Introduction
Hepatocellular carcinoma (HCC), one of the most
deadly cancers worldwide, accounts for the majority of primary
liver cancer cases (1). The
overexpression of histone deacetylases (HDACs) plays an important
role in almost all cancerous behaviors of HCC, including cancer
initiation, progression and chemotherapy-resistance (2). A number of HDAC inhibitors, such as
vorinostat and trichostatin A, have been tested to treat HCC both
in preclinical models and clinical trials (3–5). HDAC
inhibitors exert their anti-tumor activities by inducing cell cycle
arrest, apoptosis, autophagy and differentiation (6). Furthermore, combining HDAC inhibitors
with other agents has been identified as a potential approach to
overcoming chemotherapeutic resistance (7,8).
Hypoxia activates genetic programs that facilitate
HCC cell metastasis, proliferation, as well as chemotherapy and
radiotherapy-resistance (9).
Hypoxia-inducible transcription factor (HIF) is activated in HCC
cells under hypoxic conditions, leading to angiogenesis and poor
prognosis (10). BAY 87–2243
inhibits HIF-1α and HIF-2α protein accumulation under hypoxia, and
BAY 87–2243 also suppresses the activity of mitochondrial complex I
in non-small cell lung cancer (11).
Furthermore, BAY 87–2243 significantly reduces tumor growth in BRAF
mutant melanoma cancer cells by targeting mitochondrial complex I
(12). BAY 87–2243 induces
mitochondrial permeability transition pore opening, stimulates
autophagosome formation and leads to the activation of
necroptotic/ferroptotic cell death in melanoma cells (13). In addition, the reduction of tumor
hypoxia by application of BAY-87-2243 before the beginning of
fractionated radiotherapy can improve local tumor control (14).
The activation of HDAC and hypoxia are associated
with cancer initiation, progression and chemo-resistance in HCC
cells (9,10). The current study aimed to investigate
whether blocking both HDAC and hypoxia can be efficient to treat
HCC. Thus, the authors hypothesized that combining HIF-1α inhibitor
with HDAC inhibitor could be a logical combination regimen to treat
HCC.
Materials and methods
Cell culture and reagents
BAY 87–2243 (cat. no. S7309), CHIR-99021 (cat. no.
S2924), vorinostat (cat. no. S1047) and trichostatin A (cat. no.
S1045) were purchased from Selleck Chemicals. Hep3B cells were
purchased from the Cell Bank of the Chinese Academy of Sciences.
The cells were cultured in MEM medium containing 10% fetal bovine
serum (PAN Biotech) at 37°C in a humidified atmosphere with 5%
CO2. Propidium iodide (PI; cat. no. 70-ZF-50-0001)
solution was purchased from Multi-Sciences Biotech.
Cell proliferation assays
Hep3B cells were seeded into 96-well plates at a
density of 4×103 per well and were allowed to grow
overnight at 37°C. The cells were incubated with BAY 87–2243 (2 or
4 µM) and/or HDAC inhibitors (0.2–4 µM) for 72 h at 37°C. Cell
proliferation was detected by the SRB assay, as previously
described (9). Briefly, cells were
fixed with 10% trichoroacetic acid solution at 4°C overnight.
Subsequent to washing, 0.4% SRB solution (100 µl per well;
Sigma-Aldrich) was added into each well. After 20 min staining at
room temperature, wells were rinsed with 1% acetic acid to remove
unbound dye and then left to air dry. Subsequently, 100 µl
Tris-base lye (10 mM; NeoFROXX GmbH) was added, followed by 10-min
oscillation. The absorbance was then recorded at 515 nm using a
multiscan spectrum.
Colony formation assay
Hep3B cells were seeded onto 6-well plates at a
density of 1,000 cells per well. After 14 days, cells were stained
with 0.1% crystal violet solution for 30 min at 4°C and the
colonies were imaged by a camera.
Detection of cell death
Hep3B cells were seeded into 6 well plates and
incubated with BAY 87–2243 (10 µM; higher than the SRB assay using
96 well plates) and/or and/or HDAC inhibitors (0.25–2 µM), and PI
staining was then used to detect cell death, as previously
described (9). Sub-G1 analysis
following PI staining was used to detect apoptosis. Cells were
harvested, washed with PBS three times and fixed with pre-cooled
70% ethanol at −20°C overnight. Cells were washed and resuspended
in 500 µl PBS containing 50 µg/ml RNase at 37°C for 30 min. The
cells were then stained with 5 µg PI at room temperature for 30
min. For each sample, 2×104 cells were collected and
analyzed using a FACS-Calibur cytometer and the data were analyzed
using Cellquest Software (version 6.0; BD Biosciences).
