUUCAGAUGGUGCUA |
| miR-29b
inhibitor |
AACACUGAUUUCAAAUGGUGCUA |
| miRNA NC |
UUUGUACUACACAAAAGUACUG |
| U6-F |
CTCGCTTCGGCAGCACA |
| U6-R |
AACGCTTCACGAATTTGCGT |
| miR-29a-F |
TAGCACCATTTGAAATCAGTTT |
| miR-29a-R |
TGCGTGTCGTGGAGTC |
| miR-29b-F |
AAAATATTTGGTTTTTATTAGGGT |
| miR-29b-R |
CATAACCTCTTCCTTTACCATTAAA |
| Jagged 1 (Homo
sapiens)-F |
GTCCATGCAGAACGTGAACG |
| Jagged 1 (Homo
sapiens)-R |
GCGGGACTGATACTCCTTGA |
| Notch1 (Homo
sapiens)-F |
TGGACCAGATTGGGGAGTTC |
| Notch1 (Homo
sapiens)-R |
GCACACTCGTCTGTGTTGAC |
| Notch2 (Homo
sapiens)-F |
CTCTTCCGTATCCGCACCATCATG |
| Notch2 (Homo
sapiens)-R |
GAGCCATGCTTACGCTTTCG |
| VE-cadherin
(Homo sapiens)-F |
CTACCAGCCCAAAGTGTGTG |
| VE-cadherin
(Homo sapiens)-R |
GTGTTATCGTGATTATCCGTGA |
| FSP1 (Homo
sapiens)-F |
GCAACAGGGACAACGAGG |
| FSP1 (Homo
sapiens)-R |
CTGGGCTGCTTATCTGGG |
| FN (Homo
sapiens)-F |
GATAAATCAACAGTGGGAGC |
| FN (Homo
sapiens)-R |
CCCAGATCATGGAGTCTTTA |
| SNAI1 (Homo
sapiens)-F |
TTCCAGCAGCCCTACGAC |
| SNAI1 (Homo
sapiens)-R |
AGCCTTTCCCACTGTCCTC |
| β-actin (Homo
sapiens)-F |
CAAAGGCCAACAGAGAGAAGAT |
| β-actin (Homo
sapiens)-R |
TGAGACACACCATCACCAGAAT |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from HRMECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The RNA quality was measured using a NanoDrop 2000
spectrophotometer (Thermo Fisher, Scientific, Inc.). For the
detection of mRNA expression, cDNA was synthesized with 1 µg of
total RNA using an iScript™ cDNA synthesis kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's protocol. qPCR analysis was performed using the ABI
7000 PCR instrument (Applied Biosystems; Thermo Fisher Scientific,
Inc.) with SYBR Premix Ex Taq™ II (Takara Biotechnology Co., Ltd.,
Dalian, China). The reaction for mRNA was performed under the
following conditions: 95°C for 1 min, and 40 cycles at 95°C for 5
sec and 60°C for 30 sec. For the detection of miR-29a and miR-29b,
total cDNA was synthesized by using a miScript II RT kit (Qiagen
GmbH, Hilden, Germany), under the following conditions: 37°C for 60
min, 95°C for 5 min and 4°C for 5 min. qPCR of miRNA expression was
performed using a miScript SYBR Green PCR kit (Qiagen GmbH). The
thermocycling conditions for miRNA was pas follows: 95°C for 15
min, followed by 40 cycles at 94°C for 15 sec, 55°C for 30 sec and
70°C for 30 sec. The relative amounts of mRNA and miRNA were
calculated using the 2−ΔΔq method and normalized
to β-actin and U6 small nuclear RNA, respectively (19). The sequences of the primers are
presented in Table I.
Western blotting
HRMECs were lysed in RIPA lysis buffer and the
protein was collected and quantified with a bicinchoninic acid
assay kit (Beyotime Institute of Biotechnology, Shanghai, China). A
total of 40 µg of protein from each sample were subjected to sodium
dodecyl sulfate polyacrylamide gel electrophoresis (10% gel) and
electrotransferred onto polyvinylidene fluoride membranes.
Following blocking with 5% non-fat milk (Sigma-Aldrich) at room
temperature for 2 h, the membranes were incubated with primary
antibodies against Notch 1 (cat. no. ab8925; 1:1,000), Notch 2
(cat. no. ab8926; 1:1,000), Jagged 1 (cat. no. ab7771; 1:1,000) and
β-actin (cat. no. ab8227; 1:5,000) (all from Abcam, Cambridge, UK)
at 4°C for 16 h. The membranes were then incubated with horseradish
peroxidase-conjugated secondary antibodies (cat. no. 14708 or
14709; 1:5,000; Cell Signaling Technology, Inc., Danvers, MA, USA)
for additional 2 h at 25°C. Western blots were visualized using
Western Lighting Plus-ECL (PerkinElmer, Inc., Waltham, MA, USA) and
recorded using the Bio-Rad gel image analysis system (Bio-Rad
Laboratories, Inc.). β-actin was used as the internal control.
Immunofluorescence
HRMECs were rinsed in 1X PBS and fixed with 4%
paraformaldehyde at 25°C for 30 min and permeated with 0.2% Triton
X-100 (Beyotime Institute of Biotechnology) in PBS for 10 min.
Following blocking with 5% goat serum (cat. no. C-0005; BIOSS,
Beijing, China) for 30 min at 25°C, fixed cells were incubated with
a primary antibody against CD31 (cat. no. ab28364; 1:200) or α-SMA
(cat. no. ab5694; 1:200) (both from Abcam) overnight at 4°C. The
cells were then washed three times with PBS and incubated with
Alexa Fluor 488 AffiniPure goat anti-mouse IgG (cat. no.
115–545-062) or FITC-conjugated goat anti-rabbit IgG (cat. no.
