Introduction
Renal interstitial fibrosis is the final common
pathway of end stage renal disease (1). It is characterized by the increased
deposition of extracellular matrix (ECM) materials, including
collagen type I (Col I) and type III (Col III), fibronectin (FN),
and laminin, as well as activated renal interstitial fibroblasts
(2). Transforming growth factor
(TGF)-β1 is one of the main factors that can induce fibrosis
(3). TGF-β1 has been identified as a
central mediator in renal fibrosis (4). TGF-β initiates canonical and
non-canonical pathways to exert multiple biological effects
(5). Among them, the mothers against
decapentaplegic homolog (Smad) signaling pathway has been
recognized as a major pathway of TGF-β signaling in progressive
renal fibrosis (6). Connective
tissue growth factor (CTGF) is one of the downstream factors that
induce fibrosis (7). In a normal
physiological environment, CTGF has been demonstrated to be mainly
involved in angiogenesis and cell differentiation (8). It is worth noting that CTGF could
mediate the process of tissue repair and fibrosis under
pathological conditions (9). In the
process of myocardial injury, repair and fibrosis, CTGF has been
revealed to be a molecule that activates fibroblasts (10). The central link of renal interstitial
fibrosis is the activation of fibroblasts with the expression of
α-smooth muscle actin (SMA) serving as the main biomarker (11). α-SMA is a hallmark of a variety of
renal phenotypic transformations and has been commonly used to
detect the phenotypic transformation of fibroblasts into
myofibroblasts (12). α-SMA-positive
myofibroblasts have been demonstrated to be the main synthetic
cells that deposit ECM materials (12).
The amount of collagen secreted by myofibroblasts
has been determined four to five times greater than that of
fibroblasts; collagen has been revealed to increase the deposition
of ECM (13). Furthermore,
myofibroblasts have a strong contractile capacity, which has been
demonstrated to cause the remodeling of the kidney structure,
resulting in fibrosis (14). Smads
are involved in the TGF-β1 signal transduction pathway (15). It has been consistently demonstrated
that Smad2 and Smad3 are extensively activated in the fibrotic
kidney in patients with and animal models of chronic kidney disease
(6). The phosphorylated (p-)Smad2
and p-Smad3 form an oligomeric complex with a common Smad, Smad4,
which has been revealed to translocate into the nucleus to regulate
the transcription of target genes in collaboration with various
co-activators and co-repressors, ultimately inducing fibrosis
(16). Proteasome inhibitors can
interfere with and influence cellular functions by inhibiting the
activity of the proteasome (17).
Therefore, using a proteasome inhibitor to change the activity of
the proteasome cleavage site is the focal point of studies on
inflammation by our group (18). A
previous study demonstrated that a proteasome inhibitor, MG132,
inhibited proliferation and induced apoptosis in renal interstitial
fibroblasts that had been stimulated to differentiate into
myofibroblasts by TGF-β1 (19). In
the current study, the authors examined the influence of MG132 on
TGF-β1-induced renal interstitial fibrosis to investigate the
potential application of MG132 in slowing renal interstitial
fibrosis, as well as the potential mechanism by which it
functions.
Materials and methods
Chemicals
TGF-β1 was purchased from R&D Systems, Inc.
(Minneapolis, MN, USA). Dulbecco's modified Eagle's medium:
Nutrient Mixture F-12 (DMEM/F12) cell culture medium and
trypsin-EDTA were purchased from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Fetal calf serum (FCS) was purchased from
PAA Laboratories (GE Healthcare, Chicago, IL, USA). The proteasome
inhibitor, MG132, was purchased from Calbiochem (EMD Millipore,
Billerica, MA, USA) and dissolved in dimethyl sulfoxide as a 40 µM
stock solution stored at −20°C. Reverse transcription associated
reagents were purchased from Promega Corporation (Madison, WI,
USA). Quantitative polymerase chain reaction (qPCR) primers were
synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). Rabbit
anti-rat Smad2/3 (cat. no. 5678), p-Smad2 (cat. no. 3104), p-Smad3
(cat. no. 9520), α-SMA (cat. no. #19245) and GAPDH (cat. no. #8884)
monoclonal antibodies were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Rabbit anti-rat CTGF (cat. no. 555SR-100)
monoclonal antibodies were obtained from BioVision, Inc. (Milpitas,
CA, USA). Mouse anti-rat FN monoclonal antibodies (cat. no. AB2051)
were purchased from EMD Millipore. Mouse anti-rat β-actin (cat. no.
A1978) and rabbit anti-rat Col III (cat. no. SAB4200749) monoclonal
antibodies were bought from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Horseradish peroxidase (HRP)-conjugated immunoglobulin
(Ig)G secondary antibodies (goat anti-rabbit, cat. no. 5220-0337;
goat anti-mouse, cat. no. 5450-0011) were obtained from Kirkegaard
& Perry Laboratories (SeraCare Life Sciences Inc., Milford, MA,
USA). Enhanced chemiluminescence reagent (Western lightning
Plus-ECL; cat. no. NEL105001EA) was purchased from Edo Biotech AB
(Lindome, Sweden) and polyvinylidene fluoride (PVDF) membranes were
purchased from EMD Millipore.
Cell culture
Rat renal fibroblast NRK-49F cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in DMEM/F12
medium supplemented with 10% FCS in a humidified incubator
containing 5% CO2 at 37°C.
Reverse transcription (RT)-qPCR
analysis
Cells plated in 6-well plates
(2×106/well) were treated with TGF-β1 (5 ng/ml) with or
without MG132 at specific concentrations (0, 0.5, 1, 2.5 and 5 µM)
for 24 h. The control group was defined as untreated cells cultured
for 24 h. Another group of cells plated in 6-well plates
(2×106/well) were treated the cells with TGF-β1 (5
ng/ml) with or without MG132 (2.5 µM) for defined lengths of time
(0, 6, 12 and 24 h). In this group, the control group was defined
as cells cultured for 0 h. RNA was purified using an RNA extraction
kit (cat. no. Z3100; Promega Corporation), converted to cDNA using
TaqMan™ microRNA RT kit (cat. no. 4366596; Thermo Fisher
Scientific, Inc.) and the following genes were amplified: CTGF,
α-SMA, FN, Col III and GAPDH using the primers listed in Table I. SYBR Green reagent (Tokobo Ltd.,
London, UK) was used for PCR amplification and detection of
transcripts. The reaction conditions were as follows: Denaturation
at 94°C for 10 min, and 40 cycles of denaturation at 94°C for 15
sec, and annealing and extension at 60°C for 1 min. Each experiment
was repeated three times in triplicate. Relative mRNA expression
was calculated using the relative quantification method with GAPDH
as an internal control (20). Data
are expressed as n times the untreated group.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Type | Primer sequence
(5′-3′) | Product length
(bp) |
|---|
| GAPDH | Forward |
AGTATGACTCCACTCACGGCAA | 100 |
|
| Reverse |
TCTCGCTCCTGGAAGATGGT |
|
| α-Smooth muscle
actin | Forward |
CATCCGACCTTGCTAACG | 168 |
|
| Reverse |
TCCAGAGTCCAGCACAATAC |
|
| Connective tissue
growth factor | Forward |
ATCCCTGCGACCCACACAAG | 145 |
|
| Reverse |
CAACTGCTTTGGAAGGACTCGC |
|
| Fibronectin | Forward |
CCAGGCACTGACTACAAGAT | 146 |
|
| Reverse |
CATGATACCAGCAAGGAGT |
|
| Collagen type
III | Forward |
TGATGGGATCCAATGAGGGAGA | 143 |
|
| Reverse |
GAGTCTCATGGCCTTGCGTGTTT |
|
Treatment groups
The first collection of cells (8×106)
were treated with TGF-β1 (5 ng/ml) for defined lengths of time (0,
6, 12 and 24 h) with or without MG132 (2.5 µM) pretreatment for 0.5
h; the control group was defined as cells cultured for 0 h. The
second collection of cells were treated with TGF-β1 (5 ng/ml) for
24 h with or without 0.5 h pretreatment with MG132 at 0.5, 1, 2.5
and 5 µM. In these two collections, CTGF, α-SMA, FN, Col III and
GAPDH protein expression levels were assessed. The third collection
of cells were treated with TGF-β1 (5 ng/ml) for different times (0,
15, 30 min, 1 and 2 h); the control group was defined as cells
cultured for 0 min. The fourth collection of cells were treated
with or without 0.5 h MG132 pretreatment at 0, 0.5, 1, 2.5 and 5 µM
and TGF-β1 (5 ng/ml) treatment for 1 h; the control group was
defined as untreated cells cultured for 1 h. In these two
collections, p-Smad2, p-Smad3, Smad2/3 and β-actin protein
expression levels were assessed. The fifth collection of cells were
treated with TGF-β1 (5 ng/ml) with or without MG132 at 0.5, 1, 2.5
and 5 µM for 24 h; the control group was defined as untreated cells
cultured for 24 h. FN and β-actin protein expression levels were
assessed in this collection.