Western blot analysis
Western blot analysis was performed as previously
described (9). Proteins were
extracted with lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl,
0.1% sodium dodecyl sulfate, 1 mM ethylenediaminetetraacetic acid,
0.5% deoxycholic acid, 1% NP-40, 2.0 µg/ml aprotinin, 1 mM
phenylmethylsulfonylfluoride and 0.02% sodium azide (Beyotime
Institute of Biotechnology). The lysates were centrifuged at 10,000
× g for 30 min at 4°C, then the concentrations of protein were
determined by the BCA method using Enhanced BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Total proteins (40 µg/lane)
were fractionated on 8–15% Tris-glycine gels, and then they were
transferred to polyvinylidene fluoride membrane, blocked with 5%
(w/v) Difco Skim milk (cat. no. 232100; BD Biosciences) in PBS at
room temperature for 1 h and probed with the primary antibodies at
4°C overnight. The proteins were incubated and visualized with
horseradish peroxidase-conjugated goat anti-mouse (cat. no. GAM007)
and anti-rabbit (cat. no. GAM007) IgG secondary antibodies
(MultiSciences) at a dilution of 1:5,000 for 1 h at room
temperature. Finally, proteins were visualized using the enhanced
chemiluminescence detection system (PerkinElmer). Anti-poly
(ADP-ribose) polymerase (PARP; cat. no. sc-7150; 1:1,000), anti-AKT
(cat. no. sc-135829; 1:500), anti-phospho-AKT (Ser-473; cat. no.
sc-7985; 1:500) and anti-GAPDH (cat. no. sc-25778; 1:1,000)
antibodies were obtained from Santa Cruz Biotechnology, Inc.
Anti-phospho-glycogen synthase kinase (GSK)-3β (Ser-9; cat. no.
5558S; 1:500) and anti-snail antibodies (cat. no. 3879S; 1:1,000)
were purchased from Cell Signaling Technology, Inc. Anti-GSK3β
antibody (cat. no. 610202; 1:1,000) was obtained from BD
Biosciences.
Wound healing assay
Hep3B cells were seeded in 24-well plates and
cultured until they reached 90% confluency. Confluent monolayer
cells were gently scratched with a sterile pipette tip and then
washed three times with PBS to clear cell debris and suspended
