Macrophages, originating from monocytic precursors,
have multiple functions and are widely known for their phagocytic
capacity, antigen-presenting function and active secretory
properties. Once localized in the liver, macrophages exhibit high
phagocytic activity to remove endotoxins and pernicious substances
from the portal vein. Resident tissue macrophages and inflammatory
monocytes recruited from bone marrow have a dual role in organ
damage induced by various factors, including infection, auto-immune
disorders and mechanical or toxic injuries (1). Following liver injury, the resident
liver macrophages are activated and exert pro-inflammatory,
pro-wound healing and restorative effects at different stages of
hepatic injury and the repair response (2). Studies using animal models of
chemical-induced liver injury have identified macrophages as the
key regulators of liver repair and regeneration, or fibrosis. In
the present review, the various functions of macrophages in hepatic
toxicity are illustrated.
Macrophages are widely distributed phagocytic innate
immune cells that have essential roles in tissue homeostasis and
the host defence. The diverse tissue macrophage populations
resident in most tissues of the body mainly originate from the yolk
sac in the process of embryogenesis, and certain tissue macrophages
are developed from fetal liver and hematopoietic progenitors at
later time-points (3). For closed
tissues, resident macrophages [e.g., lung alveolar macrophages and
liver Kupffer cells (KCs)] mostly originate from fetal liver
monocytes (4). Liver macrophages,
accounting for 20–35% of hepatic non-parenchymal cells, make up the
largest proportion (80–90%) of tissue macrophages in the host and
are an essential constituent of the mononuclear phagocytic system
(5). They consist of two distinct
populations: ‘Sessile’ KCs and motile liver macrophages, named as
monocyte-derived macrophages (MoMø). The former, ‘sessile’ KCs,
function as a scavenger to remove microorganisms and cell debris
from the blood and clear aged erythrocytes. Furthermore, in adult
tissues, they undergo self-maintenance independently of
hematopoietic stem cells. Phenotypes of ‘sessile’ KCs are
characterized as F4/80hi, CD11blo,
CD169+, CD68+, Mac-2+ and
CD80lo/− (6–8). The latter, motile liver macrophages are
distinct from ‘sessile’ KCs in terms of local migration to
participate in inflammatory foci (9). The major function of motile liver
macrophages is immune surveillance. Furthermore, these cells
directly originate from circulating monocytes. Surface expression
marker profiles of motile liver macrophages include
F4/80int, CD11bhi and CD80hi
(8). These characteristics suggest
that liver macrophages have distinct liver-specific gene expression
patterns.
In spite of the widespread use of specific terms to
define macrophage activation states [i.e., classically activated
(M1) and alternatively activated (M2), no experimental standards
are currently available for describing their activation (10). The original terminology using M1 and
M2 macrophage activation states is derived from different
macrophage gene expression patterns stimulated with interferon
(IFN)-γ/lipopolysaccharide (LPS) or interleukin (IL)-4/IL-13
(11). Within this terminology,
classically activated M1 macrophages (activated by IFN-γ, LPS or
high-mobility group protein 1) are functionally pro-inflammatory,
microbicidal and tumoricidal. Furthermore, they exhibit
anti-proliferative and cytotoxic activity. Virtually all of these
features are produced by the release of numerous inflammatory
cytokines, including tumor necrosis factor (TNF)-α, IL-1, IL-6 and
IL-12/23 (p40). By contrast, alternatively activated M2 macrophages
downregulate the inflammatory response and facilitate tissue repair
by increasing the expression of IL-10, IL-4/IL-13 and transforming
growth factor (TGF)-β, as well as vascular endothelial growth
factor (VEGF)-α. Due to the complex biological characteristics of
macrophage subsets, M2 macrophages are further subdivided to
account for their differences: M2a, M2b and M2c activated by
IL-4/IL-13, LPS/IL-1β and IL-10/glucocorticoids, respectively
(12). However, the concept of the
M1 and M2 definitions requires to be revised; this should include a
reproducible experimental standard, minimal reporting standards, a
definition of the activators and markers of activation (10,13). In
fact, macrophages display variable functions (e.g., initiation and
perpetuation of inflammation, promotion of liver fibrosis and
resolution of inflammation and fibrosis) in diverse
microenvironments. The plasticity of macrophage activation may be
elucidated by analyzing macrophage expression profiles.
Furthermore, it is noteworthy that the ‘restorative macrophages’ in
the liver fibrosis resolution phase derived from recruited
lymphocyte antigen 6 complex, locus C (Ly6C)+ monocytes
have a phenotype that is distinct from the M1/M2 definitions. Thus,
the M1/M2 terminology is, at large, not applicable to liver
diseases. However, whether the resolution of liver damage only
requires newly recruited monocytes or hepatic macrophages capable
of proliferating and switching their state of activation or that
may be transformed in response to complex signals has remained to
be sufficiently elucidated. In the present review, the original
definition from the respective previous studies discussed is quoted
when referring to mononuclear phagocyte cell types.
The liver is a crucial organ of drug and toxin
metabolism in the body, making it susceptible to toxic substances.
When the liver is injured, monocytes and KCs are recruited and
activated to exert numerous functional roles (1). Carbon tetrachloride (CCl4)
toxicity has various distinct ultimate outcomes, including
hepatocyte necrosis, liver fibrosis and cirrhosis, or even cancer
(14). Tissue repair is a complex
process influenced by intricate cellular signaling pathways
consisting of various cytokines, chemokines, nuclear receptors and
growth factors that may trigger the expression of pro-mitogenic
genes and finally promote cell division (15). Hence, the mouse model of
CCl4-induced hepatic injury or fibrosis is probably the
best representative experimental model for elucidating the various
roles of liver macrophages in response to liver injury or fibrosis
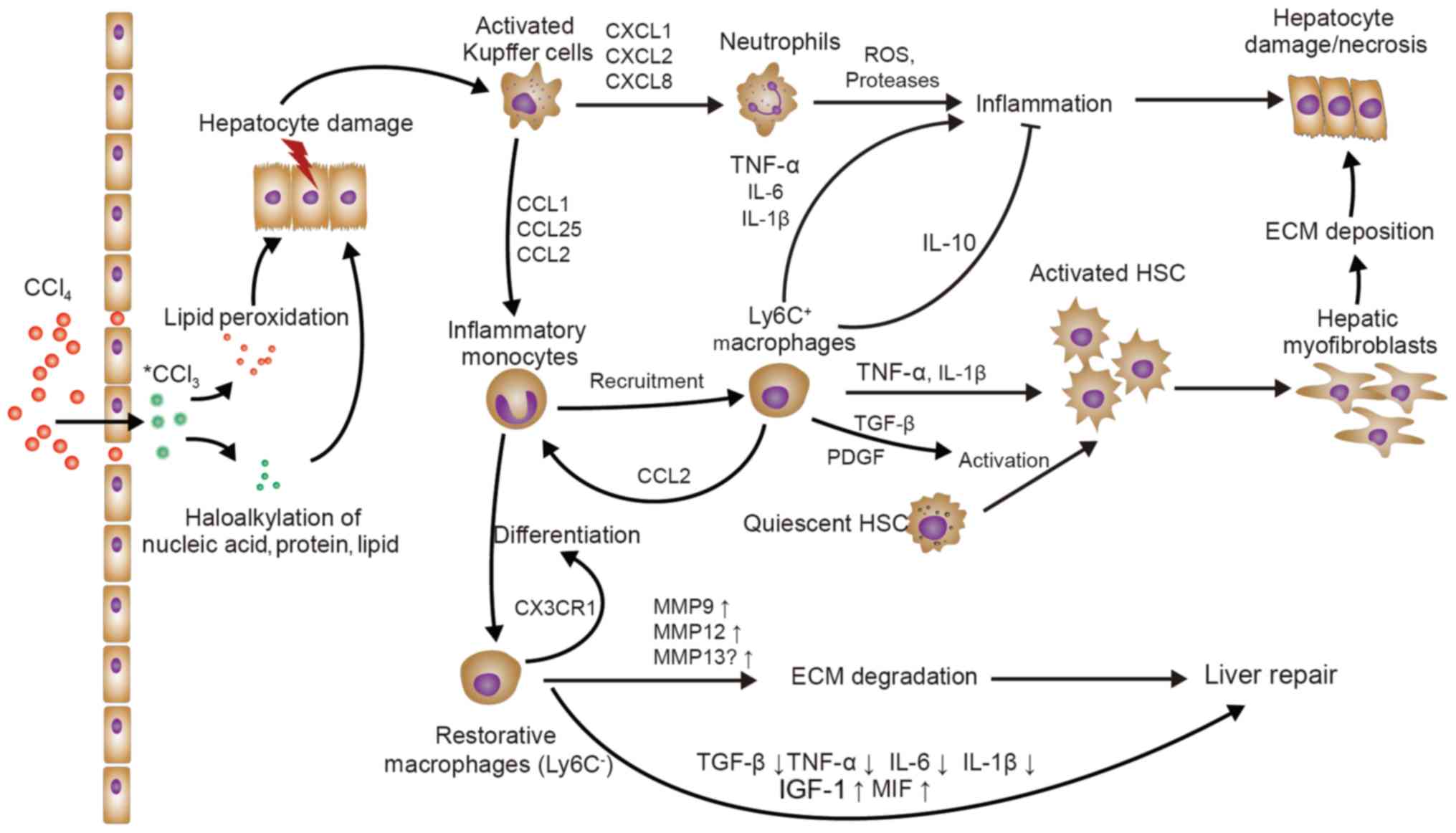
(Fig. 1).