Western blotting
Following all treatments, cells were lysed in cell
lysis solution (phenylmethylsulfonyl fluoride:
Radioimmunoprecipitation assay, 1:100; cat. no. P0013B; Shanghai
Biyuntian Bio-Technology Co., Ltd., Shanghai, China) at 4°C and
centrifuged at 4,024 × g for 30 min at 4°C. Protein concentrations
in the supernatant were determined using the Bradford method.
Protein and loading buffer were mixed at a ratio of 4:1, boiled at
100°C for 5 min and then samples (100 µg/lane) were separated on
10% SDS-PAGE gels. Bands from the gels were transferred to PVDF
membranes using 0.2 A and then blocked for 2 h at room temperature
with 5% skim milk in TBS/T buffer solution. Mouse anti-rat FN
(1:1,000), mouse anti-rat β-actin (1:500), mouse anti-rat Col III
(1:1,000) and rabbit anti-rat GPADH (1:1,000), CTGF (1:2,000),
Smad2/3 (1:1,500), p-Smad2 and p-Smad3 monoclonal antibodies
(1:5,000) were incubated overnight with the membranes at 4°C.
Membranes were then washed, and HRP-conjugated goat anti-mouse IgG
(1:2,000) or goat anti-rabbit (1:1,500) secondary antibodies were
incubated with the membranes for 2 h at 4°C. Membranes were then
covered with ECL reagent in a darkroom for 5 min and exposed to
X-ray film; the film was then developed and fixed. ImageJ software
(version 1.8.0; National Institutes of Health, Bethesda, MD, USA)
was used for the densitometric analysis of the blots.
Statistical analysis
All the data are expressed as mean ± standard
deviation, representative of three repeats and were analyzed by the
SPSS 11.0 software package (SPSS, Inc., Chicago, IL, USA).
Comparisons were made among groups using one-way analysis of
variance followed by Fisher's Least Significant Difference test.
P<0.05 indicated that the difference between groups was
statistically significant.
Results
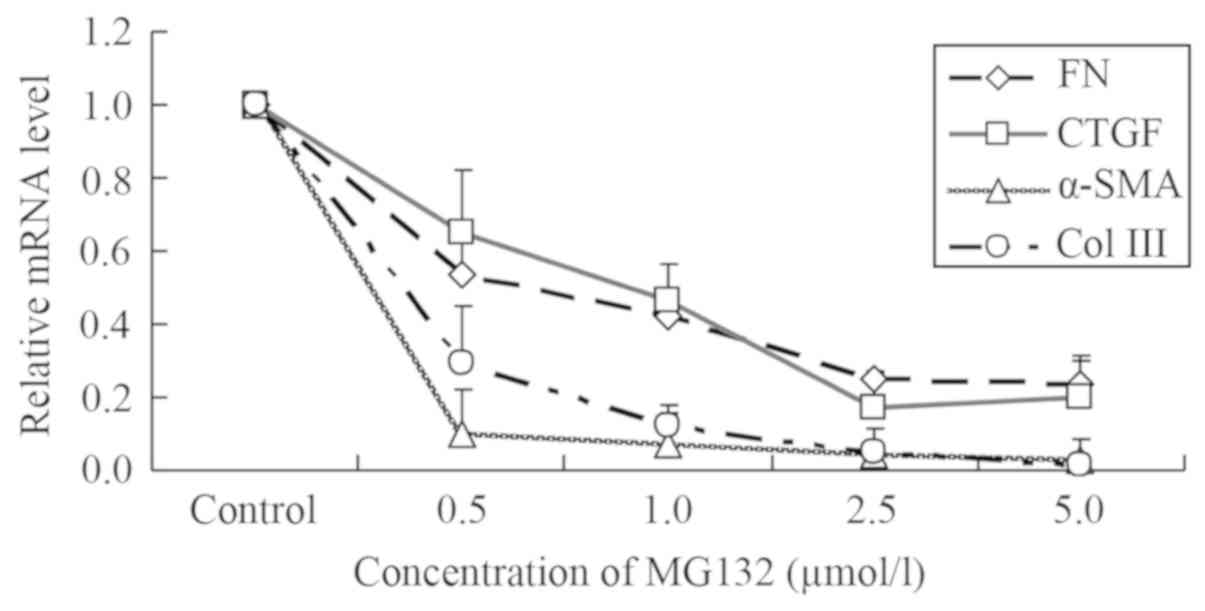
MG132 downregulates the mRNA level of
fibrosis-associated factors
NRK-49F cells have been demonstrated to express
CTGF, α-SMA, FN and Col III (6–9).
Following treatment with different concentrations of MG132, the
mRNA expression of CTGF, α-SMA, FN and Col III decreased compared
with the control group, and exhibited a potential dose-dependent
effect (Fig. 1).
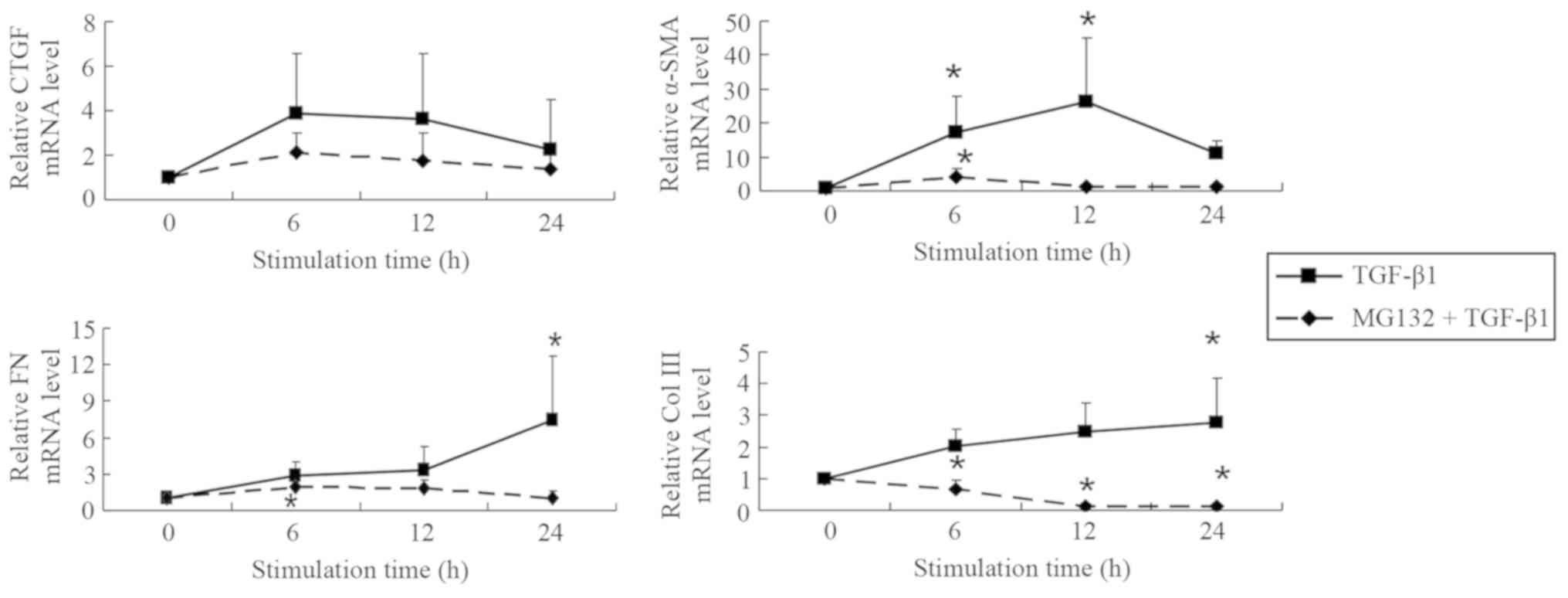
MG132 pretreatment decreases the mRNA
level of fibrosis-associated factors in NRK-49F cells simulated by
TGF-β1 in a potential time-dependent manner
It was found that 2.5 µM MG132 was effective in
reducing the TGF-β1-induced expression of fibrosis-associated genes
(CTGF, α-SMA, FN and Col III) at each time point assessed (6, 12
and 24 h; Fig. 2). Compared with the
control group, CTGF mRNA levels in the TGF-β1 group were 3.9-fold
more greatly expressed at 6 h, 3.6-fold higher at 12 h, and
2.3-fold higher at 24 h, indicating that the difference in
expression gradually declined with increasing time following
initial treatment. Following pretreatment with 2.5 µM MG132,
fold-changes decreased to 2.1, 1.8 and 1.4-fold for 6, 12 and 24 h,
respectively. The mRNA level of α-SMA was also elevated after 6 h
of TGF-β1 stimulation. α-SMA expression reached a peak at 12 h and
was 26.2-fold more highly expressed compared the control group, but
was only 10.9-fold higher at 24 h. Compared with the control group,
α-SMA expression was significantly increased in after 6 and 12 h of
TGF-β1 stimulation (both P<0.05). Following pretreatment with
2.5 µM MG132, these fold-changes in α-SMA expression decreased to
4.2, 1.2 and 1.1 at 6, 12 and 24 h, respectively, of which 6 h of
treatment increased expression significantly compared with the
control group (P<0.05).
