Introduction
Nasopharyngeal carcinoma (NPC) is the most frequent
malignant head and neck cancer in Southeast Asia, especially in
southern China. The annual incidence rate is ~20–50 cases per
100,000 individuals (1). An
estimated 42,100 new cases and 21,320 fatalities were attributed to
NPC in China in 2013 (2).
Epstein-Barr virus infection, genetic factors and environmental
conditions are involved in the pathogenesis of NPC (3). A cure rate of >90% after
radiotherapy has been reported in early stage NPC (4). However, most NPC patients are initially
diagnosed as undifferentiated and nonkeratinizing carcinoma at an
advanced stage (5).
In recent years, the local and regional control of
NPC has been markedly improved with the emergence of
intensity-modulated photon-based radiation therapy (6). Although radiotherapy is the primary
treatment modality, multidisciplinary management is the current
treatment standard, especially for patients with local advanced
disease. Concurrent chemotherapy with radiotherapy remains the
standard of care (7). At the present
time, the 5-year overall survival rate of patients with local
advanced NPC is only 50–70% and 30–40% of patients still develop
distant metastasis within 4 years with 10–20% developing
locoregional relapse (8–10). Patients with advanced disease suffer
from a worse outcome. To improve the long-term overall survival
rate of NPC, it is crucial to explore more effective treatment
modalities.
Statins are 3-hydroxy-3-methylglutaryl coenzyme A
(HMG-CoA) reductase inhibitors, which are frequently used as safe
and effective therapeutic agents for hypercholesterolemia,
contributing to a reduction in morbidity and mortality from
atherosclerosis and coronary artery disease (11,12).
Statins target mevalonate, one of the cholesterol precursors, which
is catalyzed by HMG-CoA reductase. It has been reported that
overexpression of mevalonate is associated with cell survival and
proliferation of cancer cells (13).
Statins inhibit the mevalonate pathway and reduce the synthesis of
geranylgeranyl pyrophosphate and farnesyl pyrophosphate, which are
indispensable for the posttranslational modification of certain
regulatory proteins (14). In
addition to a lipid-lowering effect, statins also exert
antiproliferative and proapoptotic effects in cancer cells by
regulating the cell cycle (15,16).
Koul et al (17) demonstrated
that inhibition of the mevalonate pathway could affect a number of
cellular functions in bone-resorbing osteoclasts and inhibit cancer
cell proliferation, viability, motility, invasion, and
angiogenesis. Statin-induced inhibition of the mevalonate pathway
attenuates the growth of mesenchymal-like cancer cells (18). Anticancer effects with in
vitro simvastatin treatment have been reported in prostate,
breast and ovarian cancer, and adenocarcinoma (15,19–21).
However, the effects of simvastatin in NPC remain unclear.
The authors' previous study demonstrated that
simvastatin induces loss of cell attachment, reduces colony forming
units and inhibits sphere formation in soft gel agar in squamous
cell carcinoma of the nasal cavity (22). The results of the present study
indicated that statin acts as a relatively safe and cost-effective
chemoadjuvant agent in the treatment of nasal malignant tumors.
However, little is known about the molecular mechanism of statins
in NPC. In the present study, the effect of simvastatin was
examined in human NPC cell line, C666-1 and investigated the
associated molecular mechanisms.
Materials and methods
Materials and cell culture
Simvastatin, cisplatin and mevalonic acid lactone
were purchased from Sigma-Aldrich (Merck KGaA). For in vitro
administration, simvastatin was dissolved in dimethyl sulfoxide
(DMSO) at a concentration of 50 mM and stored at −20°C as a stock
solution. Mevalonic acid lactone was dissolved in PBS at a
concentration of 200 mM as stock solution. C666-1 was purchased
from American Type Culture Collection (Thermo Fisher Scientific,
Inc.) and cultured in RPMI-1640 (Corning Inc.) supplemented with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin/streptomycin (Sigma-Aldrich; Merck KGaA). Cells
were cultured at 37°C in a humidified atmosphere of 5%
CO2. In the signal pathway experiments, cells were
pretreated with JNK inhibitor, SP6000125 (10 µM; BioSource; Thermo
Fisher Scientific, Inc.), 4 h before simvastatin treatment.
Cell viability assay
NPC cells were seeded in 96-well plates at a density
of 1×104 cells per well in 100 µl of medium. A total of
24 h after seeding, cells were treated with the indicated
concentrations of simvastatin (0–100 µM) for 48 h. Cell viability
was determined by alamarBlue Cell Viability Assay according to the
manufacturer's protocol (BioSource; Thermo Fisher Scientific,
Inc.). Briefly, at the end of incubation, culture supernatant was
replaced with alamarBlue reagent (1:20 v/v dilution in fresh
culture medium). Plates were incubated at 37°C for 2–4 h and the
absorbance was measured at 490 nm with an Omega microplate reader
(Imgen Technologies). Cells were treated with or without 50 µM of
simvastatin for 48 h, then were observed and photomicrographs were
captured using a Leica Microsystems light microscope at a
magnification of ×400.
Apoptosis assay
Cell apoptosis was assessed by Annexin V-APC and
propidium iodide (PI) assay (BD Biosciences). Briefly, C666-1 cells
were treated with or without 50 µM of simvastatin for 16 and 24 h.
The adherent cells and floating cells were harvested and washed
with cold PBS. Cells were resuspended in Annexin binding buffer at
a concentration of 5×105 cells/ml and incubated with
Annexin V-APC and PI for 15 min in the dark at room temperature.
After incubation, the cells were analyzed using Acuuri C6 and the
data were analyzed using CFlow Plus (Accuri Cytometers).