Blood monocytes represent circulating precursors of
tissue dendritic cells and macrophages, and may be divided into two
major subsets in mice: Ly6C+/hi and Ly6C−/low
monocytes. Ly6C+ mouse monocytes highly express the
chemokine receptors C-C motif chemokine receptor 1 (CCR1) and CCR2,
whereas murine Ly6C− monocytes mainly express CCR5 and
C-X3-C motif chemokine receptor 1 (CX3CR1) (16). The early recruitment of
Ly6C+ monocytes, but not of Ly6C− monocytes,
to the liver upon toxic injury is mediated by CCR2 [ligand: C-C
motif chemokine ligand 2 (CCL2)] and CCR8 (ligand: CCL1). Studies
using CCR2-deficient (CCR2−/−) and monocyte
chemoattractant protein (MCP)-1−/− mice or specific
blockade suggested that CCR2 mediates the early accumulation of
inflammatory Ly6C+ monocytes in the damaged murine liver
(17). CCR8 is also crucial for
Ly6C+ monocyte infiltration into the injured murine
liver (18). Furthermore,
Ly6C+ monocytes, migrating from the blood to tissues
affected by infection, may differentiate into inflammatory
macrophages in inflamed tissues (19). Of note, Ly6C− monocytes
have a more patrolling role at the endothelium in a lymphocyte
function-associated antigen-1- and CX3CR1-dependent fashion, acting
as scavengers and orchestrating tissue repair (20), without inflammatory stimuli. It was
recently noted that Ly6C− monocytes do not represent a
distinct lineage, but instead originate from Ly6C+
monocytes regulated by CCAAT/enhancer binding protein β in the bone
marrow and blood, and that the lifespan of Ly6C−
monocytes may be negatively controlled by Ly6C+
peripheral blood monocytes (21). A
systematic assessment of the differential roles of these monocyte
subsets and their recruitment dynamics in liver injury is required
prior to the development of therapeutic strategies aimed at
targeting these monocyte subsets.
KCs are phagocytic innate immune cells that clear
dead cells and cell debris, and maintain liver homeostasis;
furthermore, they are able to sense liver injury and subsequently
orchestrate pro-inflammatory processes. Similar to toll-like
receptors (TLRs), one of the critical roles of KCs is the ability
to sense liver damage via pathogen-associated molecular patterns
(PAMPs) or damage-associated molecular patterns (DAMPs).
In the liver, PAMPs (e.g., those associated with
LPS, fungal components and flagellin) mostly originate from the gut
as a result of gut bacterial translocation. However, DAMPs (e.g.,
adenosine triphosphate and DNA fragments) mainly originate from
damaged hepatocytes (27). One such
process that leads to DAMPs is CCl4-induced hepatic
toxicity.
CXCL1, CXCL2 and CXCL8 are central chemokines that
attract neutrophils mainly through the chemokine receptors CXCR1
and CXCR2. Neutrophil recruitment increases the release of reactive
oxygen species (ROS) and proteases, leading to hepatocyte necrosis
(29). In parallel, KCs secrete CCL2
to increase circulating CCR2+Ly6C+ monocytes
that massively expand the local macrophage pool (17). CCR2+Ly6C+
monocyte migration into the liver after injury is functionally
critical for the maintenance of liver inflammation and
fibrogenesis. The functionality of MoMø not only depends on CCL2,
but also on CCL1 and CCL25, which induce the migration of
pro-inflammatory monocytes/macrophages via CCR8 and CCR9,
respectively (18,30,31).
Ultimately, KCs sense initial liver injury and are responsible for
monocyte and neutrophil recruitment, further aggravating liver
inflammation.
With inflammatory stimuli, the program of cytokine
production by KCs, triggered by TLR signaling, is more complex than
the simple result from the activation of transcription factors. KCs
are poised to swiftly respond to PAMPs or DAMPs with the production
of TNF. In KCs, the production of TNF usually precedes and
generally promotes the carefully orchestrated release of many other
inflammatory mediators including IL-6, IL-12/23 (p40) and type I
interferons (e.g., IFN-γ and TNF-α) (32). IFN-γ is a hallmark cytokine of Th1
cells that greatly increases the production of inflammatory
mediators by macrophages. Consequently, KCs activated by IFN-γ
express numerous pro-inflammatory cytokines (e.g., IL-6 and TNF-α)
with the enhancement of nuclear factor (NF)-κB gene expression.
While these pro-inflammatory signals may lead to enhanced liver
inflammation and injury, they also have protective effects on the
liver. IL-6 signaling, via signal transducer and activator of
transcription (STAT)3 activation, markedly increases after acute
CCl4-induced hepatic damage and promotes liver
proliferation by upregulating the expression of growth factors
(e.g., hepatocyte growth factor), increases hepatocyte
responsiveness and also inhibits hepatocyte apoptosis. TNF-α has a
major role in the regulation of IL-6 secretion and liver
regeneration via induction of NF-κb (33,34).
Aside from TGF-β, platelet-derived growth factor
(PDGF) expressed in liver macrophages also contributes to liver
fibrogenesis (37). PDGF promotes
HSC proliferation and activation through PDGF receptor and does not
have this effect after neutralization (38). However, the fibrosis-associated TLR
signaling of KCs and HSCs remains elusive. Perugorria et al
(39) reported that once TLR is
activated, elevated Tpl2 may be identified as an essential signal
transducer in KCs and HSCs, and promotes fibrogenic gene
expression.
KCs have an important role in the initiation and
perpetuation of liver inflammation, which are necessary for the
host defence, but if not controlled, they may cause hepatic damage,
fibrosis and cirrhosis. Four cytokines with the ability to
downregulate macrophage function have been identified: IL-4, IL-10,
IL-13 and TGF-β1, of which IL-10 appears to have a broader and
deeper effect. Importantly, IL-10 is released from macrophages,
type 2 T-helper (Th2) cells and stromal cells. IL-10 inhibits the
expression of NF-κB, the production of pro-inflammatory cytokines
by Th1 cells, macrophages and neutrophils, the proliferation of
hepatocytes and fibrogenesis during liver repair (40,41).
Hepatic macrophages not only promote hepatic fibrosis by activating
HSCs in chronic hepatic damage, but also contribute to the
resolution of fibrosis by degrading the extracellular matrix (ECM)
(26). Macrophages produce
gelatinases (MMP9, MMP12 and MMP13) under different circumstances,
resulting in complex ECM degradation. During fibrosis regression,
recruited Ly6C− monocytes differentiate into Ly6C+
‘restorative’ macrophages, with upregulation of MMPs (MMP9 and
MMP12), downregulation of pro-inflammatory cytokines and
chemokines, enhanced expression of insulin-like growth factor 1
(IGF-1) and genes associated with anti-inflammatory or antifibrotic
effects, including CX3CR1, CD74 and macrophage migration inhibitory
factor, and a reduction in TGF-β, thus promoting recovery from
injury. Furthermore, SAMs produce MMP13, which may disassemble the
interstitial matrix and promote fibrosis resolution (23,42).
However, whether KCs or Ly6C− ‘restorative’ macrophages
are the source of MMP13 remains elusive. In addition, KCs are a
major source of CXCL9, which ameliorate liver fibrogenesis
(43). Furthermore, CX3CR1 is a
major regulator of monocyte differentiation and survival in the
liver and protects against liver fibrosis (44).
Bile duct ligation (BDL) is a commonly used animal
model of cholestatic liver disease. Ligation of the common bile
duct performed at a standardized site prevents bile flow and causes
bile reflux followed by cholangitis, coagulation defects and
hepatic damage, and may even result in biliary fibrosis and
cirrhosis (45). The absence of bile
salts and bile in the intestines after BDL may promote
translocation of endotoxins and growth of endotoxin-producing
bacteria, resulting in cholangitis (45). KCs are a major type of defence cell
for the liver, as they remove endotoxins (e.g., LPS) and
phagocytose bacteria under normal conditions. However, the
intracellular bactericidal function of KCs in the BDL model is
impaired as a result of high levels of hydrophobic bile acids in
the serum. The altered sensitivity of KCs to endotoxins in BDL
induces overproduction of TNF-α and IL-6, which leads to liver
injury, but KC blockade may suppress systemic cytokine production
and improve survival under these conditions (46). Furthermore, toxic bile salts directly
cause rodent hepatocyte apoptosis through activation of Fas
(47), which leads to apoptotic body
formation and results in HSC activation through phagocytosis of
apoptotic bodies and enhancement of fibrogenesis (48). The engulfment of hepatocyte apoptotic
bodies by KCs has also been reported to promote liver inflammation
and fibrogenesis, which is mediated by death ligands and cytokines,
including TNF-α, TNF-related apoptosis-inducing ligand (TRAIL), Fas
and TGF-β (49). However, later
studies demonstrated that KCs abrogate cholestatic liver injury via
IL-6 and acid sphingomyelinase-dependent mechanisms (50,51). In
conclusion, KCs may have protective and promoting roles in
cholestatic liver injury.