cells. Fresh serum-free medium was added, and the cells were
allowed to close the wound for 24 h under normal conditions at
37°C. Images of the wound in the same relative position were
captured with a computer-assisted microscope (model, CKX41SF;
Olympus Corporation).
Statistical analysis
One-way analysis of variance followed by Tukey post
hoc test was used to examine the significance of differences among
groups. The results were presented as the mean ± standard deviation
from three independent experiments and P<0.05 was used to
indicate a statistically significant difference.
Results
BAY 87–2243 enhances the HDAC
inhibitor-induced antiproliferative effect of Hep3B cells
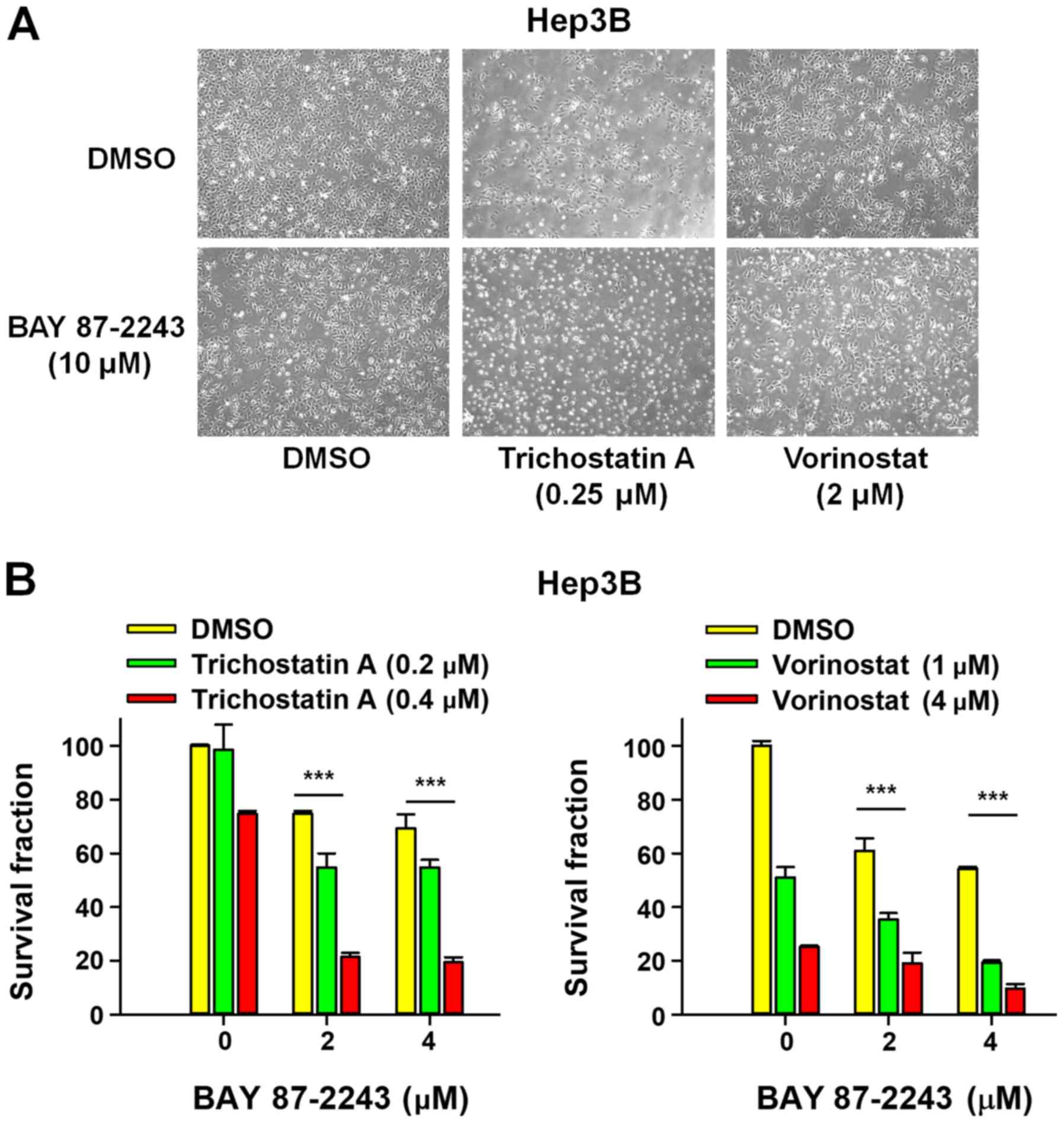
First, the morphological changes induced by the
combined treatment of BAY 87–2243 and HDAC inhibitors was detected
in Hep3B cells. Fig. 1A showed that
a very large proportion of Hep3B cells treated with BAY 87–2243
plus HDAC inhibitors (trichostatin A or vorinostat) had dislodged
from the dishes whereas the remaining adherent cells showed loss of
adhesion, shrinkage and rounding, which are typical morphological
changes associated with apoptosis (15). Furthermore, the results presented in
Fig. 1B showed that BAY 87–2243
combined with HDAC inhibitors (trichostatin A or vorinostat)
significantly decreased the proliferation ability of Hep3B cells
following incubation for 72 h, as compared with single
agent-treatment. To further confirm the enhanced antiproliferative
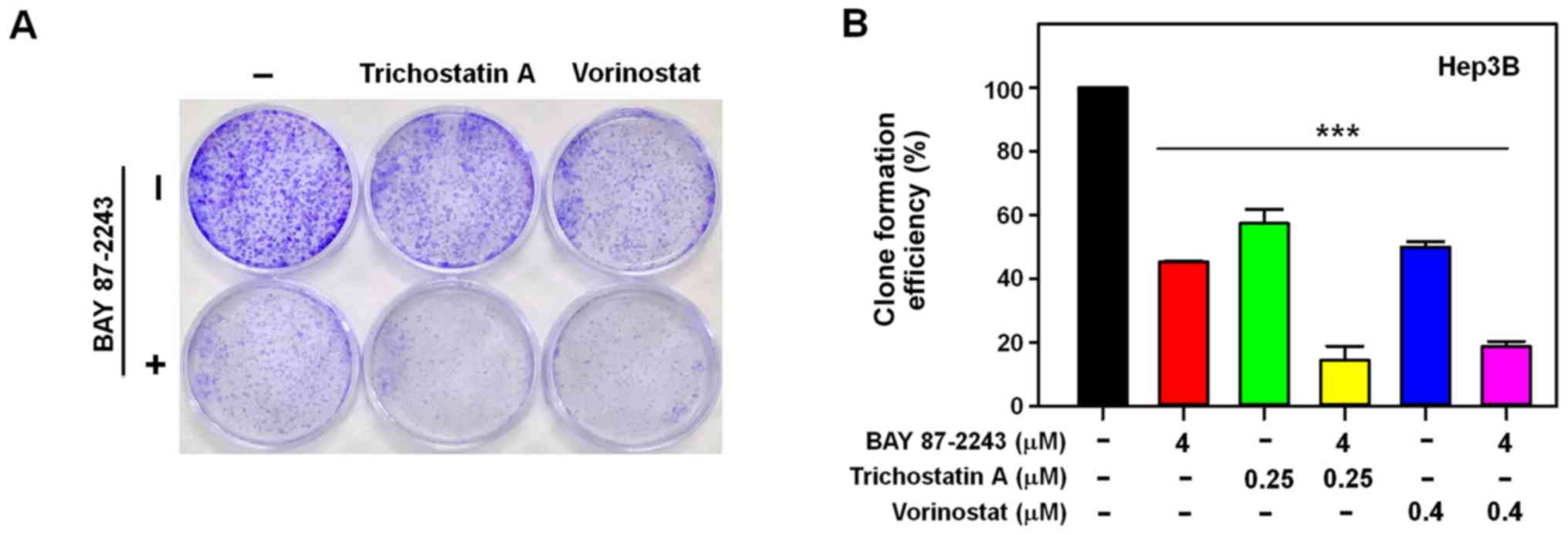
effect of the combined treatment with BAY 87–2243 and HDAC
inhibitors, whether the combination might inhibit colony formation
in Hep3B cells or not, was determined next. It was found that BAY
87–2243 combined with HDAC inhibitors-treated Hep3B cells formed
fewer and smaller colonies as compared with either the control or
single agent-groups (P<0.0001; Fig.
2A and B). These results confirmed that BAY 87–2243 enhanced
the HDAC inhibitor-induced antiproliferative effect in Hep3B
cells.