Caspase 3 fluorimetric assay
C666-1 cells were treated with simvastatin (50 µM)
or statin plus mevalonate (200 µM) for 24 h. After incubation,
cells were collected and centrifuged at 300 × g for 5 min at 4°C. A
Caspase 3 fluorimetric assay was performed using the Caspase 3 kit
(R&D Systems, Inc., Minneapolis, MN, USA). In brief, pelleted
cells were lysed using Lysis Buffer (R&D Systems, Inc.). A 100
µl portion of lysate was transferred into a 96-well plate and mixed
with 50 µl of Reaction Buffer 3 and 5 µl of Caspase-3 fluorogenic
substrate. After incubation for 1 h at 37°C, caspase activity was
determined with an Omega microplate reader.
TUNEL assay
Cells were seeded in 8-well cell culture slides at a
density of 500 cells/well. After overnight culture at 37°C, cells
were treated with or without simvastatin (50 µM) or simvastatin
plus cisplatin (35 µM) for 24 h and the TUNEL assay was performed
(R&D Systems, Inc.). The treated cells were labeled using
terminal deoxynucleotidyl transferase to transfer biotin-dUTP to
the free 3′-OH of cleaved DNA at 37°C for 1 h. Then the slides were
incubated in the dark with avidin-fluorescein isothiocyanate for 20
min at room temperature and 3 fields were visualized by
fluorescence microscopy at a magnification of ×400.
Western blot analysis
Total protein was extracted from C666-1 cells using
radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich; Merck
KGaA) and protein concentrations were quantified with the
bicinchoninic acid Protein Assay kit (Beyotime Institute of
Biotechnology, Haimen, China). Samples (10–30 µg) were separated by
10% SDS-PAGE and transferred to nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
nonfat milk in TBS-T for 1 h at room temperature and then incubated
overnight at 4°C with the following primary antibodies (1:1,000):
Polyclonal rabbit anti cleaved Caspase-3 (cat. no. 9661),
polyclonal rabbit anti Bim (cat. no. 2819), polyclonal rabbit anti
Bax (cat. no. 2772), monoclonal rabbit anti phosphorylated protein
kinase B (p-Akt; cat. no. 4058), polyclonal rabbit anti Akt (cat.