The mRNA level of FN was 2.8-, 3.3- and 7.4-fold
more highly expressed in TGF-β1-treated cells at 6, 12 and 24 h,
respectively, of which the 24 h stimulation was significantly
increased compared with the control group (P<0.05). Following
pretreatment with 2.5 µM MG132, these values decreased to 1.9, 1.8
and 1.1-fold at 6, 12 and 24 h, respectively. FN expression after 6
h of MG132 and TGF-β1 treatment was significantly increased
compared with the control group (P<0.05). The mRNA levels of Col
III were 2.0, 2.5 and 2.8-fold higher in TGF-β1-treated cells prior
to pretreatment with MG132 and were reduced to 0.7, 0.1 and
0.1-fold at 6, 12 and 24 h after pretreatment, respectively. Col
III expression was significantly increased 24 h after TGF-β1
treatment and significantly decreased 6, 12 and 24 h after MG132
and TGF-β1 treatment compared with the control group (all
P<0.05). The results indicated that 2.5 µM MG132 decreased the
mRNA levels of fibrosis-associated factors following stimulation
with 5 ng/ml TGF-β1, which causes the fibroblasts to differentiate
into myofibroblasts associated with fibrotic processes (21).
MG132 pre-treatment decreases on the
mRNA level of fibrosis-associated factors in NRK-49F cells
simulated by TGF-β1 in a potential concentration-dependent
manner
Cells pretreated with MG132 exhibited significant
decreases in α-SMA, CTGF, FN and Col III mRNA levels compared with
the TGF-β1 group (P<0.05; Fig.
3). TGF-β1 treatment significantly increased α-SMA, CTGF, FN
and Col III mRNA levels compared with the control group
(P<0.05). α-SMA mRNA levels significantly decreased in the
presence of MG132 compared with the control group (P<0.05).
TGF-β1 mRNA levels significantly decreased with increasing MG132
concentration compared with the TGF-β1 group (P<0.05); no
significant changes were observed compared with the control group
(P>0.05). FN mRNA levels significantly decreased compared with
the control group at 2.5 and 5 µM MG132 (both P<0.05). Col III
mRNA levels significantly decreased with in the presence of MG132
compared with the control group (P<0.05). All mRNA levels
exhibited a potential concentration-dependent manner.
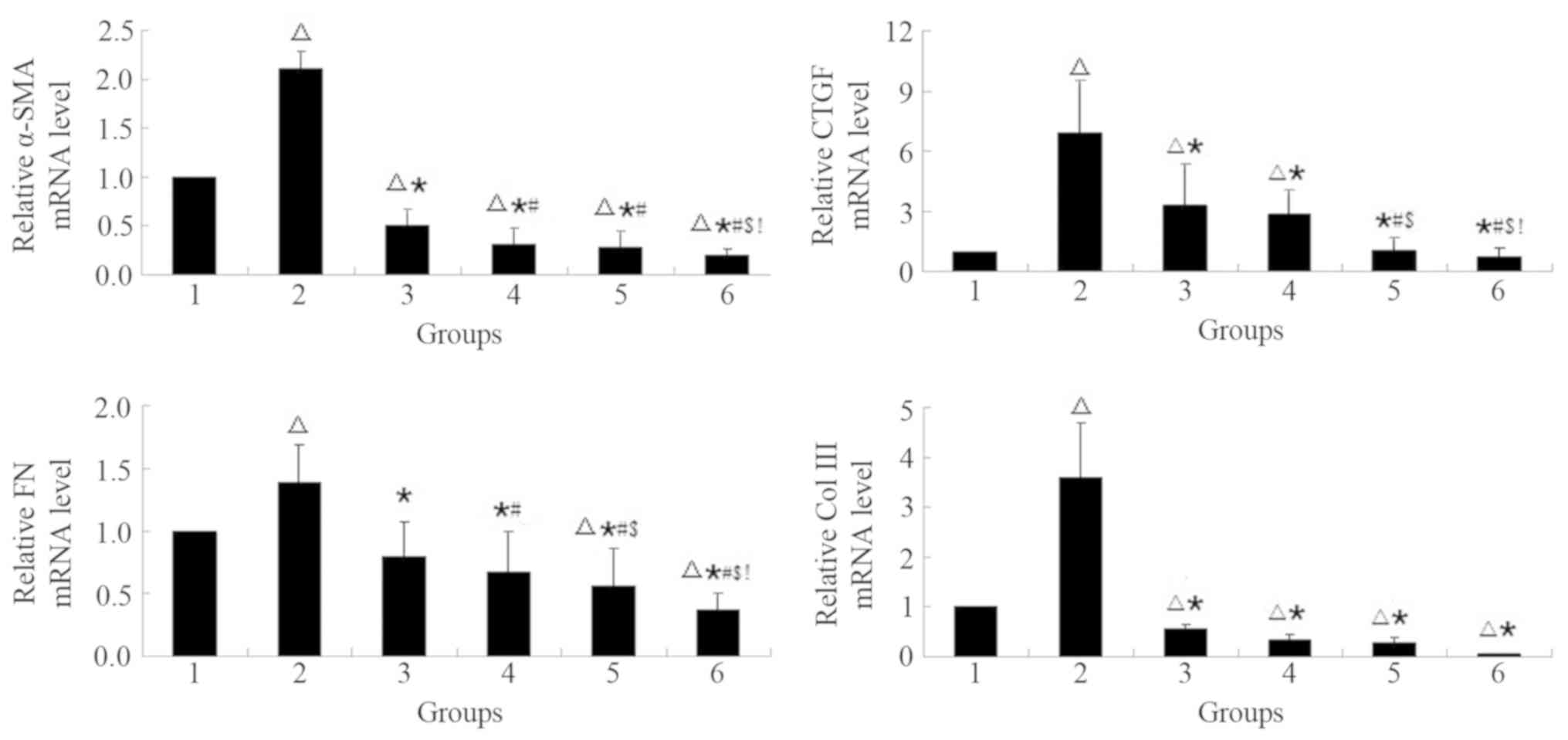 | Figure 3.MG132 pre-treatment decreases on the
mRNA level of fibrosis-associated factors in NRK-49F cells
simulated by TGF-β1 in a concentration-dependent manner. The mRNA
levels were normalized to the control group. The cells were treated
with the proteasome inhibitor, MG132, at specific concentrations
(0–5 µM) with or without TGF-β1 (5 ng/ml) for 24 h.