111–095-144) (both from Jackson ImmunoResearch Laboratories, West
Grove, PA, USA) at a dilution 1:200 for 1 h at 37°C. Nuclei were
counterstained with 1 mg/ml DAPI (cat. no. d1306; Invitrogen;
Thermo Fisher Scientific, Inc.) for 5 min at 25°C. The cells were
then visualized under a laser-scanning confocal microscope (Carl
Zeiss).
Luciferase reporter assay
The binding sites of miR-29a and miR-29b with Notch2
were predicted using the TargetScan program (http://www.targetscan.org/vert_72/). Wild-type (WT)
3′-UTR of the Notch2 gene containing the predicted miR-29a/b
binding site and relevant mutant controls (MUT) were cloned into
pGL3 vectors (Promega Corporation, Madison, WI, USA). HRMECs were
co-transfected with 100 ng of pGL3-Notch2 3′-UTR or pGL3-Notch2-mut
3′-UTR reporter plasmid and 10 ng of Renilla luciferase
expression plasmid pRL-TK (Promega Corporation), with 100 nM
miR-29a/b mimics, inhibitors or miR-NC using Lipofectamine 2000
transfection reagent (Thermo Fisher Scientific, Inc.). Cells were
harvested and lysed 48 h later and luciferase activity was
determined using a Dual-Luciferase reporter assay kit (Promega
Corporation). Firefly luciferase activity was normalized to that of
Renilla luciferase.
Statistical analysis
All data are presented as the mean ± standard
deviation and were analyzed using SPSS statistical software
(version 21.0; IBM Corp., Armonk, NY, USA). The differences were
analyzed using Student's t-test for datasets containing two groups
and one-way analysis of variance followed by Bonferroni or
Dunnett's post hoc test for multiple group comparisons.
P<0.05 was considered to indicate a statistically
significant difference.
Results
HG induces a decrease in the
expression of miR-29a/b in HRMECs
To clarify the effect of HG on the expression of
miR-29a and miR-29b, HRMECs were cultured and treated with
different concentrations of D-glucose (5–50 mM) for 7 days and HG
(30 mM) for various times (1–7 days). The results of RT-qPCR
analysis revealed that treatment with 30 mM glucose significantly
decreased the expression levels of miR-29a and miR-29b in HRMECs
(Fig. 1A). In addition, the
expression of miR-29a/b was significantly lower in the HG group
compared with that in the OSM group, but not different between the
NG group and OSM group at 7 days following stimulation (Fig. 1B). HRMECs exhibited significantly
decreased levels of miR-29a/b following exposure to HG for
33 days (Fig. 1C). These
results indicated that HG could induce the decreased expression of
miR-29a/b in HRMECs.
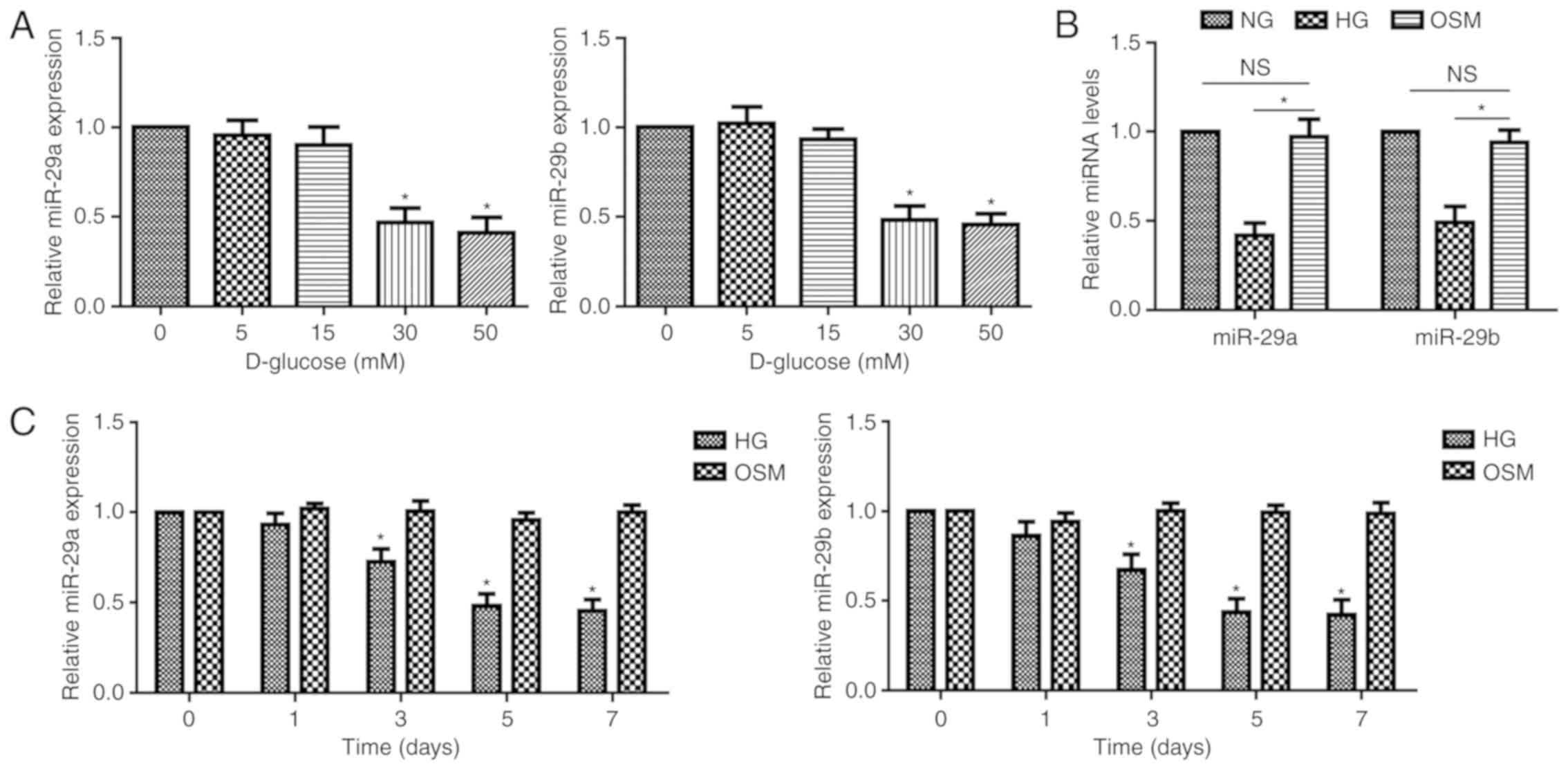 | Figure 1.Expression pattern of miR-29a/b in
HG-stimulated HMRECs. (A) RT-qPCR was used to determine the mRNA
expression of miR-29a/b in HRMECs pretreated for 7 days with 5, 15,
30 and 50 mM of D-glucose. (B) RT-qPCR was used to determine the
mRNA expression of miR-29a/b in HRMECs pretreated for 7 days with
NG (5 mM), HG (30 mM) and OSM (5 mM glucose +25 mM mannitol). (C)
RT-qPCR was used to determine the mRNA expression of miR-29a/b in
HRMECs pretreated with HG or OSM for 1, 3, 5, or 7 days. Data are
presented as the mean ± standard deviation from 3 different
experiments, each performed in triplicate. Data were analyzed by
one-way analysis of variance followed by Dunnett's post hoc test.