no. 9272), monoclonal rabbit anti p-extracellular signal regulated
kinase (Erk)1/2 (cat. no. 9106), polyclonal rabbit anti Erk1/2
(cat. no. 9102) and c-Jun (all from Cell Signaling Technology,
Inc., Danvers, MA, USA), monoclonal rabbit anti p-c-Jun (cat. no.
1527; Epitomics, Abcam, Cambridge, UK), polyclonal rabbit anti α
tubulin (cat. no. sc-5546), polyclonal rabbit anti p27 (cat. no.
sc-528), polyclonal rabbit anti cyclin-dependent kinase (CDK4; cat.
no. sc-260), and monoclonal mouse anti cyclin D1 (cat. no. sc-246;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA). After washing,
membranes were further incubated with secondary antibodies (anti
rabbit: sc-2004 and anti-mouse: sc-2005, both from Santa Cruz
Biotechnology, Inc.; 1:5,000) conjugated with horseradish
peroxidase for 1 h at room temperature. The immunoreactive signal
was detected using the enhanced chemiluminescent detection system
(GE Healthcare, Piscataway, NJ, USA).
Cell cycle analysis
After treatment with simvastatin, cell cycle
distribution and ploidy status of C666-1 cells were determined by
flow cytometry. Cells were collected and washed in PBS, then fixed
with 70% ice-cold ethanol for 24 h at −20°C. The fixed cells were
treated with 10 mg/ml RNAse for 30 min at 37°C. The DNA content of
cells was evaluated by CFlow after staining with PI (50 µg/ml).
Statistical analysis
Results were expressed as the mean ± standard
deviation. Statistical analysis was performed by Student's t-test
or analysis of variance using SPSS version 19 (IBM, Corp., Armonk,
NY, USA). SNK-q was used for comparisons among groups. P<0.05
was considered to be a statistically significant difference.
Results
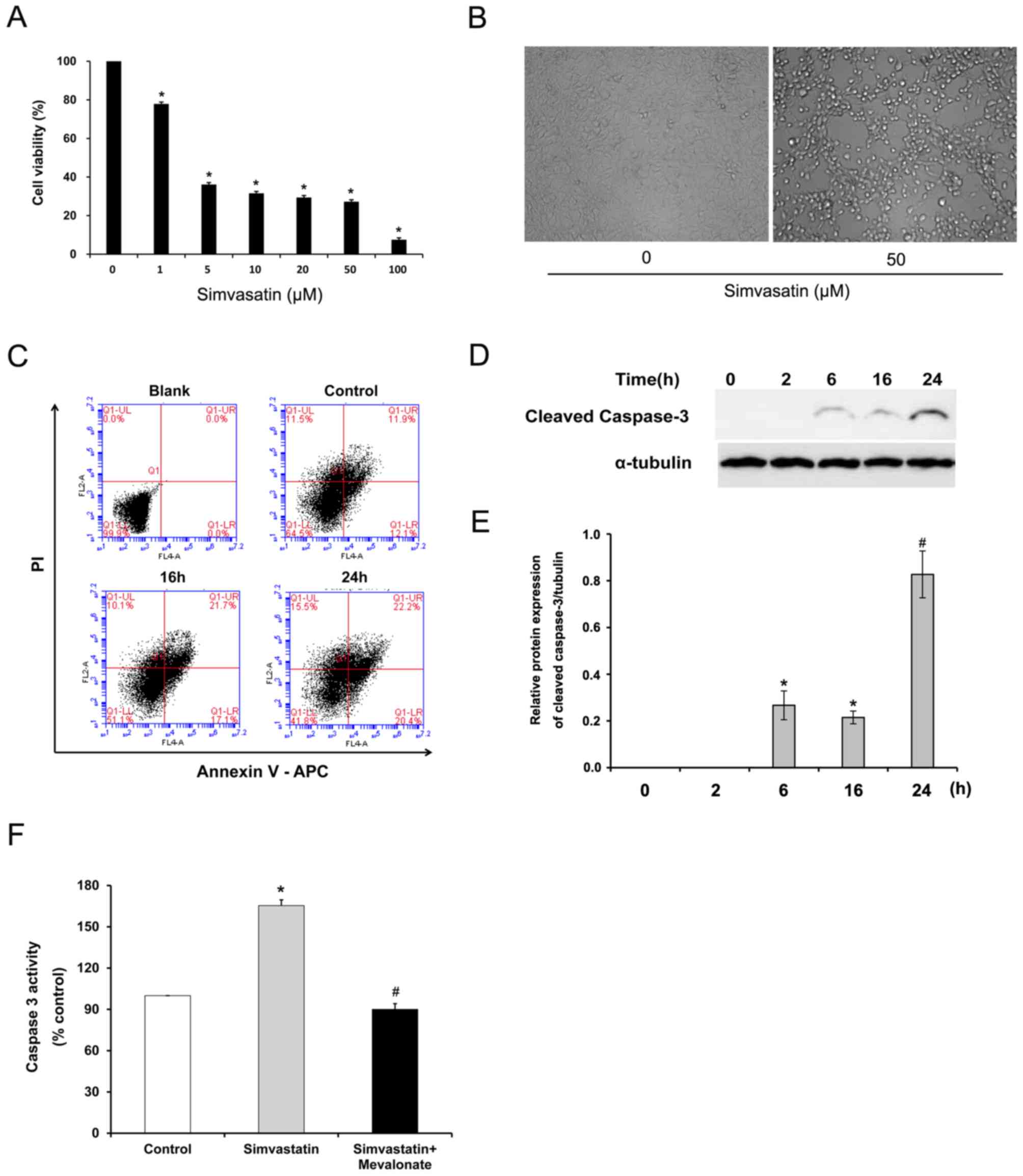
Effect of simvastatin on the viability
of C666-1 cells
To determine the inhibitory effect of simvastatin on
C666-1 cell viability, the alamarBlue Cell Viability Assay was
performed. C666-1 cells were treated with various concentrations of
simvastatin for 48 h and cell viability was measured. As presented
in Fig. 1A, simvastatin
statistically significantly decreased cell viability in a