BAY 87–2243 enhances HDAC
inhibitor-induced cell death in Hep3B cells
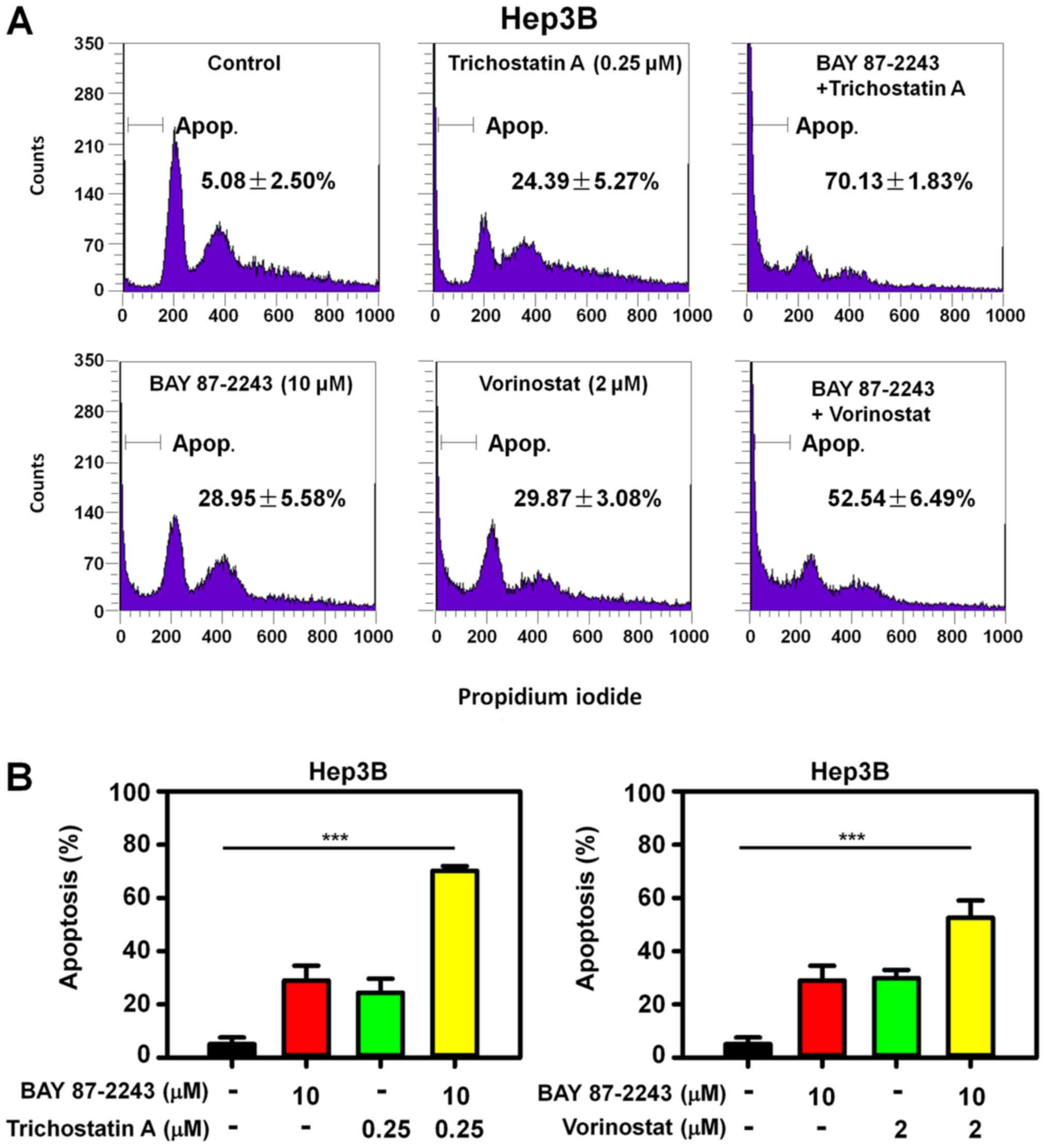
To determine whether the enhanced antiproliferative
effect of BAY 87–2243 plus HDAC inhibitors resulted from cell
death, PI staining was used to detect the cell death in Hep3B cells
treated with BAY 87–2243 and/or HDAC inhibitors. The rate of cell
death in Hep3B cells treated with BAY 87–2243 and trichostatin A
was 70.13%, and it was significantly higher than that in the BAY
87–2243 group (28.94%) and trichostatin A group only (24.39%)
following treatment for 48 h (Fig.
3A; upper panel). Similarly, the combination treatment of BAY
87–2243 and vorinostat resulted in an increased apoptotic rate, as
compared with single agent groups (Fig.