Similar to that in cholestatic liver injury, KCs
also possess a dual role in BDL-induced liver fibrosis. Although
KCs generate death ligands to promote liver inflammation (49) and produce acid sphingomyelinase to
activate AKT in hepatocytes, which is required for regeneration
(51), higher expression of TGF-β, a
fibrogenic cytokine, has also been observed (49,51).
Liver injury is associated with increased hepatic exposure to LPS.
In contrast to KCs, LPS mainly targets TLR4 in HSCs, and makes HSCs
sensitive to TGF-β via the MyD88/NF-κB-dependent pathway (52). Furthermore, high levels of IL-17A
have been detected in fibrotic livers (53). IL-17A is generated predominantly by
effector CD4+ T (Th17) cells, which differentiate from
Th0 cells, and is regulated by IL-6, TNF, TGF-β and IL-23 (54). IL-17 stimulates KCs to further
upregulate IL-17A levels, increasing IL-17 receptor A expression
and induces the production of the pro-inflammatory cytokines TNF-α,
IL-6 and IL-1β, as well as the fibrogenic cytokines TGF-β1 and PDGF
(53). IL-6 and TGF-β1 further
facilitate the differentiation and expansion of Th17 cells
(53). In addition, PDGF promotes
HSC proliferation and activation (55). However, IL-17 production may be
inhibited by activation of cannabinoid receptor 2 in macrophages
(56). Furthermore, CD68+
KCs may clear apoptotic cholangiocytes via phagocytosis, resulting
in the upregulation of MMP3, MMP8 and MMP9, which contribute to the
reversal of biliary fibrosis (57).
Thioacetamide (TAA), a centrilobular hepatotoxin, is
widely applied to induce acute or chronic hepatic disease (58). Initial lesions in the liver begin
with a cytochrome P450 family 2 subfamily E member 1-mediated
two-step bioactivation of TAA into thioacetamide sulfoxide and
further to thioacetamide-S,S-dioxide (58), which is mainly distributed in zone 3
(centrilobular area), resulting in cytotoxicity. Injured or
necrotic hepatocytes release S100 proteins, high-mobility group box
proteins (HMGBs) and heat shock proteins (HSPs) as DAMPs (59). HMGBs and S100 proteins may be
recognized by TLR-2 and TLR-4 (60)
to produce pro-inflammatory cytokines IL-6, TNFα and IL-1β.
However, HSP25 may act to protect liver cells by macrophages
invading the hepatic lesion (61).
Gadolinium chloride (GD) is used to selectively inactivate KCs,
resulting in decreased serum levels of TNF-α and IL-6, reduced
TAA-induced liver injury, as well as enhanced metallothionein and
HSP70 expression (62). Furthermore,
GD treatment decreases the numbers of CD68+ and
CD163+ macrophages and inhibits TGF-β1 expression in
macrophages (63). CD68+
cells with maintained high levels of MCP-1 expression have been
detected in injured perivascular areas for up to 20 days, whereas
CD163+ cells gradually decreased in number in the
mid-zonal areas after day 3 (64).
In addition, depletion of M1 (expressing CD68 and major
histocompatibility complex class II) and M2 (expressing CD163 and
CD204) macrophages by liposomal clodronate, may aggravate and
prolong coagulation necrosis of hepatocytes. This has been reported
to be primarily due to the depletion of M2 macrophages, revealing a
remodeling stage dominated by M2 (65). However, KCs may release nitric oxide
and trigger post-necrotic hepatocyte regeneration following TAA
treatment (66). Therefore, KCs may
also possess a dual role in TAA-induced hepatic injury.
Repeated injection of TAA or the addition of TAA to
drinking water has been widely used to induce hepatic fibrosis.
Similarly, HSCs also have a critical role in TAA-induced fibrosis
(67). PDGF-B may be the major
cytokine for HSC activation in TAA-induced liver fibrosis (68). Furthermore, galectin-3
(Gal-3)+ macrophages are also involved in the initiation
of fibrosis via the activation of HSCs (69). However, Gal-3+ macrophages
possess M1 and M2 properties in the advanced stage of liver
fibrosis (70). To remodel tissue in
the fibrotic liver, a splenectomy may be effective due causing an
accumulation of Ly6Clo macrophages and the disappearance
of hepatic progenitor-like cells (71).
Acetaminophen (APAP) overdose-induced liver injury
is the most common cause of drug-induced hepatotoxicity in Western
countries. An APAP overdose may result in mitochondrial
dysfunction, ATP depletion and DNA fragmentation, which eventually
cause cell necrosis (72). In
addition to initial hepatocyte necrosis, the release of DAMPs,
including DNA fragments, HMGB-1 and HSPs, induces KC activation and
an inflammatory response, which also contributes to the disease
(73). Although HMGB-1 has several
separate receptors, including TLR2, TLR4 and TLR9, TLR4 is a
pivotal receptor for macrophage activation and cytokine production
(TNF, CXCL2, IL-6, IL-1β and IL-10) (74). HMGB-1/TLR4-dependent activation of
IL-23 release from macrophages promotes γδT cells to produce
IL-17A, which recruits neutrophils participating in sterile
inflammation (75). Furthermore,
neutrophil infiltration to necrotic areas also depends on the
CXCR2/formyl peptide receptor 1 axis (76). However, the role of neutrophils in
liver damage is controversial, as neutrophils may either aggravate
or have no effect on liver injury (77,78).
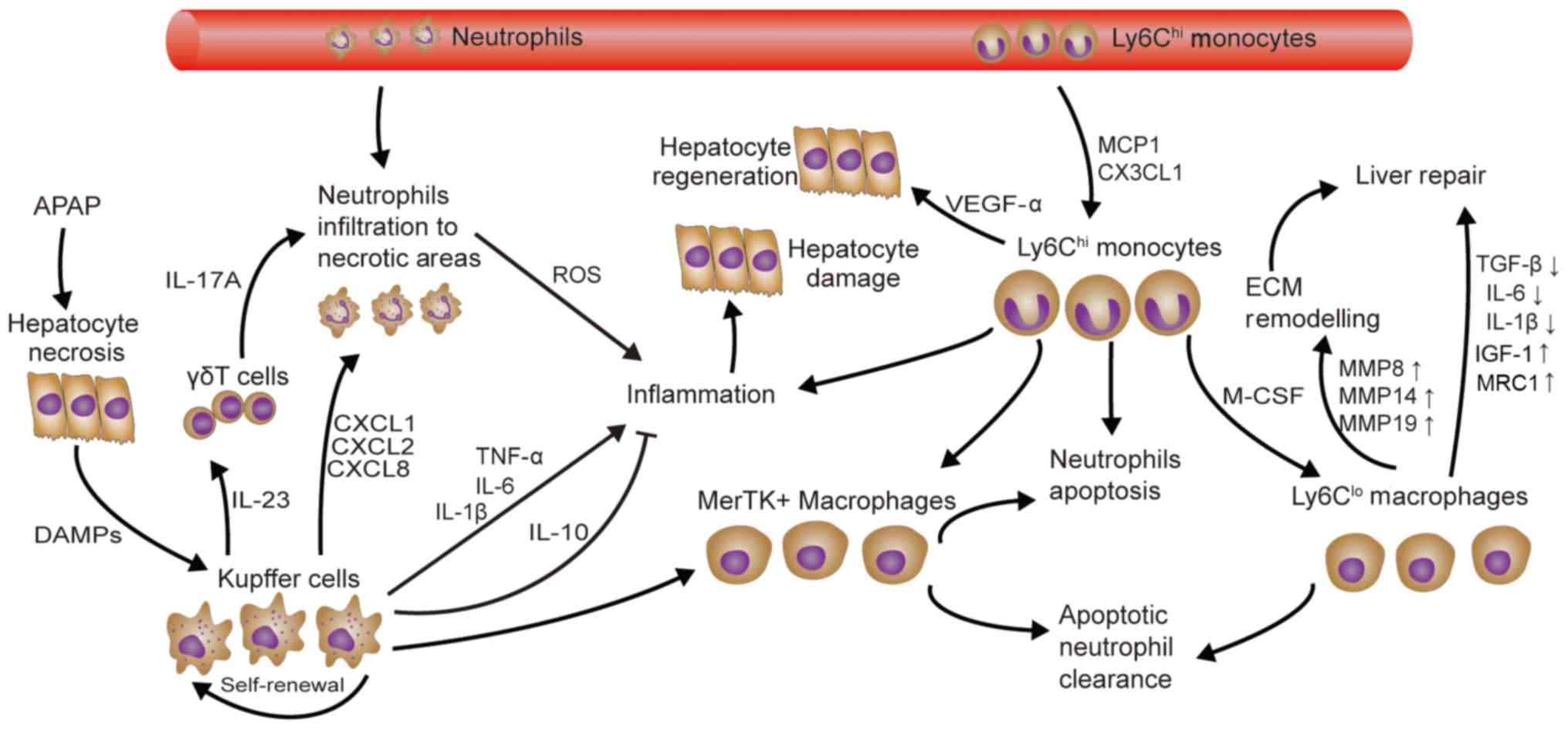
A total of three distinct macrophage subsets were
identified during APAP-induced acute liver injury and repair
(Fig. 2): Resident KCs,
Ly6Chi monocytes/macrophages and Ly6Clo MoMø.