concentration-dependent manner over the dose range of 1–100 µM
(P<0.05). Cell morphology was changed and the number of adherent
cells was reduced after statin treatment (Fig. 1B).
Effect of simvastatin on apoptosis and
caspase 3 activity in C666-1 cells
To examine whether the decreased viability of C666-1
is due to statin-induced apoptosis, cell apoptosis in C666-1 was
evaluated using flow cytometry. C666-1 cells treated with 50 µM of
simvastatin for up to 24 h were subjected to Annexin V assay.
Statin treatment increased the proportion of apoptotic cells in the
early (Annexin V+/PI−: From 12.6±0.8 to
19.5±1.8%) and late (Annexin V+/PI+: From
12.3±1.0 to 21.3±2.1%) stages (Fig.
1C). To further elucidate the statin-induced apoptosis in NPC,
the effect of simvastatin exposure on caspase 3 activity in C666-1
cells was evaluated. Simvastatin statistically significantly
increased the expression of cleaved caspase 3 after 6 h exposure
(P<0.05; Fig. 1D and E) and
induced caspase 3 activity by a 1.6-fold increase in C666-1 cells
compared with untreated cells (P<0.05; Fig. 1F). Interestingly, supplementation of
mevalonate, one of the HMG-CoA reductase downstream products,
significantly reversed the increased caspase 3 activation induced
by simvastatin (P<0.01; Fig. 1F).
These results indicate that cell apoptosis induced by simvastatin
in C666-1 cells is mediated through the HMG-CoA reductase
pathway.
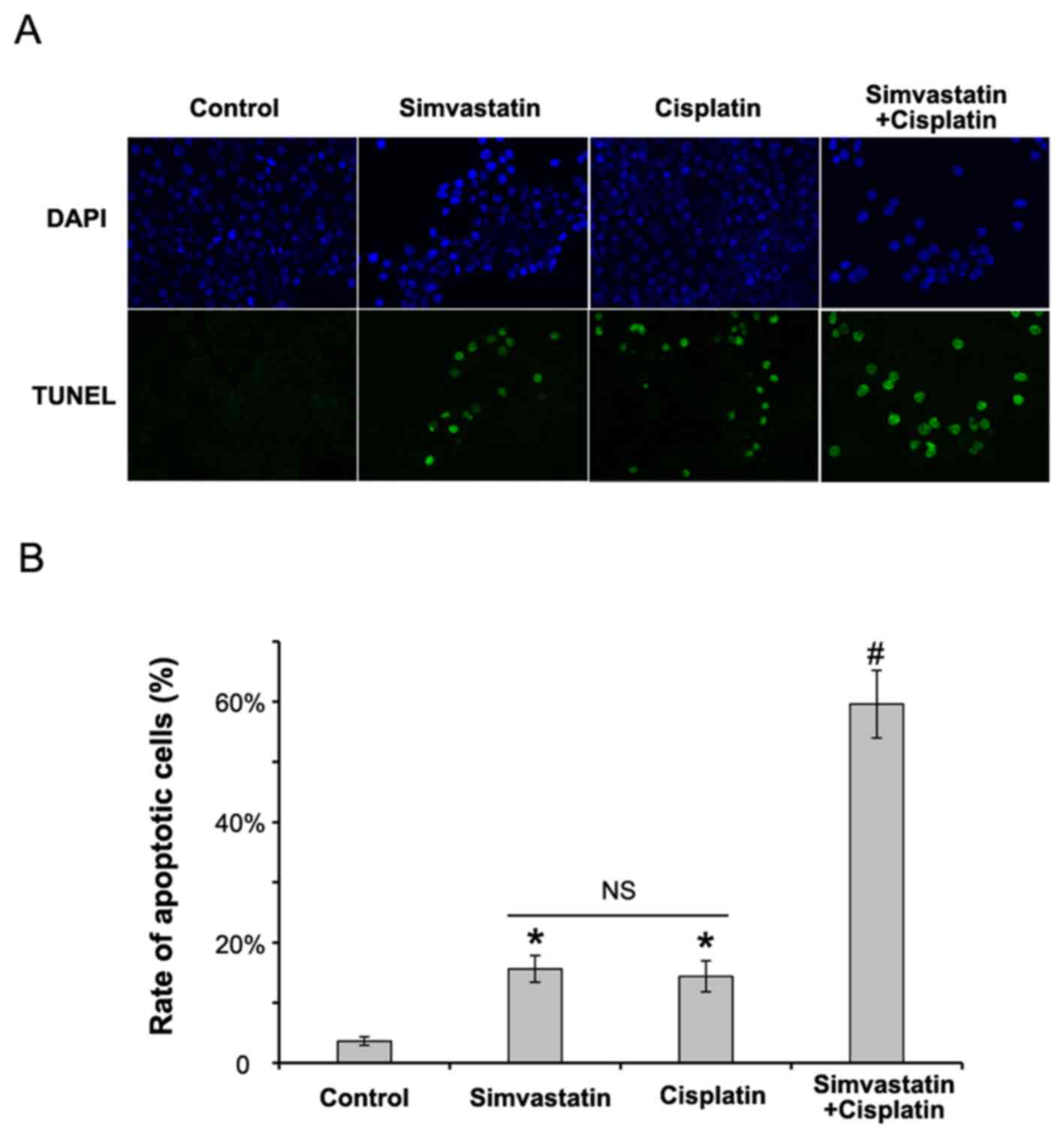
Simvastatin enhances the anti-tumor
effects of cisplatin in C666-1 cells
Combination chemotherapy is an ideal therapeutic
approach recommended in most cancers. An experiment was designed
using cisplatin and simvastatin to assess the inhibitory effects of
statin in C666-1 cells. Cells were treated with simvastatin (50 µM)
or cisplatin (35 µM) alone, or a combination of the two, for 24 h.
Cell apoptosis was assessed by TUNEL assay. As presented in
Fig. 2, treatment with a combination
of cisplatin and simvastatin statistically significantly induced
apoptosis in C666-1 cells compared with those treated with