∆P<0.05 vs. 1; *P<0.05 vs. 2;
#P<0.05 vs. 3; $P<0.05 vs. 4; and
!P<0.05 vs. 5. 1, control; 2, 5 ng/ml TGF-β1; 3, 0.5
µM MG132 + 5 ng/ml TGF-β1; 4, 1 µM MG132 + 5 ng/ml TGF-β1; 5, 2.5
µM MG132 + 5 ng/ml TGF-β1; 6, 5 µM MG132 + 5 ng/ml TGF-β1; Col III,
collagen type III; FN, fibronectin; CTGF, connective tissue growth
factor; α-SMA, α-smooth muscle actin; TGF, transforming growth
factor. |
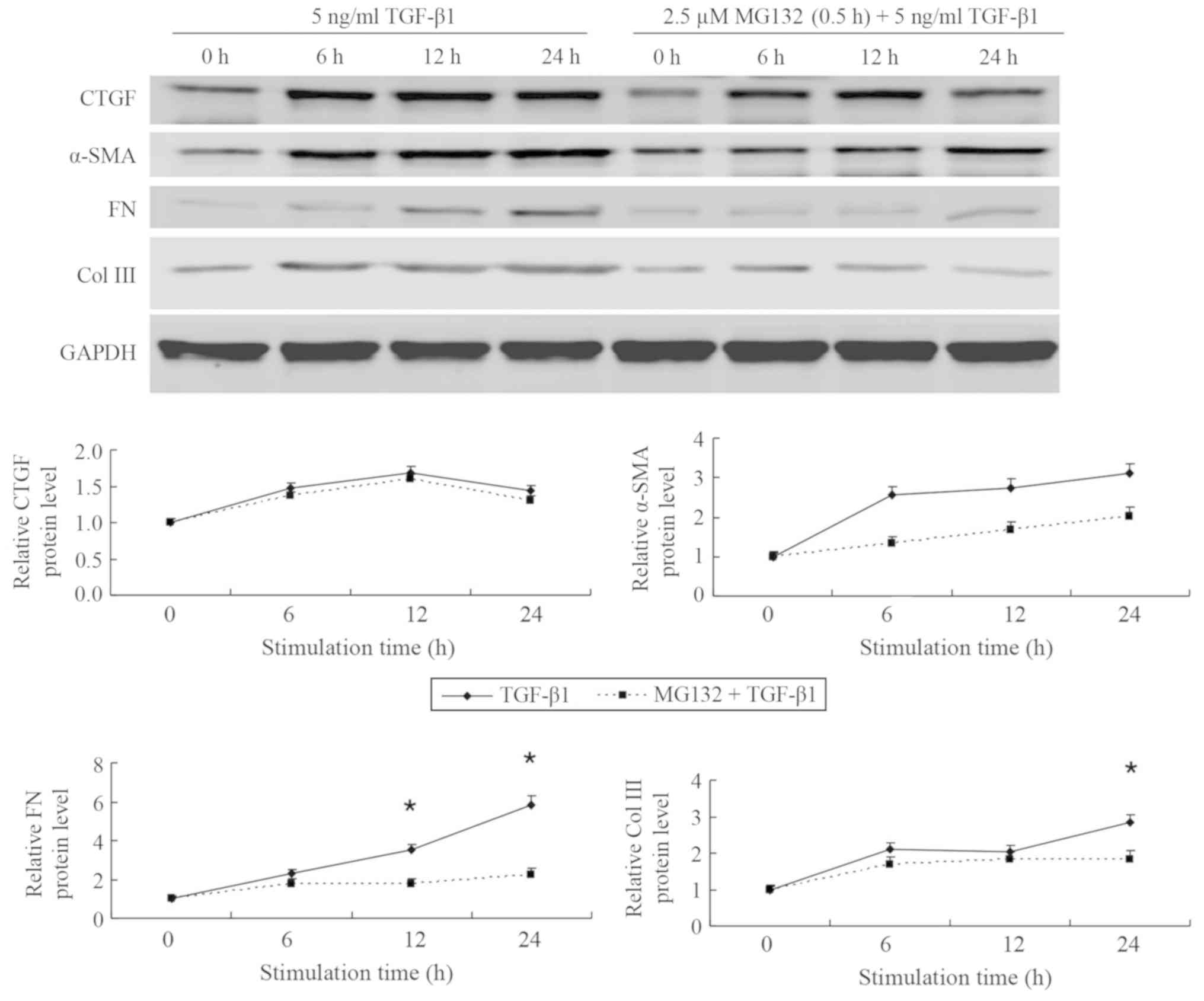
MG132 pretreatment decreases the
protein expression of fibrosis-associated factors in NRK-49F cells
simulated by TGF-β1 in a potential time-dependent manner
Compared with the control group set to 1, CTGF
expression was increased to 1.5-fold at 6 h, 1.7-fold at 12 h and
1.4-fold at 24 h in the TGF-β1 group; indicating that expression
may declined over time following an initial increase (Fig. 4). In the group pretreated with MG132,
CTGF expression of 1.4, 1.6 and 1.3-fold were measured at 6, 12 and
24 h, respectively. CTGF expression did not change significantly
over time or between the TGF-β1 and the MG132 pretreatment groups
(P>0.05). Compared with the control group set to 1, α-SMA
protein levels increased to 2.6-fold at 6 h, 2.8-fold at 12 h and
3.1-fold at 24 h. Following pretreatment with 2.5 µM MG132, these
values were 1.3, 1.7 and 2.0 at 6, 12 and 24 h, respectively;
differences between the TGF-β1 and the pretreatment groups or over
time were not significant (P>0.05). Protein levels of FN were
determined at 2.3, 3.5 and 5.8-fold TGF-β1-treated cells at 6, 12
and 24 h, respectively, compared with the control group. The values
determined at 12 and 24 h were significantly increased in the
TGF-β1 group compared with the 0 h control (P<0.05). After
pre-treatment with MG132, FN protein expression values decreased to
1.8, 1.8 and 2.2-fold at 6, 12 and 24 h, respectively; differences
over time or between the groups were not significant. Expression of
Col III was 2.1, 2.0 and 2.8-fold in the TGF-β1 group at 6, 12 and
24 h, respectively and for the MG132 pretreatment group these
values were 1.7, 1.8 and 1.8, respectively, compared with the 0 h
control; differences between the groups were not significant. Col
III protein expression was significantly increased at 24 h in the
TGF-β1 group compared with the 0 h control (P<0.05).
MG132 decreases the protein level of
fibrosis-associated factors in NRK-49F cells simulated by TGF-β1 in
a potential concentration-dependent manner
Following pretreatment with 5 µM MG132 and
stimulation with 5 ng/ml TGF-β1, the protein levels of CTGF, α-SMA,
FN and Col III were significantly decreased compared with the
TGF-β1 group (P<0.05; Fig. 5).
Compared with the TGF-β1 group, the expression of CTGF decreased to
0.6-, 0.4-, 0.3- and 0.3-fold as the MG132 concentration increased
to 0.5, 1, 2.5 and 5 µM, respectively; of which changes at 1, 2.5
and 5 µM MG132 were significant (all P<0.05). The expression of
α-SMA decreased to 0.7-, 0.6-, 0.6- and 0.5-fold as the MG132
concentration increased to 0.5, 1, 2.5 and 5 µM, respectively,
compared with the TGF-β1 group; only the 5 µM MG132 concentration
significantly decreased α-SMA expression (P<0.05). The
expression of FN decreased to 0.5-, 0.5-, 0.3- and 0.2-fold, and
Col III expression stagnated at 1.0, and decreased to 0.8-, 0.4-
and 0.3-fold as the MG132 concentration increased to 0.5, 1, 2.5
and 5 µM, respectively, compared with the TGF-β1 group. Compared
with the TGF-β1 group, Col III expression was significantly
decreased with 2.5 and 5 µM MG132 (both P<0.05).