Although KCs produce TNF-α and IL-1β, there is no credible evidence
that KCs directly cause liver cell damage (79). Furthermore, the recently discovered
Mer tyrosine kinase-expressing macrophage phenotype that has
hepatoprotective effects, including neutrophil apoptosis and
clearance, is mainly derived from the KC population (80). It is worth noting that the number of
KCs is decreased upon APAP application, and then increased by
self-renewal. In addition, Ly6Chi monocytes may be
recruited into the liver and trans-differentiated into the
Ly6Clo MoMø phenotype through CX3CL1/CX3CR1, MCP-1/CCR2
and in a monocyte colony-stimulating factor-dependent manner. In
addition, Ly6Chi monocytes and Ly6Clo MoMø
contribute to liver recovery. However, Zigmond et al
(81) and Mossanen et al
(82) demonstrated that
CCR2Ly6Chi monocytes/macrophages promote APAP
hepatotoxicity at an early stage. Another study has indicated that
Ly6Chi monocytes and Ly6Clo MoMø are likely
to work together: The former promote apoptosis of ROS-producing
neutrophils and the latter promote neutrophil clearance (83). In addition, Ly6Chi
monocytes have higher levels of VEGF-A, TGF-β1, IL-6, IL-1β and
MMP18 gene expression, whereas Ly6Clo MoMø express
higher levels of pro-restorative genes, including IGF-1 and mannose
receptor 1. In comparison with KCs, Ly6Chi monocytes and
Ly6Clo MoMø also express high levels of MMP8, MMP14 and
MMP19, which are important for ECM re-modeling (81).
D-galactosamine (D-GalN) may inhibit the synthesis
of mRNA and proteins that cause hepatocyte necrosis. Furthermore,
D-GalN-induced necrosis was reported to increase gut permeability
and cause endotoxemia in a rat model but not in a mouse model
(84). This is also thought to
contribute to hepatotoxicity. Thus, D-GalN/LPS co-administration is
commonly used to induce liver injury in mice (85). In fact, D-GalN increases the
sensitivity of rabbits, rats and mice to LPS, possibly by
upregulating TLR4 expression in liver macrophages and producing
lethal toxicity (86,87). Along these lines, depletion of TLR4
leads to a decreased inflammatory response and hepatic injury
(88). Under LPS stimulation, KCs
secrete TNF-α, and cause hepatocyte apoptosis and the release of
other cytokines (IL-1 and IL-6) (85). However, TNF-α appears to be the major
pro-inflammatory mediator, as macrophage autophagy may inhibit the
production of IL-1β. In addition, IL-6 treatment was reported to
have a beneficial effect and reduce the expression of TNF-α and
MCP-1, and promote macrophage polarization towards M2 in D-GalN/LPS
induced liver injury models (89,90).
Besides LPS, cytosine-phosphate-guanine DNA and
polyinosinic:polycytidylic acid also exert hepatic toxicity
mediated by TNF-α in D-GalN-sensitized mice (91). The depletion of macrophages and the
inhibition of TNF-α production protected against D-GalN/LPS-induced
hepatic injury (92,93). Furthermore, increasing the production
of IL-10 in macrophages via treatment with IL-35 also provided a
therapeutic effect (94).
Ischemia/reperfusion injury (IRI) may be divided
into two types, namely those with ‘warm’ ischemia and ‘cold’
ischemia. Warm ischemia may occur during liver surgery, shock or
trauma, whereas cold ischemia may develop in liver transplants.
However, the two types share a common mechanism in inflammatory
immune regulation (95). HMGB1 may
be released from hepatocytes in response to hypoxia/ischemia and
remain at high levels for up to 24 h after reperfusion, resulting
in liver inflammation and injury mediated by TLR4 (96). Inhibition of TLR4 and KCs
simultaneously was reported to reduce TNF-α, IL-6, CXCL2 production
and IRI (97,98). Conversely, KCs also exert protective
effects in IRI through IL-10 and heme oxygenase-1 (HO-1) (99,100).
Furthermore, HO-1 modified MoMø may prevent IRI, possibly via the
HO-1/STAT3 axis (101,102). In addition, neutrophils recruited
by CXCL2 and CD4+ T cells, which regulate macrophage and
neutrophil function via IFN-γ and IL-10, also have an important
role in IRI (103).
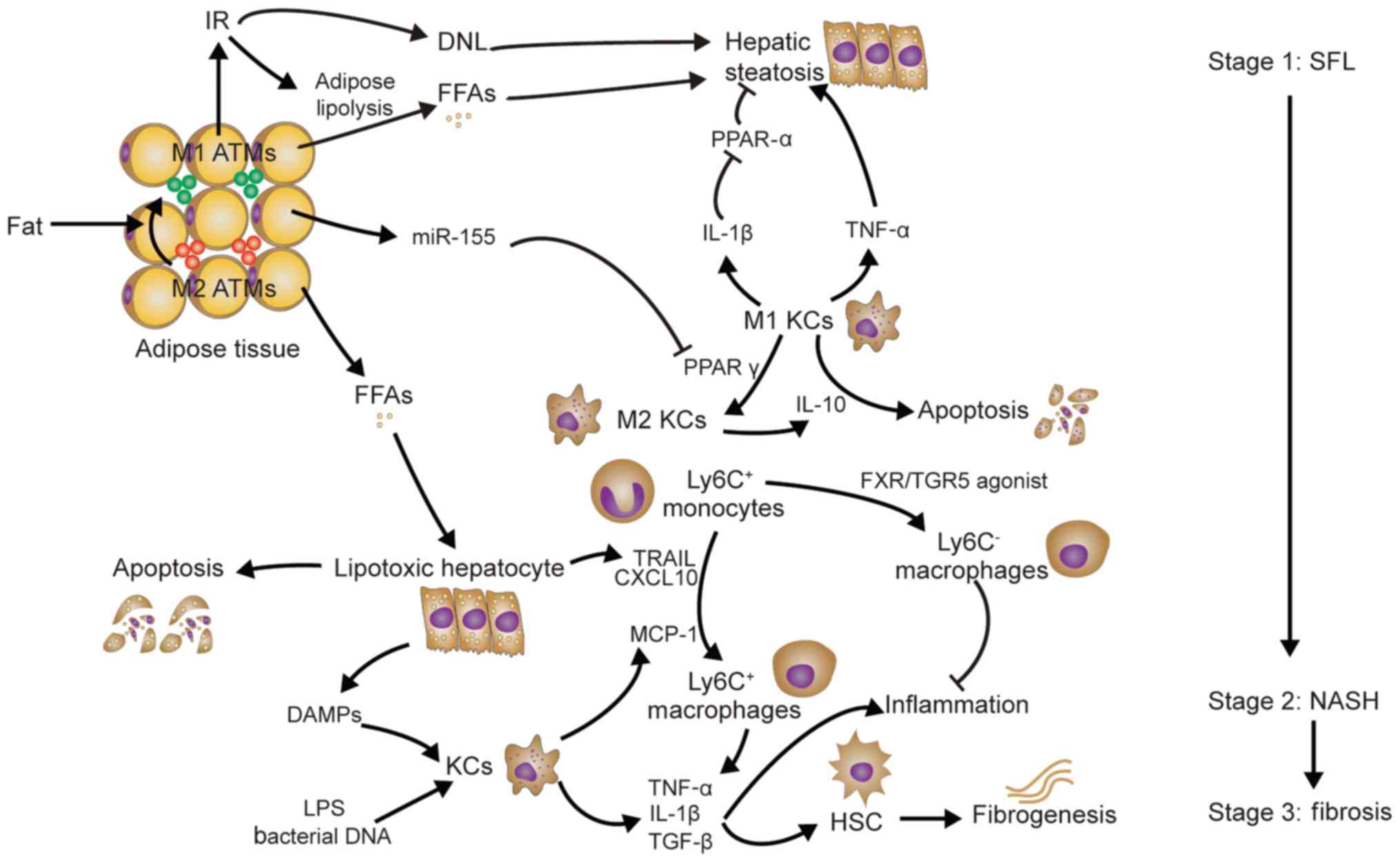
Non-alcoholic fatty liver disease (NAFLD), which
encompasses simple fatty liver (SFL), non-alcoholic steatohepatitis
(NASH) and associated cirrhosis, are usually linked to obesity,
insulin resistance and hyperlipidemia (104). The phenotypic switch of adipose
tissue macrophages (ATMs) from M2 ATMs to M1 ATMs contributes to
insulin resistance (IR) and hepatic steatosis in obese mice
(105). IR, as well as concomitant
hyperglycemia and hyperinsulinemia, may lead to increased
production of free fatty acids (FFAs), but reduced FFA oxidation
and increased de novo lipogenesis, which results in lipid
accumulation in hepatocytes and hepatic steatosis (106). In addition, KCs promote hepatic
steatosis (Fig. 3). MicroRNA-155
produced by ATMs under obesity conditions may target hepatocytes
and reduce the expression of peroxisome proliferator activated
receptor (PPAR)γ, which is required for KC polarization to the M2
phenotype (107,108); this results in an M1-predominant
phenotype of KCs. M1 KCs produce IL-1β, which inhibits PPARα
expression and leads to a suppression of fat oxidation and an
aggravation of hepatic steatosis (109). Depletion of KCs and inhibition of
TNF-α reduces hepatic steatosis in rats fed a high-fat/sucrose diet
(110). Conversely, M2 KCs produce
IL-10 to promote M1 KC apoptosis and protect against NAFLD
(111).