simvastatin or cisplatin alone (P<0.01). This result confirms
that simvastatin has a synergistic antitumor effect with a
conventional agent in NPC.
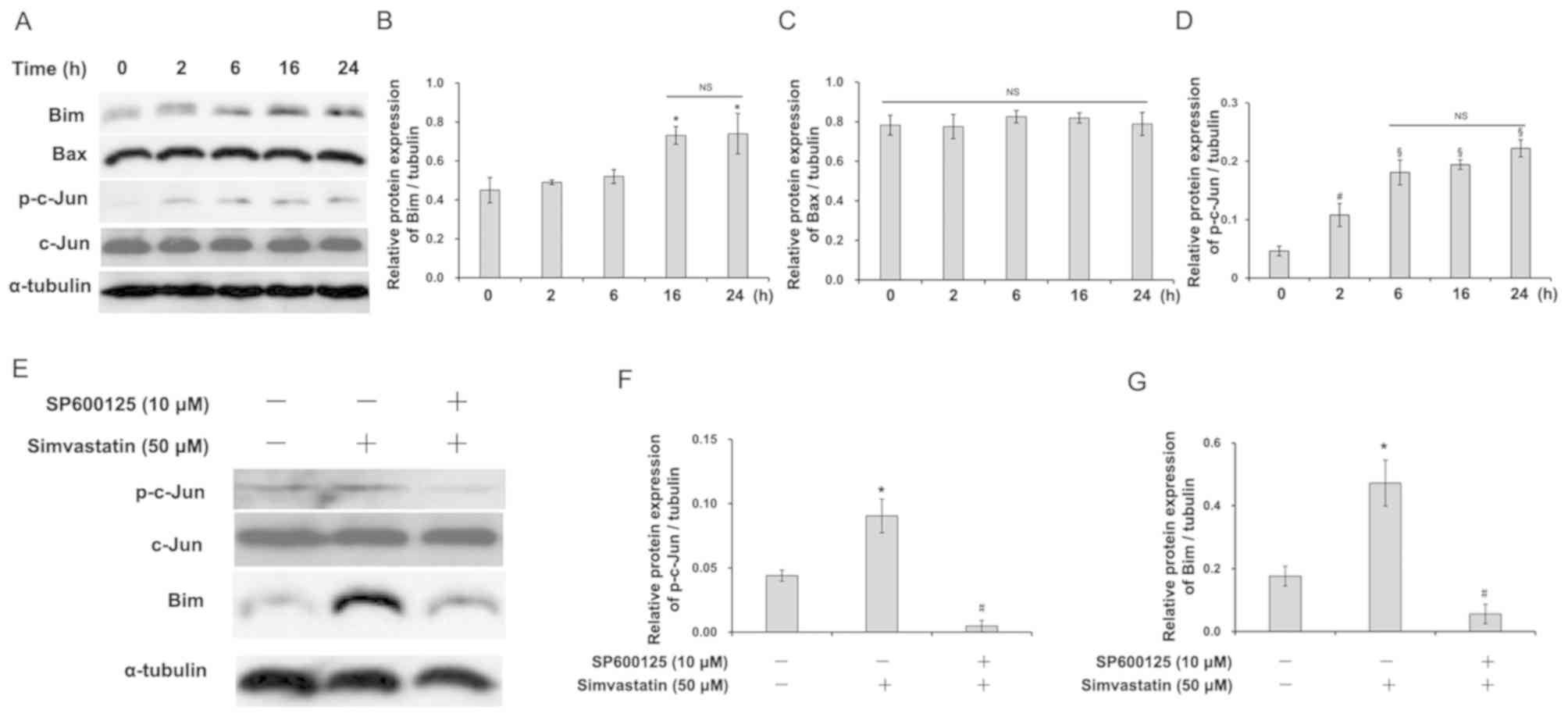
Simvastatin induces Bim expression by
activating the JNK pathway in C666-1 cells
There are two major apoptotic pathways, the
mitochondrial and death receptor pathways. The Bcl-2 family,
members of the mitochondrial apoptosis pathway, can induce
antiapoptotic and proapoptotic effects. To clarify whether the
Bcl-2 family plays a role in statin-induced apoptosis in C666-1
cells, the expression of two proapoptotic members, Bim and Bax was
observed. Simvastatin induced the expression of Bim (Fig. 3A and B), but not Bax (Fig. 3A and C), in a time-dependent manner,
suggesting that statin may induce C666-1 cell apoptosis through the
mitochondrial apoptotic pathway. Meanwhile, increased expression of
phosphorylated c-Jun in C666-1 cells was observed after simvastatin
treatment (Fig. 3A and D). As is
already known, the JNK pathway plays an important role in
regulating phosphorylation of transcriptional factor c-Jun. Then,
C666-1 cells were pretreated with a JNK inhibitor, SP6000125,
before exposing them to simvastatin. The expression of Bim
(Fig. 3E and G) and phosphorylated
c-Jun (Fig. 3E and F) was
significantly decreased by SP6000125 pretreatment (P<0.05).
These results indicate that the JNK pathway may be involved in
statin-induced apoptosis in C666-1 cells.
Effect of simvastatin on cell cycle
progress in C666-1 cells
To evaluate the effect of statin on cell cycle,
C666-1 cells were incubated with 50 µM of simvastatin for up to 48
h. The statin induced a decrease in the total S- and G2/M-phase
population in C666-1 cells (Fig. 4A
and Table I). The statin-induced
inhibition of S- and G2/M-phases appeared to be associated with a
simultaneously increasing proportion of G1 phase, suggesting that
simvastatin probably induced G1 arrest in C666-1 cells.
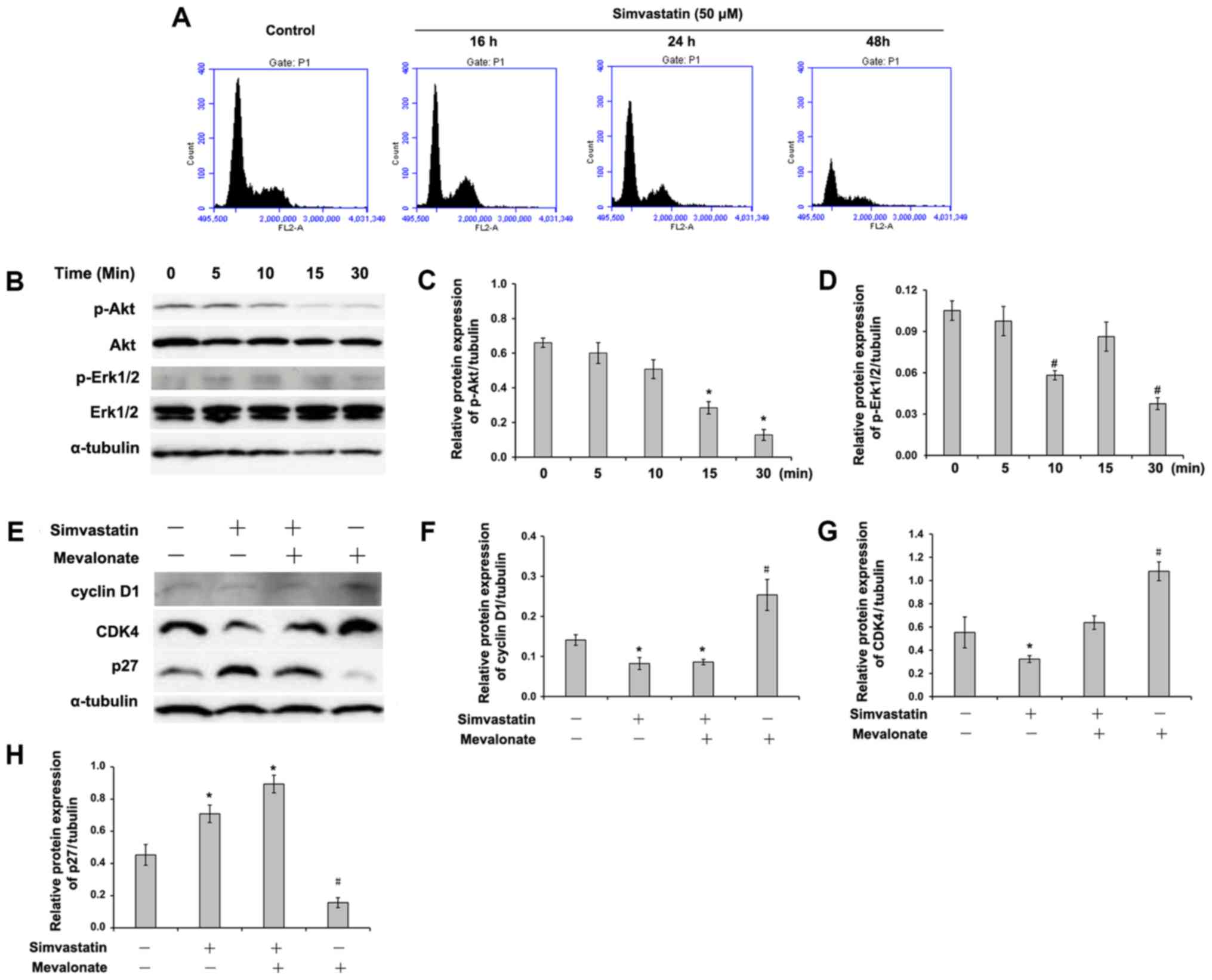 | Figure 4.Simvastatin regulates cell cycle
distribution and phosphorylation of Akt and Erk1/2 in C666-1 cells.