*P<0.05 compared with the untreated group. HG, high glucose; NG,
normal glucose; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; miR, microRNA; OSM, osmotic control;
HRMECs, human retinal microvascular endothelial cells. |
miR-29a and miR-29b inhibit HG-induced
EndMT in HRMECs
To further investigate the effects of miR-29a and
miR-29b in EndMT, miR-29a/b mimics and miR-NC were transfected into
HRMECs. RT-qPCR confirmed that the expression of miR-29a/b was
significantly increased in HRMCEs transfected with miR-29a/b mimics
compared with cells transfected with the miR-NC (Fig. 2A). HRMECs exhibited an elongated
spindle-shaped phenotype following culture with HG for 7 days
(Fig. 2B). Immunofluorescence and
RT-qPCR demonstrated that the levels of the endothelial markers
CD31 and VE-cadherin were decreased, while the levels of the
mesenchymal markers α-SMA, FSP1, FN and SNAI1 were increased in the
HG group when compared with NG group (Fig. 2B and C) (20). However, the overexpression of either
miR-29a or miR-29b reversed low levels of endothelial marker as
well as the high levels of these mesenchymal markers (Fig. 2B and C). These results suggested a
direct role of miR-29a/b in HG-induced EndMT in HRMECs.
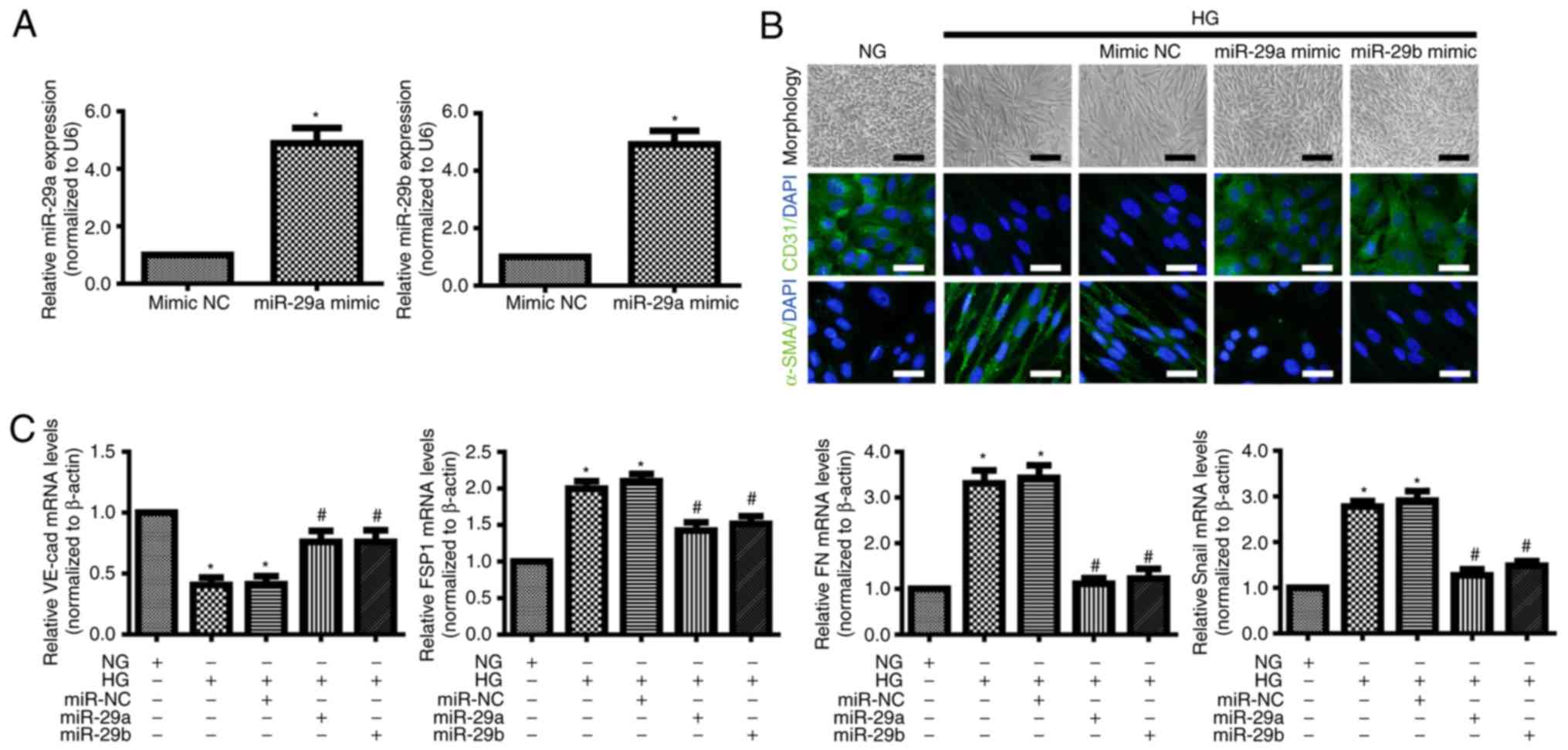 | Figure 2.miR-29a/b overexpression inhibits
HG-induced endothelial-mesenchymal transition of HRMECs. HRMECs
were transfected with miR-NC, miR-29a mimic or miR-29b. (A) RT-qPCR
analysis was performed to detect the expression of miR-29a and
miR-29b. Data were analyzed using Student's t-test. P<0.05
compared with the mimic NC group. (B) Morphological alterations and
expression of CD31 and α-SMA in HRMECs under NG (5 mM) and HG (30
mM) conditions for 7 days (black scale bar, 100 µm; white scale
bar, 50 µm). (C) RT-qPCR was used to determine the mRNA expression
of VE-cad, FSP1, FN and Snail in HRMECs under different conditions.