3A; lower panel). Therefore, BAY 87–2243 enhanced HDAC
inhibitor-induced cell death in Hep3B cells (P<0.0001; Fig. 3B). Furthermore, the BAY 87–2243 plus
HDAC inhibitor-induced enhanced apoptosis was accompanied by higher
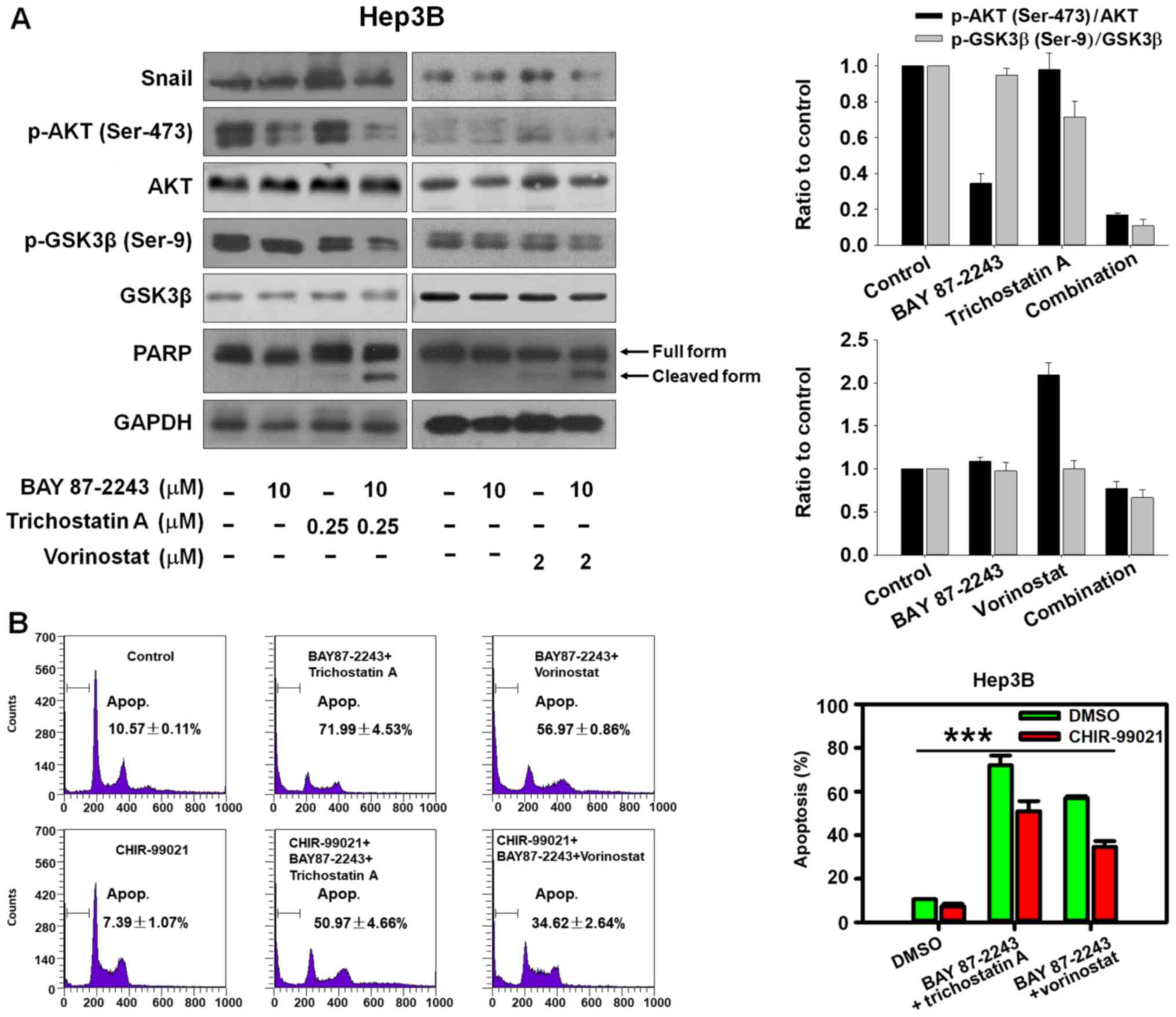
expression of the cleaved PARP, in Hep3B cells (Fig. 4A).
BAY 87–2243 combined with HDAC
inhibitors induces cell death via GSK3β activation
The AKT/GSK-3β/Snail pathway has been shown to be
involved in HCC metastasis by inducing epithelial-mesenchymal
transition (EMT) (16,17). The transcription factor Snail,
controls EMT by repressing the expressions of E-cadherin and other
epithelial genes, and GSK-3β phosphorylates Snail inducing its
degradation (18). The present data
indicated that BAY 87–2243 plus HDAC inhibitors could markedly
inhibit AKT, Snail and activate GSK-3β by suppressing
phosphorylated GSK-3β (Ser-9; Fig.
4A). Furthermore, it was found that GSK-3β inhibitor CHIR-99021
could partially reverse the cell death induced by BAY 87–2243 plus
HDAC inhibitors (Fig. 4B;
P<0.001). Thus, the potential ability of BAY 87–2243 plus HDAC
inhibitors to inhibit the AKT/GSK-3β/Snail pathway makes it an
attractive chemotherapy strategy to hinder HCC metastasis, but this
needs to be investigated further.
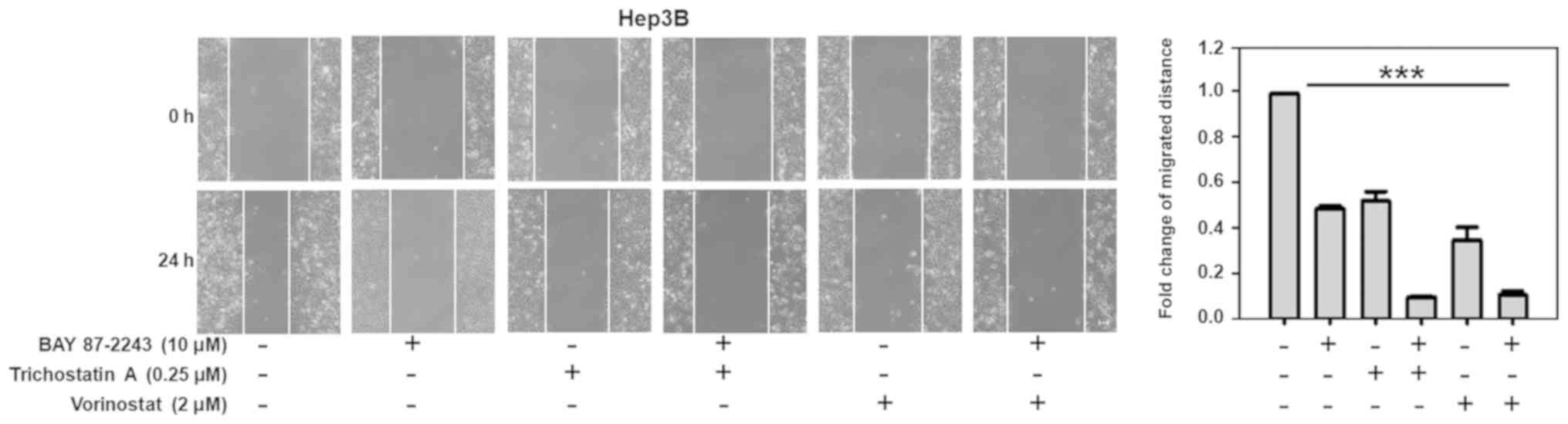
BAY 87–2243 combined with HDAC
inhibitors suppresses the migration of Hep3B cells
As shown in Fig. 5,
untreated Hep3B cells migrated within 24 h after wounding, whereas
there was moderate inhibition of cell migration when the Hep3B
cells were treated with BAY 87–2243 (10 µM) or Trichostatin A (0.25
µM). However, BAY 87–2243 plus Trichostatin A could significantly
suppress the migration of Hep3B cells (P<0.001). Similarly, BAY
87–2243 plus Vorinostat could also significantly inhibit the
migration of Hep3B cells.