The mechanisms of NASH have been described by two
different hypotheses: The ‘two-hit’ hypothesis (112) and the ‘multiple parallel hits’
hypothesis (113). However, the
identification of KCs as promoters of hepatic steatosis (as
mentioned above) challenges the latter hypothesis. Indeed, palmitic
acid and stearic acid (saturated fatty acids), and oleic acid (a
monounsaturated fatty acid) may all prompt hepatocyte apoptosis
(lipotoxicity) (114). Furthermore,
lipotoxic hepatocytes may release TRAIL and CXCL10 to induce
macrophage chemotaxis (115,116).
Furthermore, FFAs may also promote hepatocytes to release HMGB1
(117). However, Ly6C+
macrophage infiltration may be mainly dependent on MCP-1 produced
by KCs, as only early depletion of KCs prevents the development of
NASH (118). In addition,
inhibition of MCP-1 may also inhibit the development of
steatohepatitis (119). Ultimately,
macrophage accumulation aggravates liver inflammation via the
release of TNF-α and IL-1β (120).
However, bile acid receptor farnesoid X receptor/Takeda G-protein
receptor 5 agonist was reported to contribute to the intrahepatic
monocyte phenotype Ly6Clow (121). Endotoxin and bacterial DNA derived
from the intestine may also activate the release of TLR4 and TLR9
from KCs separately, resulting in IL-1β expression. In addition to
promoting liver inflammation, TNF-α and IL-1β activate HSCs to
upregulate the production of tissue inhibitor of
metallopeptidases-1, which inhibits MMPs and causes fibrogenesis
(122,123). Leptin produced by activated HSCs
may also further promote liver fibrosis, which is possibly mediated
by TGF-β1 expression (124).
Understanding the different roles and mechanisms of
macrophages in hepatic damage, fibrosis and repair is critical for
the development of novel therapies for hepatic diseases. When the
liver is injured, KCs sense initial liver injury and initiate
inflammatory cascades. Subsequently, inflammatory monocytes
accumulate in the liver via chemokine-chemokine receptor
interactions. When liver injury ceases, restorative macrophages
promote the resolution of hepatic damage and fibrosis. Thus, novel
methods to improve liver injury or fibrosis focus on targeting
liver macrophages. These interventions regulate the activation of
KCs (e.g., by inhibiting bacterial translocation or changing the
composition of bile acid), inflammatory monocyte migration (e.g.,
against various chemokines or chemokine receptors), or macrophage
polarization and differentiation (e.g., by targeted delivery of
nanoparticles) (125). However,
different interventions lack scientific comparison and require
further evaluation of their effectiveness. Numerous studies only
provided an immune function analysis at the organ level and did not
specify the phenotypes of liver macrophages. Furthermore, the
underlying mechanisms and triggers of hepatic macrophage phenotype
switching during different stages of liver injury have not been
well studied. Therefore, an in-depth study of the genomes and
phenotypes of liver macrophages is required.
Not applicable.
The present study was supported by the National Key
Research and Development Program of China (grant no.
2016YFA0101001) and the National Natural Science Foundation of
China (grant no. 81471794).
All data generated or analyzed during this study are
included in this published article.
XD wrote the manuscript and constructed the figures.
JL and YX revised the manuscript. HC designed and revised the
manuscript. All authors have read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Wynn TA and Vannella KM: Macrophages in
tissue repair, regeneration, and fibrosis. Immunity. 44:450–462.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vannella KM and Wynn TA: Mechanisms of
organ injury and repair by macrophages. Annu Rev Physiol.
79:593–617. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Epelman S, Lavine KJ and Randolph GJ:
Origin and functions of tissue macrophages. Immunity. 41:21–35.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ginhoux F and Guilliams M: Tissue-resident
macrophage ontogeny and homeostasis. Immunity. 44:439–449. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gregory SH and Wing EJ: Neutrophil-Kupffer
cell interaction: A critical component of host defenses to systemic
bacterial infections. J Leukoc Biol. 72:239–248. 2002.PubMed/NCBI
|
|
6
|
Ganz T: Macrophages and systemic iron
homeostasis. J Innate Immun. 4:446–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schulz C, Gomez Perdiguero E, Chorro L,
Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE,
et al: A lineage of myeloid cells independent of Myb and
hematopoietic stem cells. Science. 336:86–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davies LC, Jenkins SJ, Allen JE and Taylor
PR: Tissue-resident macrophages. Nat Immunol. 14:986–995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klein I, Cornejo JC, Polakos NK, John B,
Wuensch SA, Topham DJ, Pierce RH and Crispe IN: Kupffer cell
heterogeneity: Functional properties of bone marrow derived and
sessile hepatic macrophages. Blood. 110:4077–4085. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: Nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stein M, Keshav S, Harris N and Gordon S:
Interleukin 4 potently enhances murine macrophage mannose receptor
activity: A marker of alternative immunologic macrophage
activation. J Exp Med. 176:287–292. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laskin DL, Sunil VR, Gardner CR and Laskin
JD: Macrophages and tissue injury: Agents of defense or
destruction? Annu Rev Pharmacol Toxicol. 51:267–288. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martinez FO and Gordon S: The M1 and M2
paradigm of macrophage activation: Time for reassessment.
F1000Prime Rep. 6:132014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weber LW, Boll M and Stampfl A:
Hepatotoxicity and mechanism of action of haloalkanes: Carbon
tetrachloride as a toxicological model. Crit Rev Toxicol.
33:105–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mehendale HM: Tissue repair: An important
determinant of final outcome of toxicant-induced injury. Toxicol
Pathol. 33:41–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tacke F: Functional role of intrahepatic
monocyte subsets for the progression of liver inflammation and
liver fibrosis in vivo. Fibrogenesis Tissue Repair. 5 (Suppl
1):S272012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karlmark KR, Weiskirchen R, Zimmermann HW,
Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C and
Tacke F: Hepatic recruitment of the inflammatory Gr1+
monocyte subset upon liver injury promotes hepatic fibrosis.
Hepatology. 50:261–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heymann F, Hammerich L, Storch D, Bartneck
M, Huss S, Russeler V, Gassler N, Lira SA, Luedde T, Trautwein C,
et al: Hepatic macrophage migration and differentiation critical
for liver fibrosis is mediated by the chemokine receptor C-C motif
chemokine receptor 8 in mice. Hepatology. 55:898–909. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Geissmann F, Manz MG, Jung S, Sieweke MH,
Merad M and Ley K: Development of monocytes, macrophages, and
dendritic cells. Science. 327:656–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carlin LM, Stamatiades EG, Auffray C,
Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold
S and Geissmann F: Nr4a1-dependent Ly6C(low) monocytes monitor
endothelial cells and orchestrate their disposal. Cell.
153:362–375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mildner A, Schonheit J, Giladi A, David E,
Lara-Astiaso D, Lorenzo-Vivas E, Paul F, Chappell-Maor L, Priller
J, Leutz A, et al: Genomic characterization of murine monocytes
reveals C/EBPβ transcription factor dependence of Ly6C-Cells.
