Introduction
Liver fibrosis is a reaction to chronic liver injury
resulting from various etiological factors, including alcohol
consumption, drug misuse, hepatitis B or C infection or toxic
injury (1). Liver fibrosis is a
globally widespread and substantial healthcare issue that results
in end-stage liver diseases, including cirrhosis, and ultimately
hepatocellular carcinoma. The process of liver fibrosis can be
reversible, whereas cirrhosis, the result of end-stage fibrosis, is
usually irreversible (2). Therefore,
it is necessary to develop effective anti-fibrotic therapy for the
treatment of fibrosis and hepatic injury.
The activation of hepatic stellate cells (HSCs)
serves an important role in the initiation and development of liver
fibrosis. During liver injury, quiescent HSCs are activated by
stimulating factors or fibrogenic cytokines, including transforming
growth factor (TGF)-β1, platelet-derived growth factor (PDGF) and
tumor necrosis factor (TNF)-α (3),
which induce trans-differentiation into myofibroblast-like cells.
This step is characterized by the appearance of procollagen-I and
α-smooth muscle actin (SMA), ultimately leading to the accumulation
and increased output of collagen and extracellular matrix (ECM)
components (4). Notably, TGF-β1 is
an extremely potent pro-fibrogenic cytokine associated with hepatic
fibrosis in humans and other animals (5). TGF-β1 acts though stimulating the
mothers against decapentaplegic homolog (Smad)-dependent activation
of Snail, and the Smad-independent induction of the
mitogen-activated protein kinase (MAPK) pathway (6).
Research to identify safe anti-fibrotic agents is of
high importance and is urgently required. A recently identified
water-soluble pyridone agent with anti-fibrotic properties,
fluorofenidone [1-(3-fluorophenyl)-5-methyl-2-(1H)-pyridone;
AKF-PD], can attenuate liver (7,8), renal
(9), pulmonary (10) and cardiac fibrosis (11), and has been adopted in phase I
clinical trials. However, its therapeutic benefits for hepatic
fibrosis remain unclear. The aim of the present study was to
research the anti-fibrotic effects of AKF-PD on liver fibrosis in
rats and to elucidate the underlying mechanisms of the effect of
AKF-PD on the activation of HSCs.
Materials and methods
Materials
LX-2 (12) HSCs
[provided by Professor Scott L. Friedman (Icahn School of Medicine
at Mount Sinai, New York, NY, USA)] were used in the present study.
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum
(FBS) were purchased from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). TRIzol for RNA extraction was purchased from
Invitrogen; Thermo Fisher Scientific, Inc. A quantitative
polymerase chain reaction (qPCR) kit (SYBR PremixEx Taq II) was
purchased from Takara Bio, Inc. (Otsu, Japan). Antibodies against
total extracellular signal-regulated kinase (ERK1/2; cat. no.
9102), p38 mitogen-activated protein kinase (p38 MAPK; cat. no.
9212), c-Jun N-terminal kinase (JNK; cat. no. 9252), and
phosphorylated (p)-ERK (cat. no. 9101), p-p38 (cat. no. 9211) and
p-JNK (cat. no. 4668) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Antibodies against α-SMA (cat.
no. A5228) and β-actin (cat. no. A5441) were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Antibodies against
p-Smad-3 (cat. no. ab193297), collagen I (cat. no. ab34710),
collagen III (cat. no. ab23746) and Smad-3 (cat. no. ab40854) were
purchased from Abcam (Cambridge, UK). Recombinant human TGF-β1 was
purchased from PeproTech, Inc. (Rocky Hill, NJ, USA). Horseradish
peroxide (HRP)-conjugated secondary antibodies for western blot
analysis were purchased from Jackson ImmunoResearch Laboratories,
Inc. (West Grove, PA, USA), and HRP-conjugated secondary antibodies
for immunohistochemistry were obtained from Golden Bridge
International, Inc. (Bothell, WA, USA). An enhanced
chemiluminescence (ECL) kit for western blot analysis was purchased
from GE Healthcare Life Sciences (Little Chalfont, UK). Pig serum
(PS) was purchased from Beijing YuanHeng ShengMa Biology Technology
Research Institute of Biotechnology (Beijing, China). AKF-PD was
synthesized by Sunshine Lake Pharma Co., Ltd. (Dongguan, China;
cat. no. 090601).