Data were analyzed using one-way analysis of variance followed by
Bonferroni post hoc test and are presented as the mean ± standard
deviation from 3 different experiments, each performed in
triplicate. *P<0.05 compared with the NG group;
#P<0.05 compared with the HG group. HG, high glucose;
NG, normal glucose; FSP1, fibroblast-specific protein 1; HRMECs,
human retinal microvascular endothelial cells; miR, microRNA; NC,
nontargeting control; VE-cad, vascular endothelial cadherin; FN,
fibronectin; RT-qPCR, reverse transcription-quantitative polymerase
chain reaction; α-SMA, α-smooth muscle actin; CD31, cluster of
differentiation 31. |
miR-29a and miR-29b target Notch2 to
regulate its expression in HRMECs
Notch signaling has been suggested to serve a role
in retinal vascular morphogenesis during development and in
pathological angiogenesis associated with ocular diseases,
including DR (21,22). A recent report confirmed that Notch2
functions as a regulator of myocardial fibrosis in diabetic
cardiomyopathy (23). RT-qPCR and
western blot analyses demonstrated that HG treatment increased the
mRNA and protein expression of Jagged 1, Notch1 and Notch2 in
HRMECs (Fig. 3A and B). The
TargetScan program was subsequently used to investigate the
potential association between the miR-29a/b cluster and the Notch
signaling pathway, and identified Notch2 as a target for miR-29a
and miR-29b (Fig. 3C). The
luciferase reporter assays revealed that the introduction of either
miR-29a or miR-29b in HRMECs significantly suppressed the reporter
activity of the WT but not that of MUT Notch2 3′-UTR. However, an
increase in the luciferase activity of WT 3′-UTR of Notch2 was
observed following transfection with the miR-29a/b inhibitors
(Fig. 3D). In addition, RT-qPCR
analysis demonstrated that miR-29a/b overexpression markedly
decreased Notch2 levels, which was further confirmed by western
blotting. Reciprocally, the miR-29a/b silencing was accompanied by
an increase in Notch2 expression (Fig.
3E). These results suggested that Notch2 is a direct target of
miR-29a/b in HRMECs.
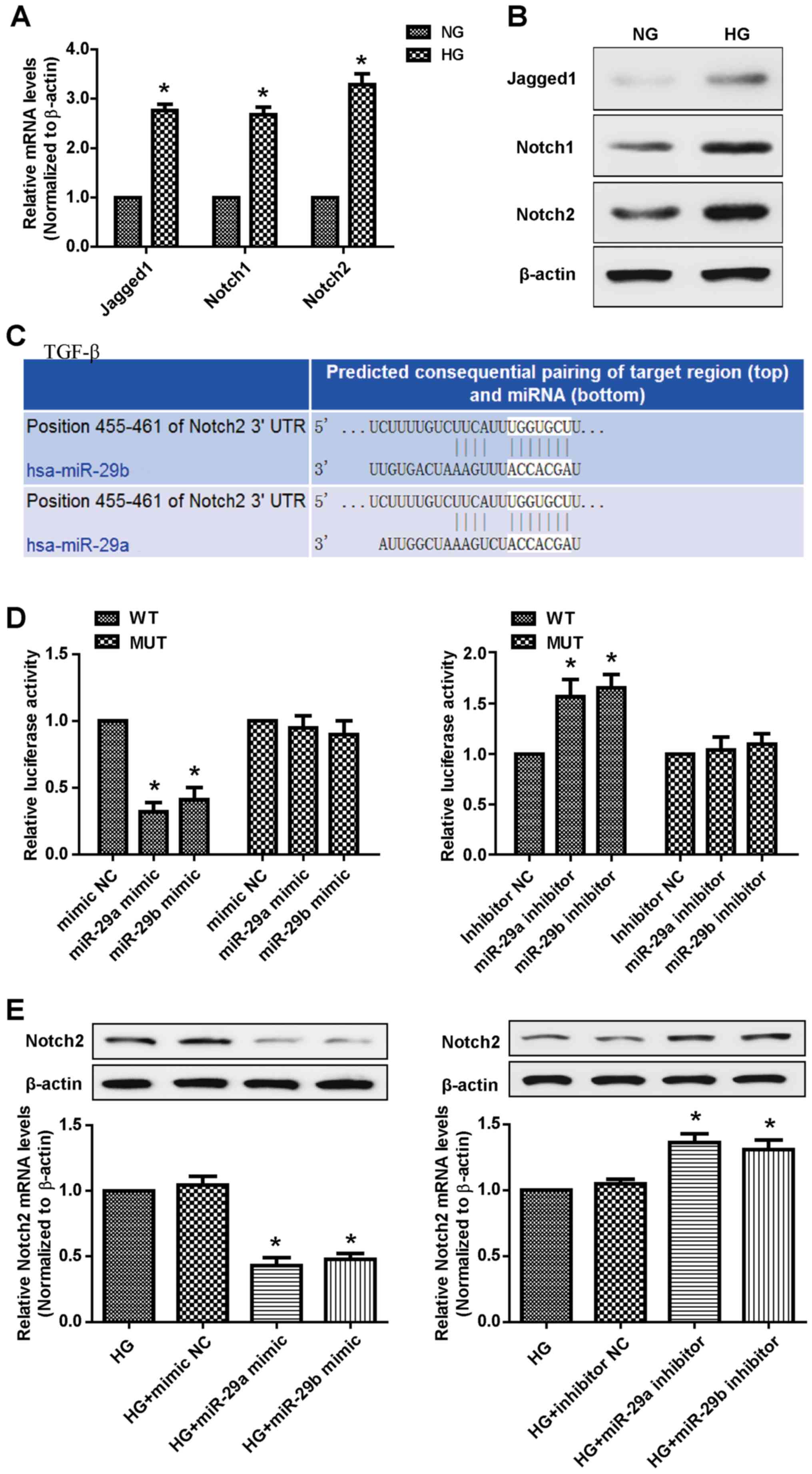 | Figure 3.Notch2 is the target gene of
miR-29a/b in HRMECs. The expression levels of Jagged 1, Notch1 and
Notch2 were determined by (A) RT-qPCR and (B) Western blotting in
HRMECs stimulated with HG (30 mM) for 7 days. Data were analyzed
using Student's t-test. (C) Online TargetScan algorithm predicted
that both miR-29a and miR-29b bound to the 3′-UTR of Notch2. (D) A