Immunity. 46:849–862 e847. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Duffield JS, Forbes SJ, Constandinou CM,
Clay S, Partolina M, Vuthoori S, Wu S, Lang R and Iredale JP:
Selective depletion of macrophages reveals distinct, opposing roles
during liver injury and repair. J Clin Invest. 115:56–65. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramachandran P, Pellicoro A, Vernon MA,
Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A,
Gordon-Walker TT, et al: Differential Ly-6C expression identifies
the recruited macrophage phenotype, which orchestrates the
regression of murine liver fibrosis. Proc Natl Acad Sci USA.
109:E3186–3195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma PF, Gao CC, Yi J, Zhao JL, Liang SQ,
Zhao Y, Ye YC, Bai J, Zheng QJ, Dou KF, et al: Cytotherapy with
M1-polarized macrophages ameliorates liver fibrosis by modulating
immune microenvironment in mice. J Hepatol. 67:770–779. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ramachandran P, Iredale JP and Fallowfield
JA: Resolution of liver fibrosis: Basic mechanisms and clinical
relevance. Semin Liver Dis. 35:119–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tacke F and Zimmermann HW: Macrophage
heterogeneity in liver injury and fibrosis. J Hepatol.
60:1090–1096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wree A and Marra F: The inflammasome in
liver disease. J Hepatol. 65:1055–1056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weber LWD, Boll M and Stampfl A:
Hepatotoxicity and mechanism of action of haloalkanes: Carbon
tetrachloride as a toxicological model. Crit Rev Toxicol.
33:105–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marra F and Tacke F: Roles for chemokines
in liver disease. Gastroenterology. 147:577–594.e571. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakamoto N, Ebinuma H, Kanai T, Chu PS,
Ono Y, Mikami Y, Ojiro K, Lipp M, Love PE, Saito H, et al:
CCR9+ macrophages are required for acute liver
inflammation in mouse models of hepatitis. Gastroenterology.
142:366–376. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chu PS, Nakamoto N, Ebinuma H, Usui S,
Saeki K, Matsumoto A, Mikami Y, Sugiyam K, Tomita K, Kanai T, et
al: C-C motif chemokine receptor 9 positive macrophages activate
hepatic stellate cells and promote liver fibrosis in mice.
Hepatology. 58:337–350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hamidzadeh K, Christensen SM, Dalby E,
Chandrasekaran P and Mosser DM: Macrophages and the Recovery from
Acute and Chronic Inflammation. Annu Rev Physiol. 79:567–592. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zimmers TA, McKillop IH, Pierce RH, Yoo JY
and Koniaris LG: Massive liver growth in mice induced by systemic
interleukin 6 administration. Hepatology. 38:326–334. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Schwabe RF, DeVries-Seimon T, Yao
PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA
and Tabas I: Free cholesterol-loaded macrophages are an abundant
source of tumor necrosis factor-alpha and interleukin-6: Model of
NF-kappaB- and map kinase-dependent inflammation in advanced
atherosclerosis. J Biol Chem. 280:21763–21772. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pradere JP, Kluwe J, De Minicis S, Jiao
JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman
R, et al: Hepatic macrophages but not dendritic cells contribute to
liver fibrosis by promoting the survival of activated hepatic
stellate cells in mice. Hepatology. 58:1461–1473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lotersztajn S, Julien B, Teixeira-Clerc F,
Grenard P and Mallat A: Hepatic fibrosis: Molecular mechanisms and
drug targets. Annu Rev Pharmacol Toxicol. 45:605–628. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Borkham-Kamphorst E, Kovalenko E, van
Roeyen CR, Gassler N, Bomble M, Ostendorf T, Floege J, Gressner AM
and Weiskirchen R: Platelet-derived growth factor isoform
expression in carbon tetrachloride-induced chronic liver injury.
Lab Invest. 88:1090–1100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hao ZM, Fan XB, Li S, Lv YF, Su HQ, Jiang
HP and Li HH: Vaccination with Platelet-Derived Growth Factor B
Kinoids Inhibits CCl4-Induced Hepatic Fibrosis in Mice. J Pharmacol
Exp Ther. 342:835–842. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Perugorria MJ, Murphy LB, Fullard N,
Chakraborty JB, Vyrla D, Wilson CL, Oakley F, Mann J and Mann DA:
Tumor progression locus 2/Cot is required for activation of
extracellular regulated kinase in liver injury and toll-like
receptor-induced TIMP-1 gene transcription in hepatic stellate
cells in mice. Hepatology. 57:1238–1249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Louis H, Van Laethem JL, Wu W, Quertinmont
E, Degraef C, Van den Berg K, Demols A, Goldman M, Le Moine O,
Geerts A and Devière J: Interleukin-10 controls neutrophilic
infiltration, hepatocyte proliferation, and liver fibrosis induced
by carbon tetrachloride in mice. Hepatology. 28:1607–1615. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thompson K, Maltby J, Fallowfield J,
McAulay M, Millward-Sadler H and Sheron N: Interleukin-10
expression and function in experimental murine liver inflammation
and fibrosis. Hepatology. 28:1597–1606. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fallowfield JA, Mizuno M, Kendall TJ,
Constandinou CM, Benyon RC, Duffield JS and Iredale JP:
Scar-associated macrophages are a major source of hepatic matrix
metalloproteinase-13 and facilitate the resolution of murine
hepatic fibrosis. J Immunol. 178:5288–5295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wasmuth HE, Lammert F, Zaldivar MM,
Weiskirchen R, Hellerbrand C, Scholten D, Berres ML, Zimmermann H,
Streetz KL, Tacke F, et al: Antifibrotic effects of CXCL9 and its
receptor CXCR3 in livers of mice and humans. Gastroenterology.
137:309–319, 319 e301-303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Karlmark KR, Zimmermann HW, Roderburg C,
Gassler N, Wasmuth HE, Luedde T, Trautwein C and Tacke F: The
fractalkine receptor CX(3)CR1 protects against liver fibrosis by
controlling differentiation and survival of infiltrating hepatic
monocytes. Hepatology. 52:1769–1782. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Scott-Conner CE and Grogan JB: The
pathophysiology of biliary obstruction and its effect on phagocytic
and immune function. J Surg Res. 57:316–336. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lazar G, Paszt A, Kaszaki J, Duda E,
Szakacs J, Tiszlavicz L, Boros M, Balogh A and Lazar G: Kupffer
cell phagocytosis blockade decreases morbidity in endotoxemic rats
with obstructive jaundice. Inflamm Res. 51:511–518. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Faubion WA, Guicciardi ME, Miyoshi H,
Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH and Gores GJ: Toxic
bile salts induce rodent hepatocyte apoptosis via direct activation
of Fas. J Clin Invest. 103:137–145. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Canbay A, Higuchi H, Bronk SF, Taniai M,
Sebo TJ and Gores GJ: Fas enhances fibrogenesis in the bile duct
ligated mouse: A link between apoptosis and fibrosis.
Gastroenterology. 123:1323–1330. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Canbay A, Feldstein AE, Higuchi H,
Werneburg N, Grambihler A, Bronk SF and Gores GJ: Kupffer cell
engulfment of apoptotic bodies stimulates death ligand and cytokine
expression. Hepatology. 38:1188–1198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gehring S, Dickson EM, San Martin ME, van
Rooijen N, Papa EF, Harty MW, Tracy TF Jr..Gregory SH: Kupffer
cells abrogate cholestatic liver injury in mice. Gastroenterology.
130:810–822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Osawa Y, Seki E, Adachi M, Suetsugu A, Ito
H, Moriwaki H, Seishima M and Nagaki M: Role of acid
sphingomyelinase of kupffer cells in cholestatic liver injury in
mice. Hepatology. 51:237–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Seki E, De Minicis S, Osterreicher CH,
Kluwe J, Osawa Y, Brenner DA and Schwabe RF: TLR4 enhances TGF-beta
signaling and hepatic fibrosis. Nat Med. 13:1324–1332. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Meng F, Wang K, Aoyama T, Grivennikov SI,
Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al:
Interleukin-17 Signaling in inflammatory, kupffer cells, and
hepatic stellate cells exacerbates liver fibrosis in mice.
Gastroenterology. 143:765–776.e3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Steinman L: A brief history of T(H)17, the
first major revision in the T(H)1/T(H)2 hypothesis of T
cell-mediated tissue damage. Nat Med. 13:139–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ying HZ, Chen Q, Zhang WY, Zhang HH, Ma Y,
Zhang SZ, Fang J and Yu CH: PDGF signaling pathway in hepatic
fibrosis pathogenesis and therapeutics. Mol Med Report.