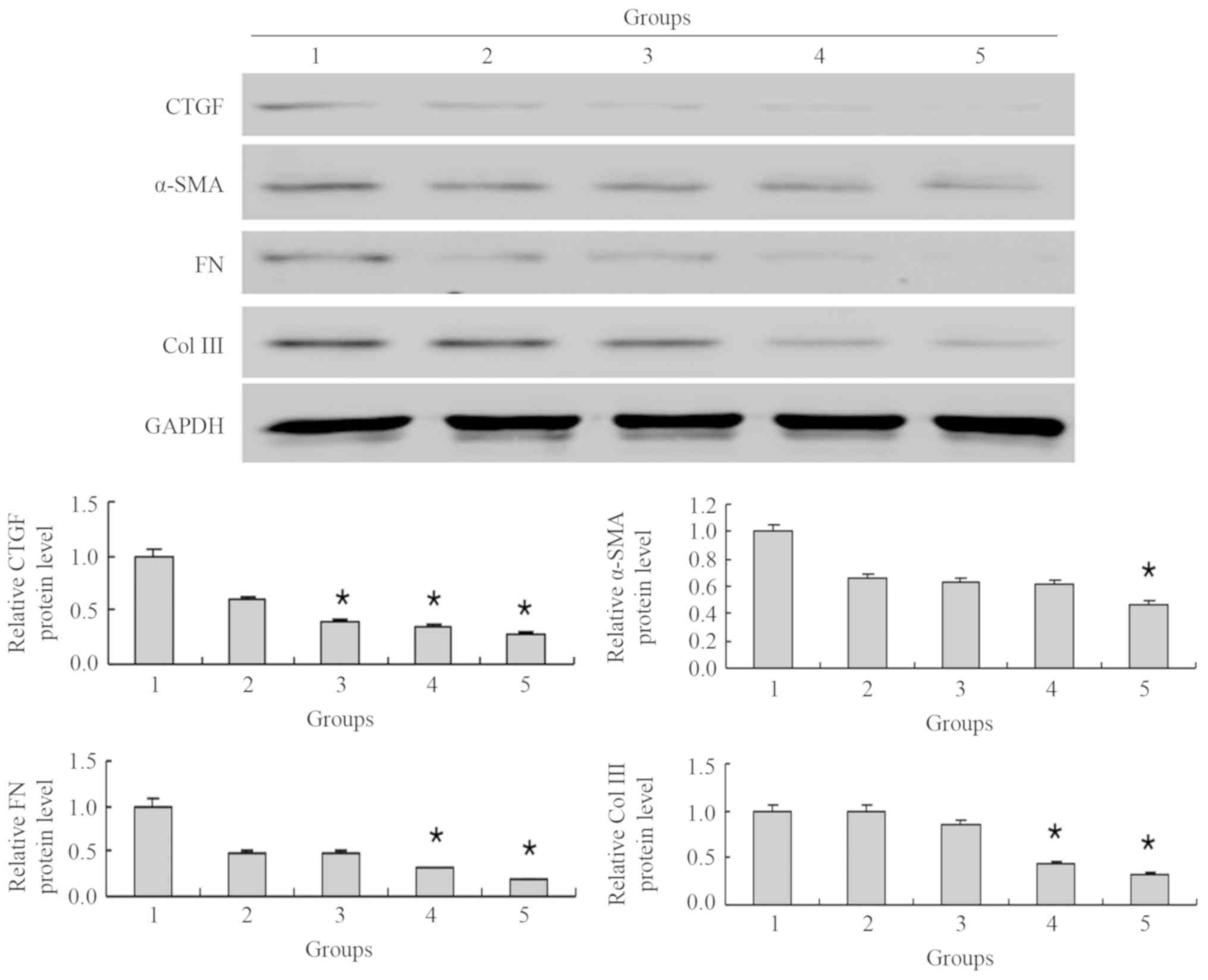 | Figure 5.MG132 decreases the protein level of
fibrosis-associated factors in NRK-49F cells simulated by TGF-β1 in
what appears to be a concentration-dependent manner. The protein
levels were normalized to the control group. The cells were treated
with the proteasome inhibitor, MG132, at specific concentrations
(0–5 µM) with TGF-β1 (5 ng/ml) for 24 h. *P<0.05 vs. 2. 1, 5
ng/ml TGF-β1; 2, 0.5 µM MG132 + 5 ng/ml TGF-β1; 3, 1 µM MG132 + 5
ng/ml TGF-β1; 4, 2.5 µM MG132 + 5 ng/ml TGF-β1; 5, 5 µM MG132 + 5
ng/ml TGF-β1; Col III, collagen type III; FN, fibronectin; CTGF,
connective tissue growth factor; α-SMA, α-smooth muscle actin; TGF,
transforming growth factor. |
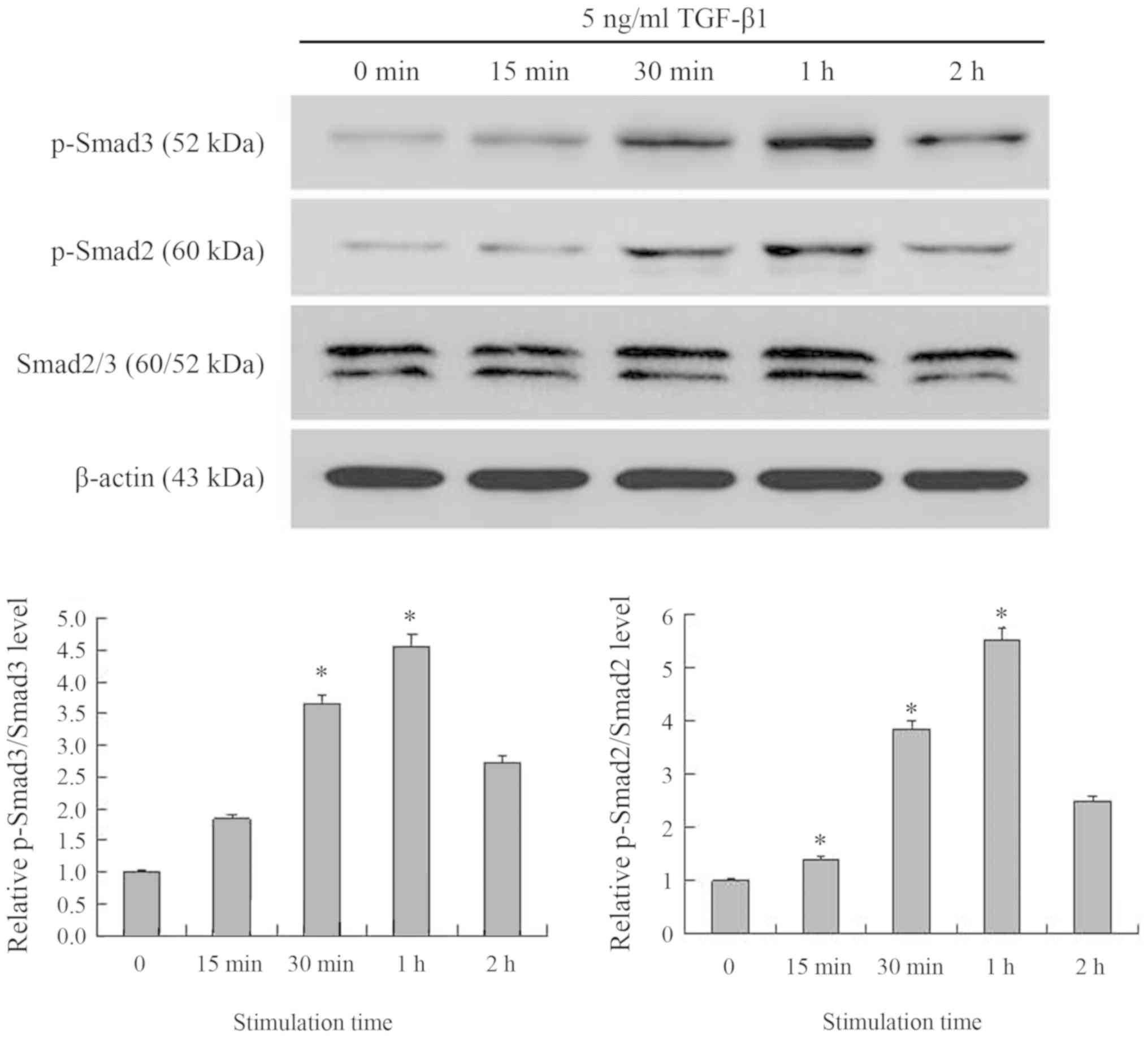
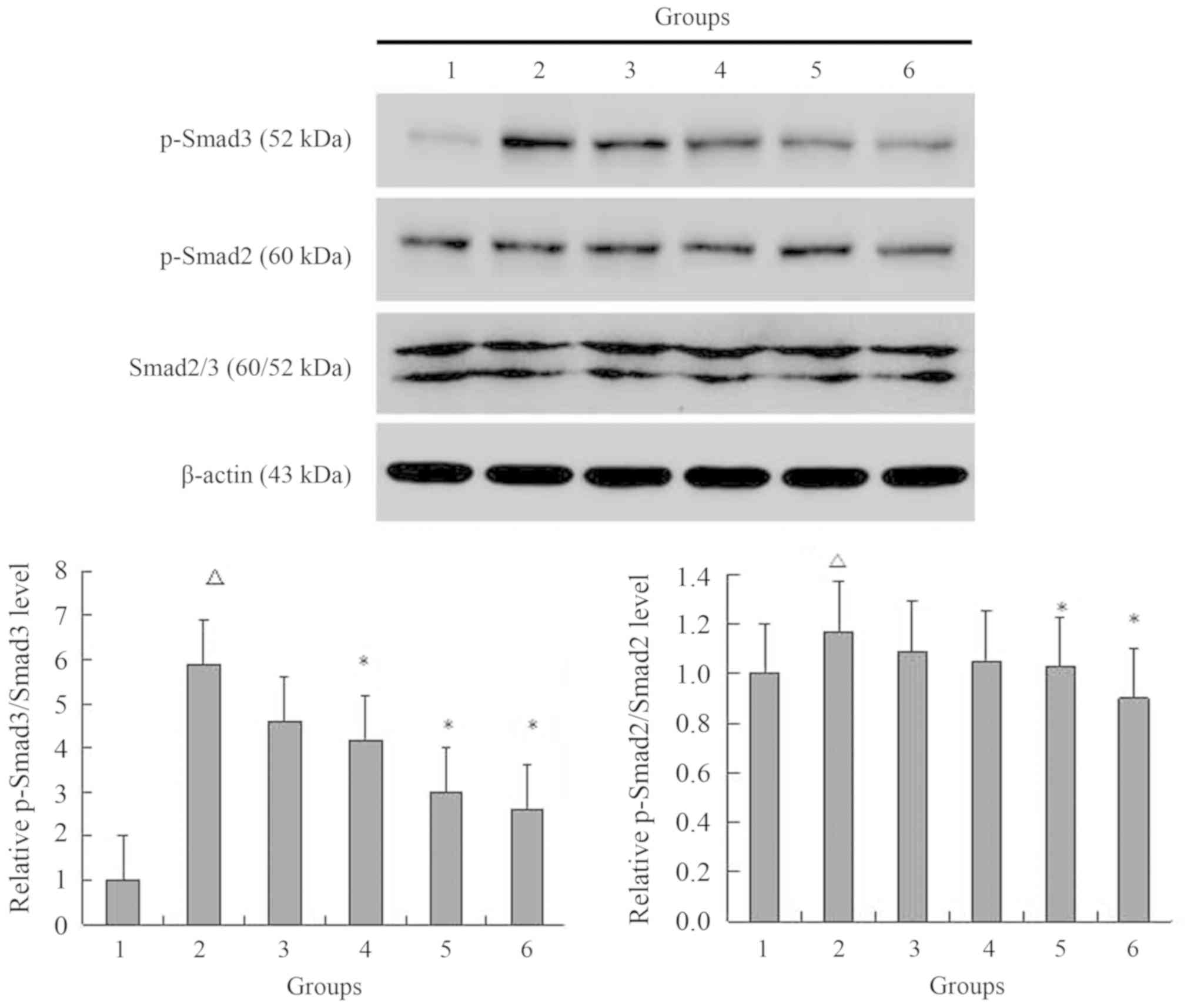
TGF-β1 increases on the
phosphorylation of Smads
The results indicated that NRK-49F cells express the
p-Smad2, and p-Smad3 β proteins (Fig.