Discussion
HCC is a metabolically heterogeneous cancer, and the
use of glucose by HCC cells can influence their tumorigenicity
(19). Therefore, increased glucose
metabolism is important for the growth of HCC cells (20). In addition, the suppression of HDACs
inhibits glucose metabolism and HCC cell growth by restoring
fructose-1,6-bisphosphatase activity (3). BAY 87–2243 inhibits cell proliferation
with low efficacy under standard conditions, but high efficacy
under glucose depletion, a condition favoring mitochondrial
generation as an energy source (11). In the present study, it was
hypothesized that HDAC inhibitors might increase the anti-HCC
efficacy of BAY 87–2243 in HCC cells. Indeed, the results
demonstrated that BAY 87–2243 may suppress the proliferation of HCC
cells, and BAY 87–2243 plus HDAC inhibitors (trichostatin A or
vorinostat) could distinctly inhibit proliferation and induce cell
death in Hep3B cells. Therefore, a combination chemotherapy regimen
incorporating BAY 87–2243 and HDAC inhibitors may be an effective
chemotherapy strategy for the treatment of HCC.
HIF-1α could be activated by hypoxia-independent
mechanisms such as the PI3K/AKT/mTOR signaling pathway (21). In the present study, BAY 87–2243 as a
HIF-1α inhibitor could decrease p-AKT (ser-473), indicating that
HIF-1α suppression could also affect PI3K/AKT/mTOR signaling
pathway. Furthermore, the present data also indicated that BAY
87–2243 plus HDAC inhibitors could suppress PI3K/AKT/mTOR
signaling. In addition, the inactivation of GSK-3β plays an
important role in mediating the improved glucose metabolism, and
GSK-3β (Ser-9) phosphorylation reduces the activity of GSK-3β
(22,23). In the present study, it was shown
that BAY 87–2243 plus HDAC inhibitors could activate GSK-3β by
suppressing phosphorylated GSK-3β (Ser-9). In addition, GSK-3β
inhibitor CHIR-99021 successfully reversed the cell death induced
by BAY 87–2243 plus HDAC inhibitors. These data indicated that BAY
87–2243 plus HDAC inhibitors might suppress glucose metabolism by
activating GSK-3β in HCC cells.
The majority of HCC patients are diagnosed in
advanced stages of the disease, and the poor survival in patients
with HCC can be largely attributed to rapid intrahepatic recurrence
due to cancer metastases (24). HDAC
inhibitors, including trichostatin A and vorinostat, induce cell
death but simultaneously augment cell migration and metastasis to
ruin therapeutic efficacy via activating protein kinase C signaling
(25). Furthermore, HDAC inhibitors
are able to promote the expression of Snail, and trigger
Snail-induced EMT which is critical for HDAC inhibitors-initiated
invasion and metastasis (26). The
findings of the present study highlighted that BAY 87–2243 could
reverse the HDAC inhibitor-induced activation of Snail in HCC
cells. Furthermore, the present data showed that BAY 87–2243 plus
HDAC inhibitor could significantly suppress the migration of HCC
cells, indicating that combining BAY 87–2243 with HDAC inhibitors
appears to be an attractive option for the patients with metastatic
HCC. However, further research is required to elucidate the
anti-metastatic activity of BAY 87–2243 plus HDAC inhibitors in
HCC.