(A) C666-1 cells were treated with or without 50 µM of simvastatin
for the indicated times and subjected to DNA analysis by flow
cytometry. The data represent one of three experiments. (B) The
expression of p-Akt, p-Erk1/2, Akt and Erk1/2 were determined by
western blotting. Quantified expression of (C) p-Akt and (D)
p-Erk1/2 protein. Data are presented as the mean ± standard
deviation (n=3). *P<0.05 vs. the simvastatin-treated cells for
0, 5 and 10 min; #P<0.05 vs. the simvastatin-treated
cells for 0, 5 and 15 min. (E) C666-1 cells were treated with or
without 50 µM of simvastatin in the presence or absence of 200 µM
of mevalonate for 24 h. The expression of cyclin D1, CDK4 and p27
was determined by western blotting. Quantified expression of (F)
cyclin D1, (G) CDK4 and (H) p27 protein. Data are presented as the
mean ± standard deviation (n=3). *P<0.05 vs. the control cells;
#P<0.05 vs. the simvastatin-treated or simvastatin-
and mevalonate-treated cells. P-Akt, phosphorylated protein kinase
B; Erk, extracellular signal regulated kinase; CDK,
cyclin-dependent kinase; NS, not significant. |
 | Table I.Cell cycle distribution induced by
simvastatin in C666-1 cells. |
Table I.
Cell cycle distribution induced by
simvastatin in C666-1 cells.
|
| Control (%) | Simvastatin (50 µM)
(%) |
|---|
| G1 | 53.90±3.33 |
76.40±3.65a |
| S | 24.83±2.83 |
9.37±2.02a |
| G2/M | 12.90±1.93 | 12.87±1.83 |
Effects of simvastatin on the
activation of mitogen-activated protein kinase 1 (MAPK) and Akt and
the expression of cell cycle regulatory proteins
The present study sought to identify the molecular
mechanism involved in statin-induced inhibitory effects in C666-1
cells. Western blot analysis demonstrated that the phosphorylated
levels of Akt and Erk1/2 were downregulated by simvastatin
(Fig. 4B-D). As described above,
statin treatment induced cell cycle arrest in the G1 phase.
Therefore, the effects of simvastatin on the expression of cell
cycle regulatory proteins was examined. The results of the present
study demonstrated that simvastatin induced a markedly increase in
p27 expression (Fig. 4E and H) and
decreased the expression of cyclin D1 (Fig. 4E and F) and CDK 4 (Fig. 4E and G) in C666-1 cells.
Interestingly, mevalonate partially reversed these effects induced
by statin.
Discussion
The results of the present study demonstrated that
simvastatin decreased cell viability in C666-1 cells and the
statin-induced inhibitory effects were associated with HMG-CoA
reductase-dependent apoptosis and cell cycle arrest. It was
demonstrated that simvastatin has a synergistic anti-tumor effect
with cisplatin in NPC cells and suggest the potential utility of
statins as a chemopreventive agent in the treatment of NPC.
Simvastatin induces apoptosis in NPC cells through
the mitochondrial apoptotic pathway. There are a variety of
molecules involved in apoptosis. A well-known group of these
molecules is the Bcl-2 family, which is the important member of the
mitochondrial apoptotic pathway. The proapoptotic Bcl-2 family
members induce cell death by translocating from the mitochondrial
membrane to cytosol (23). Signaling
via the JNK pathway has been linked to altered expression of Bcl-2
family members in ovarian cancer and melanoma (19,24). The
results of the present study demonstrated that Bim, but not Bax, a
proapoptotic Bcl-2 family member, was induced in NPC cells after
simvastatin treatment. Bim initiates mitochondrial dysfunction and
activates the mitochondrial apoptotic pathway by releasing
cytochrome c and activating Caspase 3. Transcription factor
c-Jun regulates Bim expression in neurons by nerve growth factor
(25). The result of the present
study is consistent with a previous study (19). An increase in the levels of active,
cleaved Caspase-3 and phosphorylated c-Jun in C666-1 cells was
observed after simvastatin treatment, suggesting that simvastatin
promotes apoptosis in C666-1 cells by inducing Bim expression.
Statin-induced cell cycle arrest in G1 probably
results from decreased expression of cyclin D1 and a corresponding
increase in p27. The present study's DNA ploidy analysis revealed
that the treatment of C666-1 cells with simvastatin resulted in
cell cycle arrest in the G1 phase. Mitogenic stimuli may increase
the intracellular level of cyclin D1, which then forms an complex
with CDK4 and 6. Active cyclin D1-CDK4/6 complexes sequentially
phosphorylate and inactivate the retinoblastoma (Rb) protein to
initiate E2F-dependent transcription, a requirement for cell cycle
progression into the S-phase (26,27).
Exogenous cyclin D1 has been demonstrated to increase cyclin
D1/CDK4 activity and cause hyperphosphorylation of the Rb protein.