6). After the cells were stimulated with TGF-β1 for 15 min, the
expression of p-Smad2 and p-Smad3 increased, reaching their peak
expression after 1 h of treatment, but decreasing after 2 h.
Compared with the control group, p-Smad3 levels were 1.8-fold more
greatly expressed at 15 min, 3.6-fold higher at 30 min, 4.6-fold
higher at 1 h and 2.7-fold higher at 2 h. p-Smad3 levels were
significantly increased after 30 min and 1 h of TGF-β1 treatment
compared with the control group (both P<0.05). The levels of
p-Smad2 expression were 1.4-, 3.9-, 5.5- and 2.5-fold more highly
expressed at 15, 30 min, 1 and 2 h, respectively. p-Smad2 levels
were significantly increased after 15, 30 min and 1 h of TGF-β1
treatment compared with the control group (all P<0.05).
MG132 decreases the phosphorylation of
Smads in NRK-49F cells simulated by TGF-β1
The results indicated that 5 ng/ml TGF-β1
significantly increased the expression of p-Smad2 and p-Smad3
compared with the control group (P<0.05; Fig. 7). After pretreatment with different
concentrations of MG132 and then stimulation with 5 ng/ml TGF-β1,
the expression levels of p-Smad2 and p-Smad3 decreased. TGF-β1
increased the p-Smad3 protein expression levels to 5.9-fold
compared with the control group. This change was decreased to 4.6-,
4.2-, 3.0- and 2.6-fold as the MG132 concentration increased to
0.5, 1, 2.5 and 5 µM, respectively, compared with the control
group. p-Smad3 levels were significantly decreased with 1, 2.5 and
5 µM MG132 pretreatments compared with the TGF-β1 group (all
P<0.05). TGF-β1 increased the p-Smad2 levels to 1.2-fold
compared with the control group. Compared with the control group,
fold-changes decreased to 1.1-, 1.0-, 1.0- and 0.9-fold, as the
MG132 concentration increased to 0.5, 1, 2.5 and 5 µM,
respectively. p-Smad2 levels were significantly decreased with 2.5
and 5 µM MG132 pretreatments compared with the TGF-β1 group (both
P<0.05).
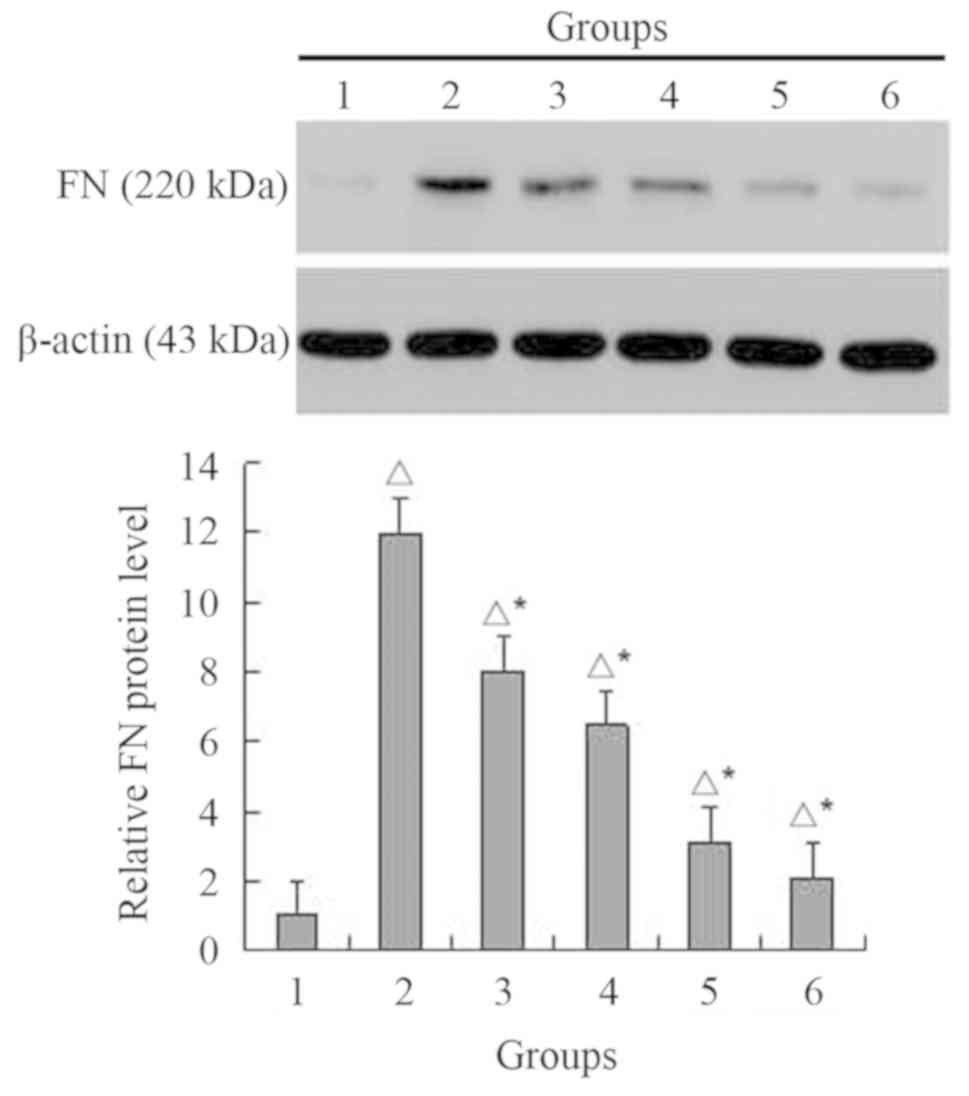
MG132 decreases the expression of FN
in NRK-49F cells simulated by TGF-β1
TGF-β1 (5 ng/ml) induced a significant, 11.9-fold
increase in the FN protein expression levels compared with control
group (P<0.05; Fig. 8).
Fold-changes decreased to 8.0, 6.5, 3.1 and 2.1-fold as the MG132
concentration increased to 0.5, 1, 2.5 and 5 µM, respectively. FN
levels decreased significantly compared with the control and TGF-β1
groups, respectively (both P<0.05).
Discussion
Renal interstitial fibroblasts are a subtype of
renal interstitium intrinsic cells, which constitute the main
ECM-secreting cell type during fibrosis (22). Due to their ability to proliferate
quickly, interstitial fibroblasts can produce copious amounts of FN
and collagen, specifically Col I and Col III (23). These cells serve a very important
role in renal interstitial fibrosis, overproducing ECM materials
that lead to scarring and fibrosis (24). Therefore, the authors of the current
study monitored the expression of ECM materials and the signal
transduction pathways regulating their production when renal
interstitial fibroblasts were stimulated with pro-inflammatory
signals (e.g. TGF-β1). TGF-β1 is closely connected with renal
interstitial fibrosis, as it promotes the production of ECM
materials (Col I, III and IV, and FN), while decreasing the
expression of matrix metalloproteinases, thereby increasing the
generation of plasminogen activator inhibitor and tissue inhibitor
of metalloproteinases, which altogether slow ECM degradation
(25). When the TGF-β1 signal is
transduced, it connects with TGF-β1 receptor I and II, causing
phosphorylation of the signal transduction factors, Smad2 and
Smad3; these factors translocate to the cell nucleus with Smad4 to
initiate the transcription of genes involved in fibrosis, cell
proliferation and inflammation (26). The Smad signaling pathway has been
implicated in several renal diseases and pathophysiologic
reactions, including diabetic nephropathy, glomerular nephritis and
glomerular sclerosis (27).