Animal model
Male albino Wistar rats weighing 120–150 g (5–6
weeks old) were obtained from Shanghai SLAC Laboratory Animal Co.,
Ltd. (Shanghai, China). Rats were bred and maintained in an
air-conditioned environment (temperature 23–25°C; humidity 50±2%;
12 h light/dark cycle), with a commercial diet and water available
ad libitum. The experimental protocol was approved by the
Ethics Review Committee for Animal Experimentation of Central South
University (Changsha, China), and all rats received humane care in
compliance with the university's guidelines. For the purpose of the
present study, rats were randomly divided into the following
groups: Normal group (n=15), PS model group (n=15) and PS+AKF-PD
treatment group (n=15). Hepatic fibrosis was induced via
intraperitoneal injections of 0.5 ml PS twice weekly for 8 weeks
(13), whereas the normal group
received 0.5 ml sterile saline. From the ninth week, the treatment
group was administered AKF-PD intragastrically (240 mg/kg/day) once
daily for 4 weeks. The normal and PS model groups were
simultaneously administered with 0.5% carboxymethyl cellulose
sodium intragastrically every day for 4 weeks. At the end of week
12, all rats were sacrificed. Livers were rapidly harvested, rinsed
in cold saline and weighed while wet. A portion of liver was fixed
(room temperature) in 10% neutral-buffered formalin for
histopathology and the remaining tissue was stored at −70°C until
assayed.
Histological and immunohistochemical
analysis
The formalin-fixed rat liver tissue was dehydrated
in a graded alcohol series and embedded in paraffin. The paraffin
sections (4-µm thick) were stained (room temperature for 10 min)
with hematoxylin and eosin or Masson's trichrome. To determine the
degree of necroinflammatory liver injury, histological grading and
the quantification of infiltrating inflammatory cells were blindly
performed by an independent pathologist, as described previously
(14,15). To further analyze the degree of
interstitial collagen deposition, Masson's trichrome-stained
sections were graded as previously reported (16). 3,3′-Diaminobenzidine
immunohistochemical staining was performed using the DAKO EnVision
system (Dako; Agilent Technologies, Inc., Santa Clara, CA, USA), as
previously detailed (17).
Subsequent to blocking non-specific binding with the included
blocking solution, the slides were incubated with primary
antibodies against collagen I (1:100), collagen III (1:150) and
α-SMA (1:250) overnight at 4°C, followed by the corresponding
secondary antibodies (1:100) at room temperature for 1 h.
HRP-labeled polymer and a chromogen substrate were then employed to
develop the staining. As a negative control, the primary antibody
was replaced with PBS. Samples were visualized using light
microscopy at a magnification of ×100. Given the homogeneity of the
staining of the target proteins, the interstitial staining of
collagen I, collagen III and α-SMA was measured by a blinded
observer using computerized morphometry (QWin 2.8 software; Leica
Microsystems GmbH, Wetzlar, Germany) (18).
Analysis of mRNA expression
Total RNA was isolated from the fresh liver tissue
using TRIzol reagent, according to the manufacturer's protocol.
First-strand cDNA was synthesized from 2 µg total RNA in a 20-µl
reaction, using a ReverseAid first strand cDNA synthesis kit
(Thermo Scientific Inc.) according to the manufacturer's protocol.
Specific primers for collagen I, collagen III, α-SMA, TGF-β1 and
β-actin were designed from their GenBank sequences (8) and synthesized by Bio Basic, Inc.
(Markham, ON, Canada; Table I). qPCR
quantitation for each target mRNA was performed on an ABI Model
7900 Detector (Applied Biosystems, Foster City, CA, USA) using the
Takara qPCR kit. The thermocycling conditions were as follows: 95°C
for 10 sec; followed by 40 cycles at 95°C for 5 sec, 60°C for 30
sec; followed by 95°C for 15 sec, 60°C for 15 sec and 95°C for 15
sec). The amount of each mRNA in the samples was calculated from
the standard curve and normalized to the β-actin mRNA. The
comparative 2−ΔΔCq method was used for quantification
and the results were expressed as fold-changes relative to the
normal controls (19).
 | Table I.Nucleotide sequences of the primers
used for reverse transcription-quantitative polymerase chain
reaction. |
Table I.