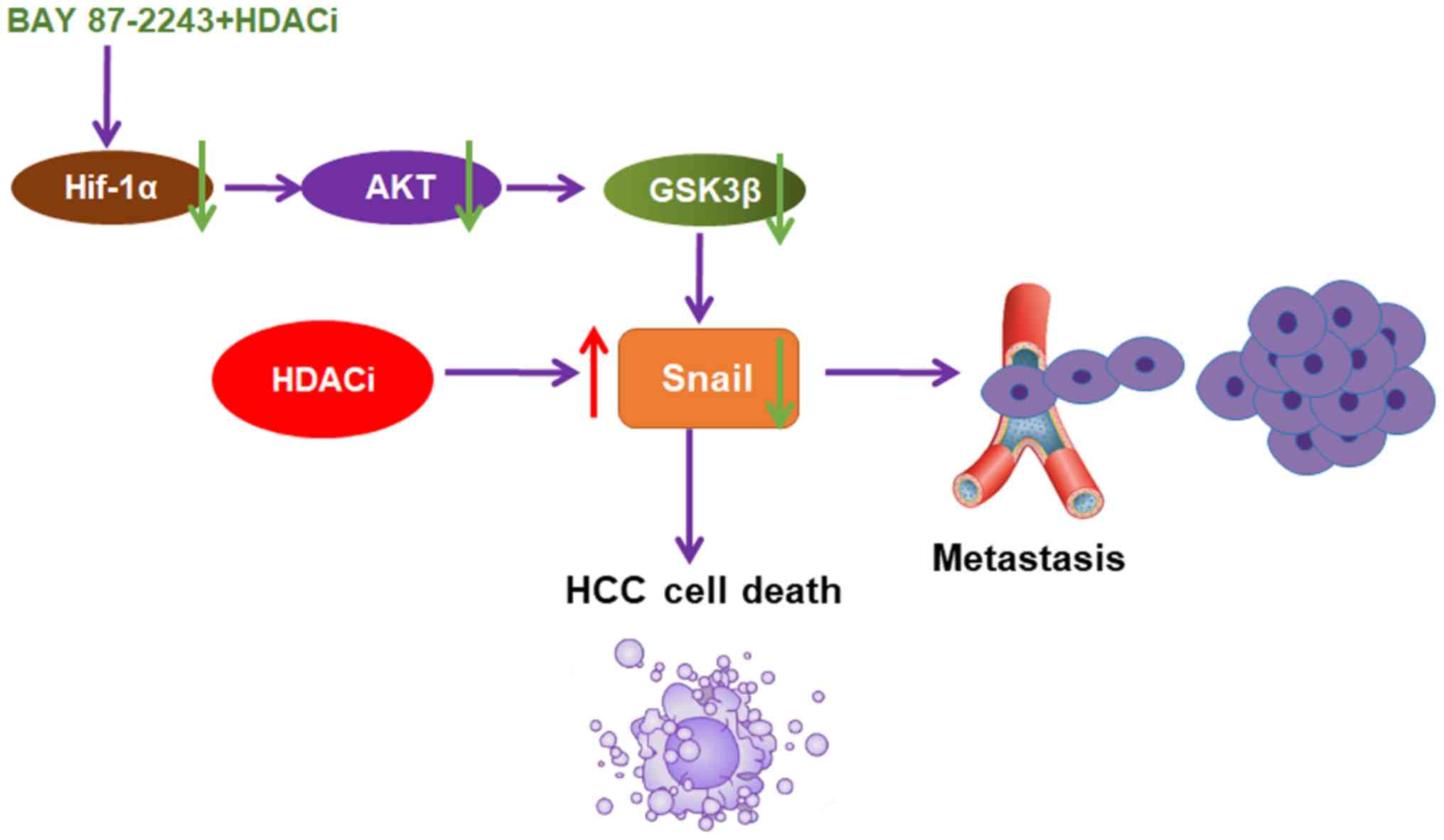
In conclusion, the present data provided, to the
best of our knowledge, the first evidence that BAY 87–2243 could
enhance the anti-HCC effect of HDAC inhibitors through the
AKT/GSK-3β/Snail signaling pathway (Fig.
6). It was also shown that BAY 87–2243 plus Vorinostat could
inhibit the migration of Hep3B cells. BAY 87–2243 plus HDAC
inhibitors might be an attractive chemotherapy strategy for HCC
therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by Zhejiang Province
Medical Key Discipline Construction (grant no. 2018-2-03, 2018),
National Natural Science Foundation of China (grant no. 81702887,
2017), Hangzhou Major Science and Technology Project (grant no.
20172016A01, 2017), Fund of Hangzhou Medical Key Discipline
Construction (grant no. 2017-51-07, 2017), and Zhejiang Provincial
Foundation of Natural Science (grant nos. LQ16H310004, 2017 and
LY19H310004, 2019).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
YL, MJR, NYZ and LWW performed the experiments. NML
and CZ conceived and designed the study. CZ wrote the manuscript.
All authors read and approved the manuscript.
Ethics approval and consent to
participate
This article does not contain any studies with
animal or human participants performed by any of the authors.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Geng Y, Michowski W, Chick JM, Wang YE,
Jecrois ME, Sweeney KE, Liu L, Han RC, Ke N, Zagozdzon A, et al:
Kinase-independent function of E-type cyclins in liver cancer. Proc
Natl Acad Sci USA. 115:1015–1020. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fu M, Shi W, Li Z and Liu H: Activation of
mPTP-dependent mitochondrial apoptosis pathway by a novel pan HDAC
inhibitor resminostat in hepatocellular carcinoma cells. Biochem
Biophys Res Commun. 477:527–533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang J, Jin X, Yan Y, Shao Y, Pan Y,
Roberts LR, Zhang J, Huang H and Jiang J: Inhibiting histone
deacetylases suppresses glucose metabolism and hepatocellular
carcinoma growth by restoring FBP1 expression. Sci Rep.