CTGF is an important downstream effector of TGF-β1
(28). It has been demonstrated to
promote TGF-β1-induced cell proliferation and ECM deposition,
inducing conglutination and chemotactic effects, while promoting
epithelial to mesenchymal transition (29). Several in vitro studies have
determined that CTGF stimulates the proliferation of cardiac
fibroblasts and increase the production of the ECM (30,31).
Resulting myofibroblasts and tubular epithelial cells have been
revealed to produce ECM materials to induce interstitial fibrosis
(32). α-SMA is a phenotypic
transformation marker that is highly expressed in myofibroblasts,
which are widely used as a marker of cell differentiation, while
its production simultaneously contributes significantly to fibrosis
(33).
Activation of the ubiquitin-proteasome pathway has
been demonstrated to lead to the selective degradation of
intracellular proteins and to the regulation of their degradation
(34). By controlling the
concentration of intracellular proteins, cells can maintain their
internal environment (35). Key
proteins in this pathway include those that control inflammation
and the cell cycle (36). Therefore,
proteasome inhibitors have potential therapeutic applications in
limiting inflammation and tumor growth (37). Clinical have demonstrated that
Bortezomib (the first proteasome inhibitor drug) can induce the
apoptosis of several haemal and solid tumors, including multiple
myeloma, mantle cell lymphoma, non-small cell lung carcinoma,
oophoroma, carcinoma of the pancreas, carcinoma of the prostate,
and head and neck neoplasms (38).
Proteasome inhibitors have been adopted in pilot studies involving
antibody-mediated renal rejection in amyloid light chain
amyloidosis with increasing scientific interest in their possible
applications in lupus, IgA nephropathy, idiopathic nephrotic
syndrome and renal fibrosis therapies (39,40). The
ubiquitin-proteasome inhibitor, MG132, is a specific inhibitor that
directly affects uridine phosphorylase (UPP) (41). When UPP is inhibited, the degradation
of intracellular abnormal proteins, such as caspase 3, reduces
(42). Activated caspase 3
decomposes substrates in the cytoplasm and nucleus, resulting in
chromosome condensation, mitochondrial swelling and ultimately
apoptosis (43). Caspase 3 can
thereby reduce extracellular matrix secretion through the lysis of
cells involved in its generation (44). Studies have revealed that MG132 can
inhibit alimentary canal neoplasms and leukemia (45–47).
The authors of the current study used TGF-β1 to
induce myofibroblast transformation in NRK-49F cells and observed
that p-Smad2 and p-Smad3 protein expression increased; these
proteins have been known to promote the signal transduction
pathways involved in fibrosis. It was demonstrated that MG132 can
decrease the effects of TGF-β1 by reducing the transcription of key
factors involved in fibrosis, including CTGF, α-SMA, FN and Col
III.
During the TGF-β1 signal transduction process, there
are no known proteins that readily switch off transcription
(48). Therefore, inhibiting
proteins involved in the TGF-β1 signaling pathway (e.g. Smad2, 3
and 4) is a plausible approach to limiting fibrosis (49). Another possible target would be the
down-regulation of the Smad7 protein, which can lead to the
inhibition of receptor-activated Smad-Smad4-complex activity,
preventing the signal from progressing, thereby also decreasing or
slowing the fibrotic process (50).
Ultimately, proteasome inhibitors possess some
efficacy in delaying or impeding the process of renal interstitial
fibrosis. They can promote cell apoptosis while down-regulating
cytokine production, inflammation and the deposition of ECM
materials, which has been determined to contribute to fibrosis
(51). Therefore, the application of
proteasome inhibitors in the treatment of fibrosis may be widely
beneficial.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 30270613
and 30771000).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH served an important role in interpreting the
results, and drafting and writing the manuscript. LH, HC and JL
performed experiments. BZ and YJ performed the statistical analyses
of the data. WW was involved in drafting and reviewing the
manuscript and contributed to the analysis and interpretation of
data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu M, Ning X, Li R, Yang Z, Yang X, Sun S
and Qian Q: Signalling pathways involved in hypoxia-induced renal
fibrosis. J Cell Mol Med. 21:1248–1259. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vega G, Alarcon S and San Martin R: The
cellular and signalling alterations conducted by TGF-nalling
competino renal fibrosis. Cytokine. 12:115–125. 2016. View Article : Google Scholar
|
|
3
|
Meng XM and Lan HY: Transforming growth
factor-β and renal fibrosis. Sheng Li Xue Bao. 70:612–622. 2018.(In
Chinese). PubMed/NCBI
|
|
4
|
Xu J, Yu TT, Zhang K, Li M, Shi HJ, Meng
XJ, Zhu LS and Zhu LK: HGF alleviates renal interstitial fibrosis
via inhibiting the TGF-β1/SMAD pathway. Eur Rev Med Pharmacol Sci.
22:7621–7627. 2018.PubMed/NCBI
|
|
5
|
Soleimani A, Pashirzad M, Avan A, Ferns
GA, Khazaei M and Hassanian SM: Role of the transforming growth
factor-β signaling pathway in the pathogenesis of colorectal
cancer. J Cell Biochem. 16:2018.
|
|
6
|
Liu P, Zhu L, Zou G and Ke H: Matrine
suppresses pancreatic fibrosis by regulating TGF-β/Smad signaling
in rats. Yonsei Med J. 60:79–87. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen W, Zhou ZQ, Ren YQ, Zhang L, Sun LN,
Man YL and Wang ZK: Effects of long non-coding RNA LINC00667 on
renal tubular epithelial cell proliferation, apoptosis and renal
fibrosis via the miR-19b-3p/LINC00667/CTGF signaling pathway in
chronic renal failure. Cell Signal. 54:102–114. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shieh JM, Tsai YJ, Chi JC and Wu WB: TGFβ
mediates collagen production in human CRSsNP nasal mucosa-derived
fibroblasts through Smad2/3-dependent pathway and CTGF induction
and secretion. J Cell Physiol. 13:2018.
|
|
9
|
Balah A, Ezzat O and Akool ES: Vitamin E
inhibits cyclosporin A-induced CTGF and TIMP-1 expression by
repressing ROS-mediated activation of TGF-β/Smad signaling pathway
in rat liver. Int Immunopharmacol. 65:493–502. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen L, Ji Q, Zhu H, Ren Y, Fan Z and Tian
N: MiR-30a attenuates cardiac fibrosis in rats with myocardial
infarction by inhibiting CTGF. Exp Ther Med. 15:4318–4324.
2018.PubMed/NCBI
|
|
11
|
Chen M, Yan T, Ma K, Lai L, Liu C, Liang L
and Fu X: Botulinum toxin type A inhibits α-smooth muscle actin and
myosin II expression in fibroblasts derived from scar contracture.
Ann Plast Surg. 77:46–49. 2016. View Article : Google Scholar
|
|
12
|
Holm Nielsen S, Willumsen N, Leeming DJ,
Daniels SJ, Brix S, Karsdal MA, Genovese F and Nielsen MJ:
Serological assessment of activated fibroblasts by alpha-smooth
muscle actin (α-SMA): A noninvasive biomarker of activated
fibroblasts in lung disorders. Transl Oncol. 12:368–374. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Masola V, Bellin G, Gambaro G and Onisto
M: Heparanase: A multitasking protein involved in extracellular
matrix (ECM) remodeling and intracellular events. Cells.
7:e2362018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meng XM, Tang PM, Li J and Lan HY:
TGF-β/Smad signaling in renal fibrosis. Front Physiol. 6:82015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Casas-Grajales S, Alvarez-Suarez D,
Ramos-Tovar E, Dayana Buendía-Montaño L, Reyes-Gordillo K, Camacho
J, Tsutsumi V and Lakshman MR: Stevioside inhibits experimental
fibrosis by down-regulating profibrotic Smad pathways and blocking
HSC activation. Basic Clin Pharmacol Toxicol. 12:123–126. 2018.
|
|
16
|
Sakairi T, Hiromura K, Takahashi S,
Hamatani H, Takeuchi S, Tomioka M, Maeshima A, Kuroiwa T and Nojima
Y: Effects of proteasome inhibitors on rat renal fibrosis in vitro
and in vivo. Neprology (Carlton). 16:76–86. 2011. View Article : Google Scholar
|
|
17
|
Costa AR, Machado N, Rego A, Sousa MJ,
Côrte-Real M and Chaves SR: Proteasome inhibition prevents cell
death induced by the chemotherapeutic agent cisplatin downstream of
DNA damage. DNA Repair (Amst). 73:28–33. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van de Donk NWCJ and Yong K: Oral
proteasome inhibitor maintenance for multiple myeloma. Lancet.