16:7879–7889. 2017. View Article : Google Scholar
|
|
56
|
Guillot A, Hamdaoui N, Bizy A, Zoltani K,
Souktani R, Zafrani ES, Mallat A, Lotersztajn S and Lafdil F:
Cannabinoid receptor 2 counteracts interleukin-17-induced immune
and fibrogenic responses in mouse liver. Hepatology. 59:296–306.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Popov Y, Sverdlov DY, Bhaskar KR, Sharma
AK, Millonig G, Patsenker E, Krahenbuhl S, Krahenbuhl L and
Schuppan D: Macrophage-mediated phagocytosis of apoptotic
cholangiocytes contributes to reversal of experimental biliary
fibrosis. Am J Physiol Gastrointest Liver Physiol. 298:G323–G334.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chilakapati J, Shankar K, Korrapati MC,
Hill RA and Mehendale HM: Saturation toxicokinetics of
thioacetamide: Role in initiation of liver injury. Drug Metab
Dispos. 33:1877–1885. 2005.PubMed/NCBI
|
|
59
|
Kuramochi M, Izawa T, Pervin M, Bondoc A,
Kuwamura M and Yamate J: The kinetics of damage-associated
molecular patterns (DAMPs) and toll-like receptors during
thioacetamide-induced acute liver injury in rats. Exp Toxicol
Pathol. 68:471–477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Erridge C: Endogenous ligands of TLR2 and
TLR4: Agonists or assistants? J Leukoc Biol. 87:989–999. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fujisawa K, Miyoshi T, Tonomura Y, Izawa
T, Kuwamura M, Torii M and Yamate J: Relationship of heat shock
protein 25 with reactive macrophages in thioacetamide-induced rat
liver injury. Exp Toxicol Pathol. 63:599–605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Andres D, Sanchez-Reus I, Bautista M and
Cascales M: Depletion of Kupffer cell function by gadolinium
chloride attenuates thioacetamide-induced hepatotoxicity-Expression
of metallothionein and HSP70. Biochem Pharmacol. 66:917–926. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ide M, Kuwamura M, Kotani T, Sawamoto O
and Yamate J: Effects of gadolinium chloride (GdCl3) on the
appearance of macrophage populations and fibrogenesis in
thioacetamide-induced rat hepatic lesions. J Comp Pathol.
133:92–102. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ide M, Yamate J, Machida Y, Nakanishi M,
Kuwamura M, Kotani T and Sawamoto O: Emergence of different
macrophage populations in hepatic fibrosis following
thioacetamide-induced acute hepatocyte injury in rats. J Comp
Pathol. 128:41–51. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Golbar HM, Izawa T, Wijesundera KK, Bondoc
A, Tennakoon AH, Kuwamura M and Yamate J: Depletion of hepatic
macrophages aggravates liver lesions induced in rats by
thioacetamide (TAA). Toxicol Pathol. 44:246–258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
DiezFernandez C, Sanz N, Bosca L,
Hortelano S and Cascales M: Involvement of nitric oxide synthesis
in hepatic perturbations induced in rats by a necrogenic dose of
thioacetamide. Br J Pharmacol. 121:820–826. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hernandez-Gea V, Ghiassi-Nejad Z,
Rozenfeld R, Gordon R, Fiel MI, Yue ZY, Czaja MJ and Friedman SL:
Autophagy releases lipid that promotes fibrogenesis by activated
hepatic stellate cells in mice and in human tissues.
Gastroenterology. 142:938–946. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Palacios RS, Roderfeld M, Hemmann S, Rath
T, Atanasova S, Tschuschner A, Gressner OA, Weiskirchen R, Graf J
and Roeb E: Activation of hepatic stellate cells is associated with
cytokine expression in thioacetamide-induced hepatic fibrosis in
mice. Lab Invest. 88:1192–1203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Traber PG, Chou H, Zomer E, Hong F,
Klyosov A, Fiel MI and Friedman SL: Regression of fibrosis and
reversal of cirrhosis in rats by galectin inhibitors in
thioacetamide-induced liver disease. PLoS One. 8:e753612013.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wijesundera KK, Izawa T, Tennakoon AH,
Murakami H, Golbar HM, Katou-Ichikawa C, Tanaka M, Kuwamura M and
Yamate J: M1- and M2-macrophage polarization in rat liver cirrhosis
induced by thioacetamide (TAA), focusing on Iba1 and galectin-3.
Exp Mol Pathol. 96:382–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yada A, Iimuro Y, Uyama N, Uda Y, Okada T
and Fujimoto J: Splenectomy attenuates murine liver fibrosis with
hypersplenism stimulating hepatic accumulation of Ly-6C(lo)
macrophages. J Hepatol. 63:905–916. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jaeschke H and Bajt ML: Intracellular
signaling mechanisms of acetaminophen-induced liver cell death.
Toxicol Sci. 89:31–41. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Krenkel O, Mossanen Jana C and Tacke F:
Immune mechanisms in acetaminophen-induced acute liver failure.
Hepatobiliary Surgery and Nutrition. 3:331–343. 2014.PubMed/NCBI
|
|
74
|
Yang H, Hreggvidsdottir HS, Palmblad K,
Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y,
et al: A critical cysteine is required for HMGB1 binding to
Toll-like receptor 4 and activation of macrophage cytokine release.
Proc Natl Acad Sci USA. 107:11942–11947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang X, Sun R, Wei H and Tian Z:
High-mobility group box 1 (HMGB1)-toll-like receptor
(TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced
damage-associated lethal hepatitis: Interaction of γδ T cells with
macrophages. Hepatology. 57:373–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Marques PE, Amaral SS, Pires DA, Nogueira
LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Melgaco JG,
et al: Chemokines and mitochondrial products activate neutrophils
to amplify organ injury during mouse acute liver failure.
Hepatology. 56:1971–1982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Marques PE, Amaral SS, Pires DA, Nogueira
LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Melgaco JG,
et al: Chemokines and mitochondrial products activate neutrophils
to amplify organ injury during mouse acute liver failure.
Hepatology. 56:1971–1982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Williams CD, Bajt ML, Sharpe MR, McGill
MR, Farhood A and Jaeschke H: Neutrophil activation during
acetaminophen hepatotoxicity and repair in mice and humans. Toxicol
Appl Pharmacol. 275:122–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jaeschke H, Williams CD, Ramachandran A
and Bajt ML: Acetaminophen hepatotoxicity and repair: The role of
sterile inflammation and innate immunity. Liver Int. 32:8–20. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Triantafyllou E, Pop OT, Possamai LA,
Wilhelm A, Liaskou E, Singanayagam A, Bernsmeier C, Khamri W, Petts
G, Dargue R, et al: MerTK expressing hepatic macrophages promote
the resolution of inflammation in acute liver failure. Gut.
67:333–347. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zigmond E, Samia-Grinberg S, Pasmanik-Chor
M, Brazowski E, Shibolet O, Halpern Z and Varol C: Infiltrating
monocyte-derived macrophages and resident kupffer cells display
different ontogeny and functions in acute liver injury. J Immunol.
193:344–353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mossanen JC, Krenkel O, Ergen C, Govaere
O, Liepelt A, Puengel T, Heymann F, Kalthoff S, Lefebvre E, Eulberg
D, et al: Chemokine (C-C motif) receptor 2-positive monocytes
aggravate the early phase of acetaminophen-induced acute liver
injury. Hepatology. 64:1667–1682. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Graubardt N, Vugman M, Mouhadeb O, Caliari
G, Pasmanik-Chor M, Reuveni D, Zigmond E, Brazowski E, David E,
Chappell-Maor L, et al: Ly6C(hi) monocytes and their macrophage
descendants regulate neutrophil function and clearance in
acetaminophen-induced liver injury. Front Immunol. 8:6262017.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Stachlewitz RF, Seabra V, Bradford B,
Bradham CA, Rusyn I, Germolec D and Thurman RG: Glycine and uridine
prevent D-galactosamine hepatotoxicity in the rat: Role of Kupffer
cells. Hepatology. 29:737–745. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Xiong QB, Hase K, Tezuka Y, Namba T and
Kadota S: Acteoside inhibits apoptosis in D-galactosamine and
lipopolysaccharide-induced liver injury. Life Sci. 65:421–430.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Galanos C, Freudenberg MA and Reutter W:
Galactosamine-induced sensitization to the lethal effects of
endotoxin. Proc Natl Acad Sci USA. 76:5939–5943. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kitazawa T, Tsujimoto T, Kawaratani H,
Fujimoto M and Fukui H: Expression of Toll-like receptor 4 in
various organs in rats with D-galactosamine-induced acute hepatic
failure. J Gastroenterol Hepatol. 23:E494–E498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ben Ari Z, Avlas O, Pappo O, Zilbermints
V, Cheporko Y, Bachmetov L, Zemel R, Shainberg A, Sharon E, Grief
F, et al: Reduced hepatic injury in toll-like receptor 4-deficient
mice following D-galactosamine/lipopolysaccharide-induced fulminant
hepatic failure. Cell Physiol Biochem. 29:41–50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ilyas G, Zhao EP, Liu K, Lin Y, Tesfa L,
Tanaka KE and Czaja MJ: Macrophage autophagy limits acute toxic
liver injury in mice through down regulation of interleukin-1β. J
Hepatol. 64:118–127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li L, Duan CL, Zhao Y, Zhang XF, Yin HY,
Wang TX, Huang CX, Liu SH, Yang SY and Li XJ: Preventive effects of
interleukin-6 in lipopolysaccharide/D-galactosamine induced acute
liver injury via regulating inflammatory response in hepatic
macrophages. Int Immunopharmacol. 51:99–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Dejager L and Libert C: Tumor necrosis
factor alpha mediates the lethal hepatotoxic effects of poly(I:C)
in D-galactosamine-sensitized mice. Cytokine. 42:55–61. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wolf AM, Wolf D, Rumpold H, Ludwiczek S,
Enrich B, Gastl G, Weiss G and Tilg H: The kinase inhibitor
imatinib mesylate inhibits TNF-alpha production in vitro and
prevents TNF-dependent acute hepatic inflammation. Proc Natl Acad
Sci USA. 102:13622–13627. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jiang W, Sun R, Wei HM and Tian ZG:
Toll-like receptor 3 ligand attenuates LPS-induced liver injury by
down-regulation of toll-like receptor 4 expression on macrophages.