7:438642017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen T, Gu C, Xue C, Yang T, Zhong Y, Liu
S, Nie Y and Yang H: LncRNA-uc002mbe.2 interacting with hnRNPA2B1
Mediates AKT deactivation and p21 Up-regulation induced by
trichostatin in liver cancer cells. Front Pharmacol. 8:6692017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang J and Zhong Q: Histone deacetylase
inhibitors and cell death. Cell Mol Life Sci. 71:3885–3901. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L
and Wu MC: Inhibition of autophagy signi fi cantly enhances
combination therapy with sorafenib and HDAC inhibitors for human
hepatoma cells. World J Gastroenterol. 20:4953–4962. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoon CY, Park MJ, Lee JS, Lee SC, Oh JJ,
Park H, Chung CW, Abdullajanov MM, Jeong SJ, Hong SK, et al: The
histone deacetylase inhibitor trichostatin A synergistically
resensitizes a cisplatin resistant human bladder cancer cell line.
J Urol. 185:1102–1111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li YL, Zhang NY, Hu X, Chen JL, Rao MJ, Wu
LW, Li QY, Zhang B, Yan W and Zhang C: Evodiamine induces apoptosis
and promotes hepatocellular carcinoma cell death induced by
vorinostat via downregulating HIF-1α under hypoxia. Biochem Biophys
Res Commun. 498:481–486. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang GW, Yang LY and Lu WQ: Expression of
hypoxia-inducible factor 1alpha and vascular endothelial growth
factor in hepatocellular carcinoma: Impact on neovascularization
and survival. World J Gastroenterol. 11:1705–1708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ellinghaus P, Heisler I, Unterschemmann K,
Haerter M, Beck H, Greschat S, Ehrmann A, Summer H, Flamme I, Oehme
F, et al: BAY 87-2243, a highly potent and selective inhibitor of
hypoxia-induced gene activation has antitumor activities by
inhibition of mitochondrial complex I. Cancer Med. 2:611–624.
2013.PubMed/NCBI
|
|
12
|
Schöckel L, Glasauer A, Basit F, Bitschar
K, Truong H, Erdmann G, Algire C, Hägebarth A, Willems PH, Kopitz
C, et al: Targeting mitochondrial complex I using BAY 87-2243
reduces melanoma tumor growth. Cancer Metab. 3:112015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Basit F, van Oppen LM, Schöckel L,
Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S,
Kopitz C, Heroult M, Hgm Willems P and Koopman WJ: Mitochondrial
complex I inhibition triggers a mitophagy-dependent ROS increase
leading to necroptosis and ferroptosis in melanoma cells. Cell
Death Dis. 8:e27162017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Helbig L, Koi L, Brüchner K, Gurtner K,
Hess-Stumpp H, Unterschemmann K, Baumann M, Zips D and Yaromina A:
BAY 87-2243, a novel inhibitor of hypoxia-induced gene activation,
improves local tumor control after fractionated irradiation in a
schedule-dependent manner in head and neck human xenografts. Radiat
Oncol. 9:2072014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Häcker G: The morphology of apoptosis.
Cell Tissue Res. 301:5–17. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lan Y, Han J, Wang Y, Wang J, Yang G, Li
K, Song R, Zheng T, Liang Y, Pan S, et al: STK17B promotes
carcinogenesis and metastasis via AKT/GSK-3β/Snail signaling in
hepatocellular carcinoma. Cell Death Dis. 9:2362018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu L, Dai Y, Chen J, Zeng T, Li Y, Chen
L, Zhu YH, Li J, Li Y, Ma S, et al: Maelstrom promotes
hepatocellular carcinoma metastasis by inducing
epithelial-mesenchymal transition by way of Akt/GSK-3β/Snail
signaling. Hepatology. 59:531–543. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peinado H, Portillo F and Cano A:
Switching on-off Snail: LOXL2 versus GSK3beta. Cell Cycle.
4:1749–1752. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cassim S, Raymond VA, Dehbidi-Assadzadeh
L, Lapierre P and Bilodeau M: Metabolic reprogramming enables
hepatocarcinoma cells to efficiently adapt and survive to a
nutrient-restricted microenvironment. Cell Cycle. 17:903–916. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Amann T, Maegdefrau U, Hartmann A, Agaimy
A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Schölmerich
J, Oefner PJ, et al: GLUT1 expression is increased in
hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol.
174:1544–1552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitajima Y and Miyazaki K: The critical
impact of HIF-1a on gastric cancer biology. Cancers (Basel).
5:15–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hall JL, Chatham JC, Eldar-Finkelman H and
Gibbons GH: Upregulation of glucose metabolism during intimal
lesion formation is coupled to the inhibition of vascular smooth
muscle cell apoptosis. Role of GSK3beta. Diabetes. 50:1171–1179.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu KY, Wang LT and Hsu SH: Modification
of epigenetic histone acetylation in hepatocellular carcinoma.
Cancers (Basel). 10(pii): E82018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin KT, Wang YW, Chen CT, Ho CM, Su WH and
Jou YS: HDAC inhibitors augmented cell migration and metastasis
through induction of PKCs leading to identification of low toxicity
modalities for combination cancer therapy. Clin Cancer Res.
18:4691–4701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu W, Liu H, Liu ZG, Wang HS, Zhang F,
Wang H, Zhang J, Chen JJ, Huang HJ, Tan Y, et al: Histone
deacetylase inhibitors upregulate Snail via Smad2/3 phosphorylation
and stabilization of Snail to promote metastasis of hepatoma cells.
Cancer Lett. 420:1–13. 2018. View Article : Google Scholar : PubMed/NCBI
|