Nucleotide sequences of the primers
used for reverse transcription-quantitative polymerase chain
reaction.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| Collagen I |
TCAGGGGCGAAGGCAACAGT |
TTGGGATGGAGGGAGTTTACACGA |
| Collagen III |
CGTCCTGCAGGTAACAGTGGTTC |
TGCTCCAGTTAGCCCTGCAA |
| α-SMA |
CTAAGGCCAACCGGGAGAAA |
CCAGAGTCCAGCACAATACCA |
| TGF-β1 |
CAACAATTCCTGGCGTTACCTT |
AAGCCCTGTATTCCGTCTCCTT |
| β-actin |
GGAGATTACTGCCCTGGCTCCTA |
GACTCATCGTACTCCTGCTTGCTG |
Cell culture and treatment
LX-2 cells were maintained in DMEM supplemented with
10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc.). The LX-2 cells were
seeded onto 6-well culture plates to 60–70% confluence in DMEM
containing 10% FBS for 24–48 h at 37°C. The medium was replaced
with serum-free DMEM for 24 h prior to treatment with recombinant
human TGF-β1 at 37°C at a final concentration of 5 ng/ml. For the
detection of α-SMA and collagen I, cells were pretreated at 37°C
with 2 mM AKF-PD as reported in a previous study (7), for 24 h. TGF-β1 was subsequently added
to the medium, for 24 h. Cells cultured in DMEM alone were used as
a control. The cells were treated for 48 h at 37°C, as determined
by a preliminary experiment, prior to protein isolation. For
detection of p-Smad-3, p-ERK1/2, p38 MAPK and p-JNK, cells were
pretreated with 2 mM AKF-PD for 24 h at 37°C, and TGF-β1 (5 ng/ml)
was subsequently added to the medium at 5, 15, 30 and 60 min prior
to the collection of cellular proteins. Each experiment was
replicated three times.
Western blot analysis
Total protein (30 µg) from the fresh liver tissue or
cultured cells was extracted using sodium dodecyl sulfate
(Sigma-Aldrich; Merck KGaA; cat. no. L3771) and determined using a
Pierce™ BCA Protein assay kit (cat. no. 23227; Thermo Fisher
Scientific Inc.). Samples (20 µg per lane) were separated by 8 or
10% SDS-PAGE under reducing conditions, and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Non-specific binding was blocked with TBS-T buffer [10 mM
Tris/HCl, 150 mM NaCl, 0.1% (v/v) Tween 20, pH 7.6] containing 5%
(w/v) skimmed milk for 1 h at room temperature. The membranes were
incubated overnight at 4°C with primary antibodies against α-SMA
(dilution, 1:2,500), collagen I (dilution, 1:1,000), p-ERK
(dilution, 1:1,000), p-p38 (dilution, 1:1,000), p-JNK (dilution,
1:1,000), p-Smad-3 (dilution, 1:1,000), ERK (dilution, 1:1,000),
p38 (dilution, 1:1,000), JNK (dilution, 1:1,000), Smad-3 (dilution,
1:1,000) and β-actin (dilution, 1:5,000), and were subsequently
incubated with HRP-conjugated secondary antibodies (dilution,
1:5,000) for 1 h at room temperature. The bands were visualized
using an ECL kit and quantified using Bandscan 5.0 software (Glyko,
Inc., Novato, CA, USA). Phosphorylated proteins were normalized to
the expression of the total protein and the loading control.
Statistical analysis
Data are expressed as the mean + standard deviation.
Statistical analyses were performed using SPSS 19.0 software (IBM
Corp., Armonk, NY, USA). The comparison among groups was made with
a one-way analysis of variance (ANOVA). Multiple-comparison tests
were applied only when a significant difference was determined by
ANOVA followed by an LSD post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
AKF-PD ameliorates the histological
injury induced by PS
As demonstrated in Fig.
1A, PS-gavaged rats displayed impaired hepatic lobules, a
reduced quantity of hepatic sinusoids, wide hemorrhagic necrosis
and lobular architecture with narrow strips of reticulin connecting
central zones. AKF-PD administration at the concentration of 240
mg/kg/day attenuated these pathological modifications (P<0.05).
Although the accumulation of collagen fibers between the portal
region and pseudolobules was augmented in the PS model group,
AKF-PD therapy significantly attenuated the increase in collagen
expression (P<0.05; Fig. 1B).