luciferase reporter assay was performed to detect the relative
luciferase reporter activity of the 3′-UTR of Notch2. Data were
analyzed by one-way analysis of variance followed by Dunnett's post
hoc test. *P<0.01 compared with the mimic NC group. (E) RT-qPCR
and western blot analyses were used to detect the expression levels
of Notch2 in HRMECs transfected with mimic/inhibitor NC, miR-29a
mimic/inhibitor and miR-29b mimic/inhibitor. Data were analyzed by
one-way analysis of variance followed by Dunnett's post hoc test
and are presented as the mean ± standard deviation from 3 different
experiments, each performed in triplicate. *P<0.01 compared with
the HG group. Notch2, neurogenic locus notch homolog protein 2; HG,
high glucose; HRMECs, human retinal microvascular endothelial
cells; RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; UTR, untranslated region; miR/miRNA, microRNA; NC,
nontargeting control; WT, wild-type; MUT, mutant control. |
miR-29a and miR-29b suppress the
HG-induced EndMT via the Notch2 signaling pathway in HRMECs
The present study further investigated whether
Notch2 contributed to inducing EndMT following treatment with HG.
EGFP was used to detect pcDNA-Notch2 plasmid transfection
efficiency and the results demonstrated that the rate of
fluorescent events was ~50% (Fig.
4A). RT-qPCR and western blot analyses revealed that transient
Notch2 expression in HRMECs led to substantial upregulation of the
Notch2 level (Fig. 4C).
Co-transfection with miR-29a or miR-29b mimic and the Notch2
expression plasmid (but not the pcDNA-vector plasmid) could partly
reverse the effect of miR-29a/b mimics on the expression of
endothelial markers CD31 and VE-cadherin, and mesenchymal markers
α-SMA, FSP1, FN and Snail. These were consistent with previous
results and further demonstrated that the downregulation of
miR-29a/b was concurrent with the upregulation of Notch2 during
EndMT induced by HG in HRMECs (Fig. 4B
and D). Therefore, the suppressed miR-29a/b levels appeared to
be essential in inducing the upregulation of Notch2 in the EndMT
process. Additionally, Notch2 overexpression alone or SB431542
pretreatment could alter the phenotype of HRMECs in NG and HG
conditions (Fig. 4E). However,
SB431542 pretreatment could not alter the phenotype of Notch2
overexpressed HRMECs in HG conditions. Taken together, these
results confirmed that miR-29a/b suppressed the HG-induced EndMT
via the Notch2 signaling pathway, which is independent of TGF-β
signaling.