Inhibition of cyclin D1 activation with siRNA results in a
significant reduction in Rb hyperphosphorylation and is associated
with G1 cell cycle arrest in breast cancer cells (15,28).
Suppression of cyclin D1 activity has been demonstrated to
significantly reduce mammary tumorigenesis (29). In the present study, simvastatin
decreased the expression of cyclin D1 and CDK4 in C666-1 cells.
Notably, statin treatment induced an increase in the expression of
one of the CDK inhibitors, p27. Wali et al (30), demonstrated that statins promoted p27
upregulation via promoter activation in mammary tumors. The present
study provides the first evidence that simvastatin also increases
the expression of a CDK inhibitor in NPC cells. Upregulation of p27
induced by statins is significantly associated with overcoming
anoikis resistance in head and neck squamous cell carcinoma cells
(16). P27 could be a potential
target for the anticancer effect of statins in NPC.
Statins can inhibit phosphorylation and activation
of Akt and MAPK mitogenic signaling in mammary tumor cells
(31). It has been reported that
activation of these pathways simulated cell cycle progression from
G1 to S phase through activating cyclin D1 (26,32).
Activation of Akt can enhance cyclin D1 transcription and protein
expression, phosphorylate glycogen synthase kinase 3β (GSK3β), and
thereby prevent GSK3β from phosphorylating and destabilizing cyclin
D1 protein (26,33). In addition, FOXO transcription
factors, which inhibit cyclin D1 expression and induce p27
expression, are also phosphorylated and inhibited by Akt pathway
activation (34). Similarly, MAPK
activation results in phosphorylation and stabilization of c-myc,
which induces cyclin D1 and inhibits CKI expression (35,36).
Taken together, the results of the present study indicate that
statin-induced changes in endogenous cyclin and CDK could result
from the inhibition of Akt and Erk1/2 in C666-1 cells.
In conclusion, in this study, it was demonstrated
that simvastatin induced apoptosis by upregulating Bim expression
and arrested the cell cycle in the G1 phase by decreasing the
expression of cyclin D1 and CDK4, and increasing p27 expression in
C666-1 cells. Treatment with simvastatin inhibited Akt and Erk1/2
activation. Mitogenic signaling can regulate cell cycle progression
by activating cyclin D1 and CDK4, while inhibiting p27 expression.
The results of the present study indicate that simvastatin is a
potential chemotherapy agent in NPC treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300809) and the
Shanghai Pudong New Area Science & Technology Development Fund
(grant nos. PKJ2016-Y02 and PW2016D-11).
Availability of data and materials
The datasets for this study are available from the
corresponding authors on reasonable request.
Authors' contributions
YZ, MY, LY and LZ performed the experiments, and
carried out the data analysis. ZM and WW were responsible for
manuscript writing and revision, and experimental design.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CDK
|
cyclin-dependent kinase
|
|
CKI
|
cyclin-dependent kinase inhibitor
|
|
HMG-CoA
|
3-hydroxy-3-methylglutaryl coenzyme
A
|
|
NPC
|
nasopharyngeal carcinoma
|
|
PI
|
propidium iodide
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
biotin-dUTP nick end labeling
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei KR, Zheng RS, Zhang SW, Liang ZH, Li
ZM and Chen WQ: Nasopharyngeal carcinoma incidence and mortality in
China, 2013. Chin J Cancer. 36:902017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nor Hashim NA, Ramzi NH, Velapasamy S,
Alex L, Chahil JK, Lye SH, Munretnam K, Haron MR and Ler LW:
Identification of genetic and non-genetic risk factors for
nasopharyngeal carcinoma in a Southeast Asian population. Asian Pac
J Cancer Prev. 13:6005–6010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chua DT, Sham JS, Kwong DL and Au GK:
Treatment outcome after radiotherapy alone for patients with Stage
I–II nasopharyngeal carcinoma. Cancer. 98:74–80. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yau TK, Lee AW, Wong DH, Pang ES, Ng WT,
Yeung RM and Soong IS: Treatment of Stage IV(A-B) nasopharyngeal
carcinoma by induction-concurrent chemoradiotherapy and accelerated
fractionation: Impact of chemotherapy schemes. Int J Radiat Oncol
Biol Phys. 66:1004–1010. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qiu S, Lin S, Tham IW, Pan J, Lu J and Lu
JJ: Intensity-modulated radiation therapy in the salvage of locally
recurrent nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys.
83:676–683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chua MLK, Wee JTS, Hui EP and Chan ATC:
Nasopharyngeal carcinoma. Lancet. 387:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ng WT, Lee MC, Hung WM, Choi CW, Lee KC,
Chan OS and Lee AW: Clinical outcomes and patterns of failure after
intensity-modulated radiotherapy for nasopharyngeal carcinoma. Int
J Radiat Oncol Biol Phys. 79:420–428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ou X, Zhou X, Shi Q, Xing X, Yang Y, Xu T,
Shen C, Wang X, He X, Kong L, et al: Treatment outcomes and late
toxicities of 869 patients with nasopharyngeal carcinoma treated
with definitive intensity modulated radiation therapy: New insight
into the value of total dose of cisplatin and radiation boost.