Proc Natl Acad Sci USA. 102:17077–17082. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zheng XF, Hu XY, Ma B, Fang H, Zhang F,
Mao YF, Yang FY, Xiao SC and Xia ZF: Interleukin-35 attenuates
D-galactosamine/lipopolysaccharide-induced liver injury via
enhancing interleukin-10 production in kupffer cells. Front
Pharmacol. 9:9592018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lu L, Zhou HM, Ni M, Wang XH, Busuttil R,
Kupiec-Weglinski J and Zhai Y: Innate immune regulations and liver
ischemia-reperfusion injury. Transplantation. 100:2601–2610. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Tsung A, Sahai R, Tanaka H, Nakao A, Fink
MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, et al: The
nuclear factor HMGB1 mediates hepatic injury after murine liver
ischemia-reperfusion. J Exp Med. 201:1135–1143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mosher B, Dean R, Harkema J, Remick D,
Palma J and Crockett E: Inhibition of Kupffer cells reduced CXC
chemokine production and liver injury. J Surg Res. 99:201–210.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Jiang W, Tang W, Geng Q and Xu X:
Inhibition of toll-like receptor 4 with vasoactive intestinal
peptide attenuates liver ischemia-reperfusion injury. Transplant
Proc. 43:1462–1467. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Devey L, Ferenbach D, Mohr E, Sangster K,
Bellamy CO, Hughes J and Wigmore SJ: Tissue-resident macrophages
protect the liver from ischemia reperfusion injury via a heme
oxygenase-1-dependent mechanism. Mol Ther. 17:65–72. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ellett JD, Atkinson C, Evans ZP, Amani Z,
Balish E, Schmidt MG, van Rooijen N, Schnellmann RG and Chavin KD:
Murine Kupffer cells are protective in total hepatic
ischemia/reperfusion injury with bowel congestion through IL-10. J
Immunol. 184:5849–5858. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ke B, Shen XD, Gao F, Ji HF, Qiao B, Zhai
Y, Farmer DG, Busuttil RW and Kupiec-Weglinski JW: Adoptive
transfer of Ex Vivo HO-1 modified bone marrow-derived macrophages
prevents liver ischemia and reperfusion injury. Mol Ther.
18:1019–1025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ke B, Shen XD, Ji H, Kamo N, Gao F,
Freitas MC, Busuttil RW and Kupiec-Weglinski JW: HO-1-STAT3 axis in
mouse liver ischemia/reperfusion injury: Regulation of TLR4 innate
responses through PI3K/PTEN signaling. J Hepatol. 56:359–366. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Ji H, Shen X, Gao F, Ke B, Freitas MC,
Uchida Y, Busuttil RW, Zhai Y and Kupiec-Weglinski JW: Programmed
death-1/B7-H1 negative costimulation protects mouse liver against
ischemia and reperfusion injury. Hepatology. 52:1380–1389. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Devisscher L, Verhelst X, Colle I, Van
Vlierberghe H and Geerts A: The role of macrophages in
obesity-driven chronic liver disease. J Leukoc Biol. 99:693–698.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lumeng Carey N, Bodzin Jennifer L and
Saltiel Alan R: Obesity induces a phenotypic switch in adipose
tissue macrophage polarization. J Clin Invest. 117:175–184. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Neuschwander-Tetri Brent A: Hepatic
lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis:
The central role of nontriglyceride fatty acid metabolites.
Hepatology. 52:774–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ying W, Riopel M, Bandyopadhyay G, Dong Y,
Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A,
Fu W, et al: Adipose tissue macrophage-derived exosomal miRNAs can
modulate in vivo and in vitro insulin sensitivity. Cell.
171:372–384 e312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Luo W, Xu Q, Wang Q, Wu H and Hua J:
Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2
polarization in the development of non-alcoholic fatty liver
disease. Sci Rep. 7:446122017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Stienstra R, Saudale F, Duval C, Keshtkar
S, Groener JE, van Rooijen N, Staels B, Kersten S and Mueller M:
Kupffer cells promote hepatic steatosis via interleukin-1
beta-dependent suppression of peroxisome proliferator-activated
receptor alpha activity. Hepatology. 51:511–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Huang W, Metlakunta A, Dedousis N, Zhang
P, Sipula I, Dube John J, Scott Donald K and O'Doherty Robert M:
Depletion of liver kupffer cells prevents the development of
diet-induced hepatic steatosis and insulin resistance. Diabetes.
59:347–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wan J, Benkdane M, Teixeira-Clerc F,
Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat
A, et al: M2 kupffer cells promote M1 kupffer cell apoptosis: A
protective mechanism against alcoholic and nonalcoholic fatty liver
disease. Hepatology. 59:130–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Day CP and James OF: Steatohepatitis: A
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tilg H and Moschen AR: Evolution of
inflammation in nonalcoholic fatty liver disease: The multiple
parallel hits hypothesis. Hepatology. 52:1836–1846. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Malhi H and Gores GJ: Molecular mechanisms
of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver
Dis. 28:360–369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ibrahim SH, Hirsova P, Tomita K, Bronk SF,
Werneburg NW, Harrison SA, Goodfellow VS, Malhi H and Gores GJ:
Mixed lineage kinase 3 mediates release of C-X-C motif ligand
10-bearing chemotactic extracellular vesicles from lipotoxic
hepatocytes. Hepatology. 63:731–744. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Idrissova L, Malhi H, Werneburg NW,
LeBrasseur NK, Bronk SF, Fingas C, Tchkonia T, Pirtskhalava T,
White TA, Stout MB, et al: TRAIL receptor deletion in mice
suppresses the inflammation of nutrient excess. J Hepatol.
62:1156–1163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J,
Hou YJ, Chang YX, Tu QQ, Feng GS, et al: Nuclear factor
high-mobility group box1 mediating the activation of toll-like
receptor 4 signaling in hepatocytes in the early stage of
nonalcoholic fatty liver disease in mice. Hepatology. 54:1620–1630.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Reid DT, Reyes JL, McDonald BA, Vo T,
Reimer RA and Eksteen B: Kupffer cells undergo fundamental changes
during the development of experimental NASH and are critical in
initiating liver damage and inflammation. PLoS One.
11:e01595242016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Baeck C, Wehr A, Karlmark Karlin R,
Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D,
Luedde T, et al: Pharmacological inhibition of the chemokine CCL2
(MCP-1) diminishes liver macrophage infiltration and
steatohepatitis in chronic hepatic injury. Gut. 61:416–426. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Miura K, Yang L, van Rooijen N, Ohnishi H
and Seki E: Hepatic recruitment of macrophages promotes
nonalcoholic steatohepatitis through CCR2. Am J Physiol-Gastroint
Liver Physiol. 302:G1310–G1321. 2012. View Article : Google Scholar
|
|
121
|
McMahan RH, Wang XXX, Cheng LL, Krisko T,
Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden-Mason L, Levi
M, et al: Bile acid receptor activation modulates hepatic monocyte
activity and improves nonalcoholic fatty liver disease. J Biol
Chem. 288:11761–11770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Miura K, Kodama Y, Inokuchi S, Schnabl B,
Aoyama T, Ohnishi H, Olefsky JM, Brenner DA and Seki E: Toll-like
receptor 9 promotes steatohepatitis by induction of interleukin-1
beta in mice. Gastroenterology. 139:323–334.e7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Tomita K, Tamiya G, Ando S, Ohsumi K,
Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, et al:
Tumour necrosis factor alpha signalling through activation of
Kupffer cells plays an essential role in liver fibrosis of
non-alcoholic steatohepatitis in mice. Gut. 55:415–424. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Wang J, Leclercq I, Brymora JM, Xu N,
Ramezani-Moghadam M, London RM, Brigstock D and George J: Kupffer
cells mediate leptin-induced liver fibrosis. Gastroenterology.
137:713–723. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Tacke F: Targeting hepatic macrophages to
treat liver diseases. J Hepatol. 66:1300–1312. 2017. View Article : Google Scholar : PubMed/NCBI
|