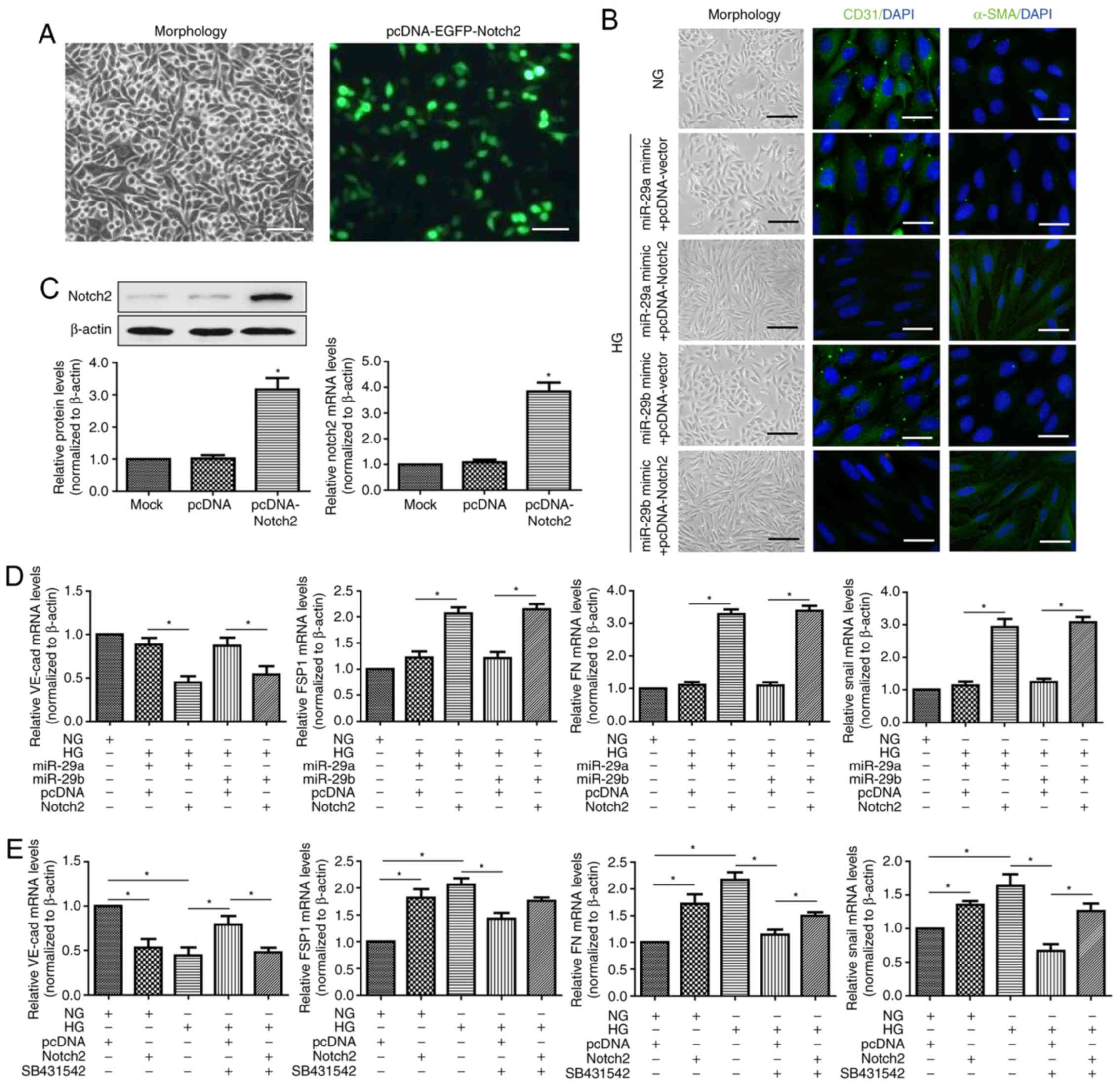 | Figure 4.Notch2 overexpression rescues the
inhibitory effect of miR-29a/b on endothelial-mesenchymal
transition. (A) Transfection efficiency of pcDNA-Notch2 plasmid in
HRMECs. Cells were analyzed under a fluorescence microscope after
48 h of transfection (scale bar, 100 µm). (B) Morphological
alterations and expression of CD31 and α-SMA (black scale bar, 100
µm; white scale bar, 50 µm). (C) RT-qPCR and western blot analyses
were performed to detect the expression of Notch2 in HRMECs
transfected with blank pcDNA-vector and pcDNA-Notch2 plasmids. Mock
group was set by adding transfection reagent only. Data were
analyzed using one-way analysis of variance followed by Dunnett's
post hoc test. *P<0.05 vs. the mock group. (D) mRNA expression
of VE-cad, FSP1, FN and Snail in HRMECs co-transfected with miR-29a
or miR-29b mimics and pcDNA-vector or pcDNA-Notch2 plasmid. Data
were analyzed by one-way analysis of variance followed by
Bonferroni post hoc test. Cells were incubated inNG (5 mM) or HG
(30 mM) conditions for 7 days. (E) RT-qPCR was used to determine
the mRNA expression of VE-cad, FSP1, FN and Snail in transfected
HRMECs with or without SB431542 treatment. Cells were incubated
with NG (5 mM) or HG (30 mM) conditions for 7 days. Data were
analyzed by one-way analysis of variance followed by Bonferroni
post hoc test and are presented as the mean ± standard deviation
from 3 different experiments, each performed in triplicate.
*P<0.05. Notch2, neurogenic locus notch homolog protein 2; HG,
high glucose; NG, normal glucose; FSP1, fibroblast-specific protein
1; HRMECs, human retinal microvascular endothelial cells; EGFP,
enhanced green fluorescent protein; α-SMA, α-smooth muscle actin;
CD31, cluster of differentiation 31; FN, fibronectin; VE-cad,
vascular endothelial cadherin; miR, microRNA; SB431542,
transforming growth factor β1 inhibitor. |
Discussion
Endothelial dysfunction is the predominant
manifestation in chronic diabetic complications including DR
(24,25). Hyperglycemia induces endothelial
injury by producing altered amounts of multiple proteins, which
serve roles in EndMT (4–6). Cao et al (6) identified that glucose-induced EndMT in
the retinal endothelial cells is mediated through TGF-β1 and is
regulated by miR-200b. Abu et al (5) demonstrated that EndMT serves a role in
creating myofibroblasts, which are responsible for the progression
of fibrosis associated with PDR. Recently, Chang et al
(4) confirmed EndMT in PDR
epiretinal membranes obtained from patients undergoing pars
plana vitrectomy and suggested that endothelin-1 served a role
in promoting this transformative process. The results of the
present study demonstrated that HG-induced a significant decrease
in the expression of miR-29a and miR-29b in HRMECs, and further
demonstrated that miR-29a/b overexpression inhibited HG-induced
EndMT by targeting Notch2.