393:204–205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu B, Jin Y, Han L, Chen H, Zhong F, Wang
W and Chen N: Proteasome inhibitor inhibits proliferation and
induces apoptosis in renal interstitial fibroblasts. Phamacol Rep.
65:1357–1365. 2013.
|
|
20
|
Arocho A, Chen B, Ladanyi M and Pan Q:
Validation of the 2-DeltaDeltaCt calculation as an alternate method
of data analysis for quantitative PCR of BCR-ABL P210 transcripts.
Diagn Mol Pathol. 15:56–61. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Johnston EF and Gillis TE: Transforming
growth factor-β1 induces differentiation of rainbow trout
(Oncorhynchus mykiss) cardiac fibroblasts into myofibroblasts. J
Exp Biol. 17:2212018.
|
|
22
|
Waasdorp M, de Rooij DM, Florquin S,
Duitman J and Spek CA: Protease-activated receptor-1 contributes to
renal injury and interstitial fibrosis during chronic obstructive
nephropathy. J Cell Mol Med. 23:1268–1279. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang C, Luo H, Xu Y, Tao L, Chang C and
Shen X: Salvianolic acid B-alleviated angiotensin II induces
cardiac fibrosis by suppressing NF-κB pathway in vitro. Med Sci
Monit. 24:7654–7664. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shao S and Zhang X, Duan L, Fang H, Rao S,
Liu W, Guo B and Zhang X: Lysyl hydroxylase inhibition by minoxidil
blocks collagen deposition and prevents pulmonary fibrosis via
TGF-β1/Smad3 signaling pathway. Med Sci Monit. 24:8592–8601. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kothapalli D and Grotendorst GR: CTGF
modulates cell cycle progression in cAMP-arrested NRK fibroblasts.
J Cell Physiol. 182:119–126. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang W, Koka V and Lan HY: Transforming
growth factor-beta and Smad signaling in kidney diseases.
Nephrology (Carlton). 10:48–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu Y, Zhou J and Tao G: Molecular aspects
of chronic radiation enterits. Clin Invest Med. 34:E119–E124. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie Y, Ostriker AC, Jin Y, Hu H, Sizer AJ,
Peng G, Morris AH, Ryu C, Herzog EL, Kyriakides T, et al: MO7 is a
negative feedback regulator of TGF-β signaling and fibrosis.
Circulation. 10:121–125. 2018.
|
|
30
|
Petrosino JM, Leask A and Accornero F:
Genetic manipulation of CCN2/CTGF unveils cell-specific
ECM-remodeling effects in injured skeletal muscle. FASEB J.
14:2018.
|
|
31
|
Feng Jian, Zuo Yumei, Xu Liang, et al:
Study of oleanolic acid in inhibiting rat cardiac fibroblasts
proliferation induced by angiotensin II through ROS-CTGF pathway.
Zhong Yao Xin Yao Yu Lin Chuang Bing Li. 5:78–82. 2014.(In
Chinese).
|
|
32
|
Park J, Choi G, Yim MJ, Lee JM, Yoo JS,
Park WS, Park SK, Park S, Seo SK, Kim TG, et al: Effect of
phlorotannins on myofibroblast differentiation and ECM protein
expression in transforming growth factor β1induced nasal
polypderived fibroblasts. Int J Mol Med. 42:2213–2220.
2018.PubMed/NCBI
|
|
33
|
Kalekou H, Kostopoulos I, Milias S and
Papadimitriou CS: Comparative study of CD34, alpha-SMA and
h-caldesmon expression in the stroma of gynaecomastia and male
breast carcinoma. Histopathology. 47:74–81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang X, Wei S, Ni S, Huang Y and Qin Q:
Ubiquitin-proteasome system is required for efficient replication
of singapore grouper iridovirus. Front Microbiol. 26:27982018.
View Article : Google Scholar
|
|
35
|
Delpire E and Gagnon KB: Water homeostasis
and cell volume maintenance and regulation. Curr Top Membr.
81:3–52. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Della Sala G, Agriesti F, Mazzoccoli C,
Tataranni T, Costantino V and Piccoli C: Clogging the
ubiquitin-proteasome machinery with marine natural products: Last
decade update. Mar Drugs. 16:E4672018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang W, Luo J, Sheng W, Xue J, Li M, Ji J,
Liu P, Zhang X, Cao J and Zhang S: Proteomic profiling of
radiation-induced skin fibrosis in rats: Targeting the
ubiquitin-proteasome system. Int J Radiat Oncol Biol Phys.
95:751–760. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang M, Cai X, Yang J, Wang C, Tong L,
Xiao J and Li L: A targeted and pH-responsive bortezomib
nanomedicine in the treatment of metastatic bone tumors. ACS Appl
Mater Interfaces. 19:2018.
|
|
39
|
Coppo R: Proteasome inhibitors in
progressive renal diseases. Nephrol Dial Transplant. 29 (Suppl
1):i25–i30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oliva L, Orfanelli U, Resnati M, Raimondi
A, Orsi A, Milan E, Palladini G, Milani P, Cerruti F, Cascio P, et
al: The amyloidogenic light chain is a stressor that sensitizes
plasma cells to proteasome inhibitor toxicity. Blood.
129:2132–2142. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Luo D, Dong XW, Yan B, Liu M, Xue TH, Liu
H, You JH, Li F, Wang ZL and Chen ZN: MG132 selectively upregulates
MICB through the DNA damage response pathway in A549 cells. Mol Med
Rep. 20:2018.
|
|
42
|
Luo DW, Zheng Z, Wang H, Fan Y, Chen F,
Sun Y, Wang WJ, Sun T and Xu X: UPP mediated diabetic retinopathy
via ROS/PARP and NF-κB inflammatory factor pathways. Curr Mol Med.
15:790–799. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cogo F, Poreba M, Rut W, Groborz K, Smyth
P, Johnston MC, Williams R, Longley DB, Burden RE, Salvesen GS, et
al: Development of an advanced nanoformulation for the
intracellular delivery of a caspase-3 selective activity-based
probe. Nanoscale. 11:742–751. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao C, Qiu L, Lv P Han A, Fang G, Liu J
and Wang S: AuNP-peptide probe for caspase-3 detection in living
cells by SERS. Analyst. 14:2018.
|
|
45
|
Ortiz-Lazareno PC, Bravo-Cuellar A,
Lerma-Díaz JM, Jave-Suárez LF, Aguilar-Lemarroy A,
Domínguez-Rodríguez JR, González-Ramella O, De Célis R,
Gómez-Lomelí P and Hernández-Flores G: Sensitization of U937
leukemia cells to doxorubicin by the MG132 proteasome inhibitor
induces an increase in apoptosis by suppressing NF-kappa B and
mitochondrial membrane potential loss. Cancer Cell Int.
14:14752014. View Article : Google Scholar
|
|
46
|
Ding WX, Ni HM, Chen X, Yu J, Zhang L and
Yin XM: A coordinated action of Bax, PUMA, and p53 promotes
MG132-induced mitochondria activation and apoptosis in colon cancer
cells. Mol Cancer Ther. 6:1062–1069. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ustundag Y, Bronk SF and Gores GJ:
Proteasome inhibition-induces endoplasmic reticulum dysfunctin and
cell death of human cholangiocarcinoma cells. World J
Gastroenterol. 13:851–857. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang F, Zhang ZF, He YR, Wu HY and Wei SS:
Effects of dipeptidyl peptidase-4 inhibitors on transforming growth
factor-β1 signal transduction pathways in the ovarian fibrosis of
polycystic ovary syndrome rats. J Obstet Gynaecol Res. 4:2018.
|
|
49
|
Sun Q, Wang Y, Zhang J and Lu J: ENMD-1068
inhibits liver fibrosis through attenuation of TGF-β1/Smad2/3
signaling in mice. Sci Rep. 7:54982017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hu ZC, Shi F, Liu P, Zhang J, Guo D, Cao
XL, Chen CF, Qu SQ, Zhu JY and Tang B: TIEG1 represses
smad7-mediated activation of TGF-β1/Smad signaling in keloid
pathogenesis. J Invest Dermatol. 137:1051–1059. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zulato E, Favaretto F, Veronese C,
Campanaro S, Marshall JD, Romano S, Cabrelle A, Collin GB, Zavan B,
Belloni AS, et al: ALMS1-deficient fibroblasts over-express
extra-cellular matrix components, display cell cycle delay and are
resistant to apoptosis. PLoS One. 6:e190812011. View Article : Google Scholar : PubMed/NCBI
|