Oncotarget. 6:38381–38397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun X, Su S, Chen C, Han F, Zhao C, Xiao
W, Deng X, Huang S, Lin C and Lu T: Long-term outcomes of
intensity-modulated radiotherapy for 868 patients with
nasopharyngeal carcinoma: An analysis of survival and treatment
toxicities. Radiother Oncol. 110:398–403. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Causevic-Ramosevac A and Semiz S: Drug
interactions with statins. Acta Pharm. 63:277–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reiner Z: Resistance and intolerance to
statins. Nutr Metab Cardiovasc Dis. 24:1057–1066. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duncan RE, El-Sohemy A and Archer MC:
Mevalonate promotes the growth of tumors derived from human cancer
cells in vivo and stimulates proliferation in vitro with enhanced
cyclin-dependent kinase-2 activity. J Biol Chem. 279:33079–33084.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bockorny B and Dasanu CA: HMG-CoA
reductase inhibitors as adjuvant treatment for hematologic
malignancies: What is the current evidence? Ann Hematol. 94:1–12.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feldt M, Bjarnadottir O, Kimbung S,
Jirström K, Bendahl PO, Veerla S, Grabau D, Hedenfalk I and
Borgquist S: Statin-induced anti-proliferative effects via cyclin
D1 and p27 in a window-of-opportunity breast cancer trial. J Transl
Med. 13:1332015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takeda I, Maruya S, Shirasaki T, Mizukami
H, Takahata T, Myers JN, Kakehata S, Yagihashi S and Shinkawa H:
Simvastatin inactivates beta1-integrin and extracellular
signal-related kinase signaling and inhibits cell proliferation in
head and neck squamous cell carcinoma cells. Cancer Sci.
98:890–899. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koul HK, Koul S and Meacham RB: New role
for an established drug? Bisphosphonates as potential anticancer
agents. Prostate Cancer Prostatic Dis. 15:111–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Warita K, Warita T, Beckwitt CH, Schurdak
ME, Vazquez A, Wells A and Oltvai ZN: Statin-induced mevalonate
pathway inhibition attenuates the growth of mesenchymal-like cancer
cells that lack functional E-cadherin mediated cell cohesion. Sci
Rep. 4:75932014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu H, Liang SL, Kumar S, Weyman CM, Liu W
and Zhou A: Statins induce apoptosis in ovarian cancer cells
through activation of JNK and enhancement of Bim expression. Cancer
Chemother Pharmacol. 63:997–1005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Parikh A, Childress C, Deitrick K, Lin Q,
Rukstalis D and Yang W: Statin-induced autophagy by inhibition of
geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate.
70:971–981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ogunwobi OO and Beales IL: Statins inhibit
proliferation and induce apoptosis in Barrett's esophageal
adenocarcinoma cells. Am J Gastroenterol. 103:825–837. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang W, Le W, Cho DY, Hwang PH and
Upadhyay D: Novel effects of statins in enhancing efficacy of
chemotherapy in vitro in nasopharyngeal carcinoma. Int Forum
Allergy Rhinol. 1:284–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hiura TS, Li N, Kaplan R, Horwitz M,
Seagrave JC and Nel AE: The role of a mitochondrial pathway in the
induction of apoptosis by chemicals extracted from diesel exhaust
particles. J Immunol. 165:2703–2711. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu BK, Wang P, Zhang XD, Jiang CC, Chen
LH, Avery-Kiejda KA, Watts R and Hersey P: Activation of Jun
N-terminal kinase is a mediator of vincristine-induced apoptosis of
melanoma cells. Anticancer Drugs. 19:189–200. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Biswas SC, Shi Y, Sproul A and Greene LA:
Pro-apoptotic Bim induction in response to nerve growth factor
deprivation requires simultaneous activation of three different
death signaling pathways. J Biol Chem. 282:29368–29374. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Massague J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Musgrove EA, Lee CS, Buckley MF and
Sutherland RL: Cyclin D1 induction in breast cancer cells shortens
G1 and is sufficient for cells arrested in G1 to complete the cell
cycle. Proc Natl Acad Sci USA. 91:8022–8026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grillo M, Bott MJ, Khandke N, McGinnis JP,
Miranda M, Meyyappan M, Rosfjord EC and Rabindran SK: Validation of
cyclin D1/CDK4 as an anticancer drug target in MCF-7 breast cancer
cells: Effect of regulated overexpression of cyclin D1 and
siRNA-mediated inhibition of endogenous cyclin D1 and CDK4
expression. Breast Cancer Res Treat. 95:185–194. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arber N, Doki Y, Han EK, Sgambato A, Zhou
P, Kim NH, Delohery T, Klein MG, Holt PR and Weinstein IB:
Antisense to cyclin D1 inhibits the growth and tumorigenicity of
human colon cancer cells. Cancer Res. 57:1569–1574. 1997.PubMed/NCBI
|
|
30
|
Wali VB, Bachawal SV and Sylvester PW:
Combined treatment of gamma-tocotrienol with statins induce mammary
tumor cell cycle arrest in G1. Exp Biol Med (Maywood). 234:639–650.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wali VB and Sylvester PW: Synergistic
antiproliferative effects of gamma-tocotrienol and statin treatment
on mammary tumor cells. Lipids. 42:1113–1123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Torii S, Yamamoto T, Tsuchiya Y and
Nishida E: ERK MAP kinase in G cell cycle progression and cancer.
Cancer Sci. 97:697–702. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liang J and Slingerland JM: Multiple roles
of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell
Cycle. 2:339–345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tran H, Brunet A, Griffith EC and
Greenberg ME: The many forks in FOXO's road. Sci STKE.
2003:RE52003.PubMed/NCBI
|
|
35
|
Reagan-Shaw S, Afaq F, Aziz MH and Ahmad
N: Modulations of critical cell cycle regulatory events during
chemoprevention of ultraviolet B-mediated responses by resveratrol
in SKH-1 hairless mouse skin. Oncogene. 23:5151–5160. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Subramaniam G, Campsteijn C and Thompson
EM: Co-expressed Cyclin D variants cooperate to regulate
proliferation of germline nuclei in a syncytium. Cell Cycle.
14:2129–2141. 2015. View Article : Google Scholar : PubMed/NCBI
|