The miR-29 family, which includes miR-29a, miR-29b
and miR-29c, has been demonstrated to regulate multiple
physiological and pathological processes (26–28). For
instance, miR-29a is involved in the pathogenesis of type 2
diabetes (29), and miR-29a
overexpression counteracted the insulin inhibition of the
phosphoenolpyruvate carboxykinase 2, mitochondrial gene by
targeting phosphoinositide 3-kinase regulatory subunit p85α in
HepG2 cells (30). In addition,
inhibition of the expression of the miR-29a/b/c family via TGF-β1
promoted the synthesis and deposition of ECM components in cardiac
or renal fibrosis (31,32). Our previous research revealed that
the expression of miR-29a and miR-29b was decreased by HG
stimulation and the downregulation of miR-29a/b exacerbated DR by
impairing the function of Müller cells (14,15). The
present study, to the best of our knowledge, demonstrated for the
first time that miR-29a/b in HRMECs may suppress HG-mediated EndMT.
These results provide a potential novel mechanism for the
antifibrosis effect of miR-29a/b on PDR and other chronic diabetic
complications.
It has become increasingly apparent that the Notch
signaling pathway may initiate EndMT, which contributes to the
aggravation of cardiac fibrosis (23,33). A
previous study demonstrated that Jagged 1-Notch interactions
induced EndMT in microvascular endothelial cells (34). Therefore, the present study
investigated the association between miR-29a/b and the Notch
signaling pathway in EndMT physiopathology under HG conditions.
RT-qPCR and western blot analyses revealed that the mRNA and
protein levels of Jagged 1, Notch1 and Notch2 in the HG-treated
HRMECs were significantly higher compared with the NG group. In
addition, TargetScan predicted that miR-29a and miR-29b bound to
the 3′-UTR of Notch2, with Luciferase reporter assays and western
blotting further confirming that miR-29a and miR-29b negatively
regulated the expression of Notch2. In addition, the overexpression
of Notch2 reversed the inhibitory effect of miR-29a/b on EndMT, and
was accompanied by the decreased expression of endothelial markers
and the increased expression of mesenchymal markers. It has been
demonstrated that TGF-β is a key inducer of EndMT (35) and that HG concentration increases the
expression of TGF-β in various cells (36,37).
However, the results of the current study revealed that
pretreatment with the TGF-β inhibitor, SB431542, could not inhibit
the HG-induced EndMT in Notch2 overexpressed HRMECs. Therefore, we
hypothesize that the miR-29a/b cluster inhibits HG-induced EndMT of
HRMECs by suppressing Notch2, irrespective of the status of the
TGF-β signaling pathway.
In conclusion, data obtained in the present study
indicated that a miR-29a/b/Notch2-mediated mechanism may be
responsible for the process of EndMT in the context of PDR.
Therapies based on controlling the expression of miR-29a/b may
represent a promising direction for the treatment of PDR in the
future.
Acknowledgements
Not applicable.
Funding
The research was supported by Natural Science
Foundation Project of Zhejiang (grant no. LY18H120002).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JZ and ZC designed the study; JZ, YZ, JC, DC and CC
performed the experiments; JZ and SZ analyzed the data; JZ, YZ and
ZC drafted and revised the manuscript. All authors approved the
final version for publication and agree to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EndMT
|
endothelial-mesenchymal transition
|
|
PDR
|
proliferative diabetic retinopathy
|
|
HRMEC
|
human retinal microvascular
endothelial cell
|
|
DR
|
diabetic retinopathy
|
|
DM
|
diabetic mellitus
|
|
3′-UTR
|
3′-untranslated region
|
|
NG
|
normal glucose
|
|
miRNA
|
microRNA
|
|
TGF-β1
|
transforming growth factor β1
|
|
HG
|
high glucose
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Cheung N, Mitchell P and Wong TY: Diabetic
retinopathy. Lancet. 376:124–136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sabanayagam C, Yip W, Ting DS, Tan G and
Wong TY: Ten emerging trends in the epidemiology of diabetic
retinopathy. Ophthalmic Epidemiol. 23:209–222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Michels RG: Proliferative diabetic
retinopathy: Pathophysiology of extraretinal complications and
principles of vitreous surgery. Retina. 1:1–17. 1981.PubMed/NCBI
|
|
4
|
Chang W, Lajko M and Fawzi AA:
Endothelin-1 is associated with fibrosis in proliferative diabetic
retinopathy membranes. PLoS One. 13:e01912852018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abu El-Asrar AM, De Hertogh G, van den
Eynde K, Alam K, Van Raemdonck K, Opdenakker G, Van Damme J, Geboes
K and Struyf S: Myofibroblasts in proliferative diabetic
retinopathy can originate from infiltrating fibrocytes and through
endothelial-to-mesenchymal transition (EndoMT). Exp Eye Res.
132:179–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao Y, Feng B, Chen S, Chu Y and
Chakrabarti S: Mechanisms of endothelial to mesenchymal transition
in the retina in diabetes. Invest Ophthalmol Vis Sci. 55:7321–7331.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pardali E, Sanchez-Duffhues G,
Gomez-Puerto MC and Ten Dijke P: TGF-β-induced
endothelial-mesenchymal transition in fibrotic diseases. Int J Mol
Sci. 18:E21572017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Piera-Velazquez S, Li Z and Jimenez SA:
Role of endothelial-mesenchymal transition (EndoMT) in the
pathogenesis of fibrotic disorders. Am J Pathol. 179:1074–1080.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Q, Qiu F, Zhou K, Matlock HG,
Takahashi Y, Rajala RVS, Yang Y, Moran E and Ma JX: Pathogenic Role
of microRNA-21 in diabetic retinopathy through downregulation of
PPARα. Diabetes. 66:1671–1682. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao S, Li T, Li J, Lu Q, Han C, Wang N,
Qiu Q, Cao H, Xu X, Chen H and Zheng Z: miR-23b-3p induces the
cellular metabolic memory of high glucose in diabetic retinopathy
through a SIRT1-dependent signalling pathway. Diabetologia.
59:644–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mortuza R, Feng B and Chakrabarti S:
miR-195 regulates SIRT1-mediated changes in diabetic retinopathy.
Diabetologia. 57:1037–1046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qin LL, An MX, Liu YL, Xu HC and Lu ZQ:
MicroRNA-126: A promising novel biomarker in peripheral blood for
diabetic retinopathy. Int J Ophthalmol. 10:530–534. 2017.PubMed/NCBI
|
|
14
|
Zhang J, Wu L, Chen J, Lin S, Cai D, Chen
C and Chen Z: Downregulation of MicroRNA 29a/b exacerbated diabetic
retinopathy by impairing the function of muller cells via forkhead
box protein O4. Diab Vasc Dis Res. 15:214–222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang J, Chen M, Chen J, Lin S, Cai D,
Chen C and Chen Z: Long non-coding RNA MIAT acts as a biomarker in
diabetic retinopathy by absorbing miR-29b and regulating cell
apoptosis. Biosci Rep. 37:BSR201700362017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He Y, Huang C, Lin X and Li J: MicroRNA-29
family, a crucial therapeutic target for fibrosis diseases.
Biochimie. 95:1355–1359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cushing L, Kuang P and Lu J: The role of
miR-29 in pulmonary fibrosis. Biochem Cell Biol. 93:109–118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM
and Lan HY: miR-29b as a therapeutic agent for angiotensin
II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol
Ther. 22:974–985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perez L, Munoz-Durango N, Riedel CA,
Echeverria C, Kalergis AM, Cabello-Verrugio C and Simon F:
Endothelial-to-mesenchymal transition: Cytokine-mediated pathways
that determine endothelial fibrosis under inflammatory conditions.
Cytokine Growth Factor Rev. 33:41–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen X, Xiao W, Liu X, Zeng M, Luo L, Wu
M, Ye S and Liu Y: Blockade of Jagged/Notch pathway abrogates
transforming growth factor β2-induced epithelial-mesenchymal
transition in human retinal pigment epithelium cells. Curr Mol Med.
14:523–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dou GR, Wang L, Wang YS and Han H: Notch
signaling in ocular vasculature development and diseases. Mol Med.
18:47–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Geng H and Guan J: MiR-18a-5p inhibits
endothelial-mesenchymal transition and cardiac fibrosis through the
Notch2 pathway. Biochem Biophys Res Commun. 491:329–336. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sorrentino FS, Matteini S, Bonifazzi C,
Sebastiani A and Parmeggiani F: Diabetic retinopathy and endothelin
system: Microangiopathy versus endothelial dysfunction. Eye (Lond).
32:1157–1163. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kang H, Ma X, Liu J, Fan Y and Deng X:
High glucose-induced endothelial progenitor cell dysfunction. Diab
Vasc Dis Res. 14:381–394. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmitt MJ, Margue C, Behrmann I and Kreis
S: MiRNA-29: A microRNA family with tumor-suppressing and
immune-modulating properties. Curr Mol Med. 13:572–585. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kriegel AJ, Liu Y, Fang Y, Ding X and
Liang M: The miR-29 family: Genomics, cell biology, and relevance
to renal and cardiovascular injury. Physiol Genomics. 44:237–244.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fabbri M, Garzon R, Cimmino A, Liu Z,
Zanesi N, Callegari E, Liu S, Alder H, Costinean S,
Fernandez-Cymering C, et al: MicroRNA-29 family reverts aberrant
methylation in lung cancer by targeting DNA methyltransferases 3A
and 3B. Proc Natl Acad Sci USA. 104:15805–15810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kong L, Zhu J, Han W, Jiang X, Xu M, Zhao
Y, Dong Q, Pang Z, Guan Q, Gao L, et al: Significance of serum
microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: A
clinical study. Acta Diabetol. 48:61–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pandey AK, Verma G, Vig S, Srivastava S,
Srivastava AK and Datta M: miR-29a levels are elevated in the db/db
mice liver and its overexpression leads to attenuation of insulin
action on PEPCK gene expression in HepG2 cells. Mol Cell
Endocrinol. 332:125–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang B, Komers R, Carew R, Winbanks CE, Xu
B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K,
Gregorevic P, et al: Suppression of microRNA-29 expression by
TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc
Nephrol. 23:252–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van Rooij E, Sutherland LB, Thatcher JE,
DiMaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou X, Chen X, Cai JJ, Chen LZ, Gong YS,
Wang LX, Gao Z, Zhang HQ, Huang WJ and Zhou H: Relaxin inhibits
cardiac fibrosis and endothelial-mesenchymal transition via the
Notch pathway. Drug Des Devel Ther. 9:4599–4611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Noseda M, McLean G, Niessen K, Chang L,
Pollet I, Montpetit R, Shahidi R, Dorovini-Zis K, Li L, Beckstead
B, et al: Notch activation results in phenotypic and functional
changes consistent with endothelial-to-mesenchymal transformation.
Circ Res. 94:910–917. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshimatsu Y and Watabe T: Roles of TGF-β
signals in endothelial-mesenchymal transition during cardiac
fibrosis. Int J Inflam. 2011:7240802011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu CH, Suriguga, Gong M, Liu WJ, Cui NX,
Wang Y, Du X and Yi ZC: High glucose induced endothelial to
mesenchymal transition in human umbilical vein endothelial cell.
Exp Mol Pathol. 102:377–383. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Di Paolo S, Gesualdo L, Ranieri E,
Grandaliano G and Schena FP: High glucose concentration induces the
overexpression of transforming growth factor-beta through the
activation of a platelet-derived growth factor loop in human
mesangial cells. Am J Pathol. 149:2095–2106. 1996.PubMed/NCBI
|