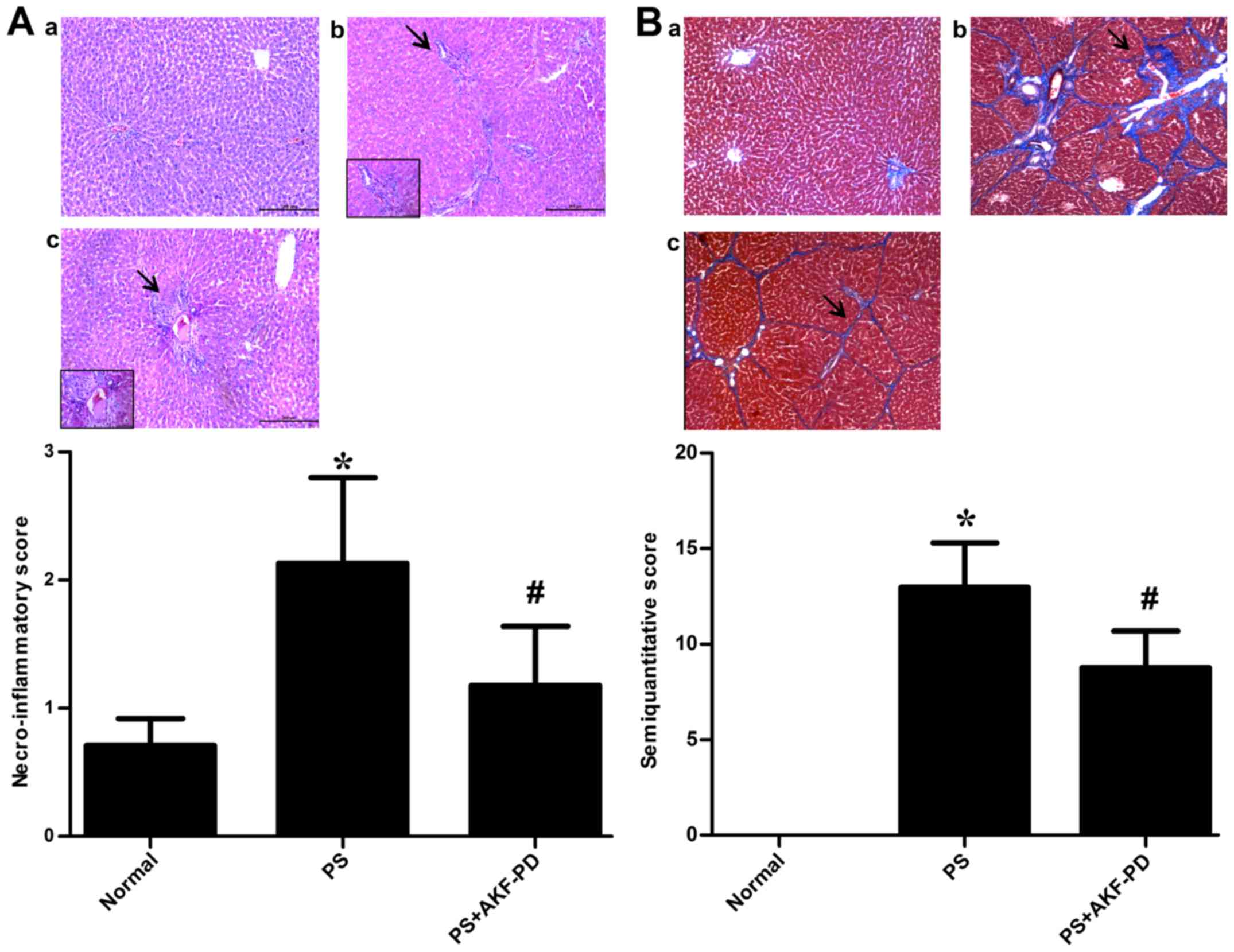 | Figure 1.AKF-PD improves the histological
injuries induced by pig serum. (A) Representative histological
images of hematoxylin and eosin-stained rat liver tissue in the (a)
normal, (b) PS and (c) PS+AKF-PD groups, respectively. Arrows
indicate impaired hepatic lobules, a reduced quantity of hepatic
sinusoids, wide hemorrhagic necrosis and lobular architecture with
narrow strips of reticulin connecting central zones. Magnification,
×200. Columns indicate the necroinflammatory score of liver
fibrosis for each group. (B) Representative images of
Masson-stained collagen deposition in rat livers in the (a) normal,
(b) PS and (c) PS+AKF-PD groups, respectively. Arrows indicate the
accumulation of collagen fibers between the portal region and
pseudolobules. Magnification, ×100. Columns indicate the
semi-quantitative score for liver fibrosis in each group. Data are
expressed as the mean ± standard deviation (15 rats/group).
*P<0.05 vs. normal group; #P<0.05 vs. PS group.
AKF-PD, fluorofenidone; PS, pig serum. |
AKF-PD attenuates ECM deposition and
HSCs activation in rat fibrotic liver
With respect to the deposition of collagen types I
and III, the main ECM components of the fibrotic liver, and α-SMA,
a marker for HSC activation, no positive staining was identified in
the control rat liver tissue (Fig.
2). The PS-injured liver exhibited a notable expression of
collagen type I, collagen type III and α-SMA, which was
significantly suppressed by AKF-PD therapy (P<0.05; Fig. 2). Furthermore, these observations
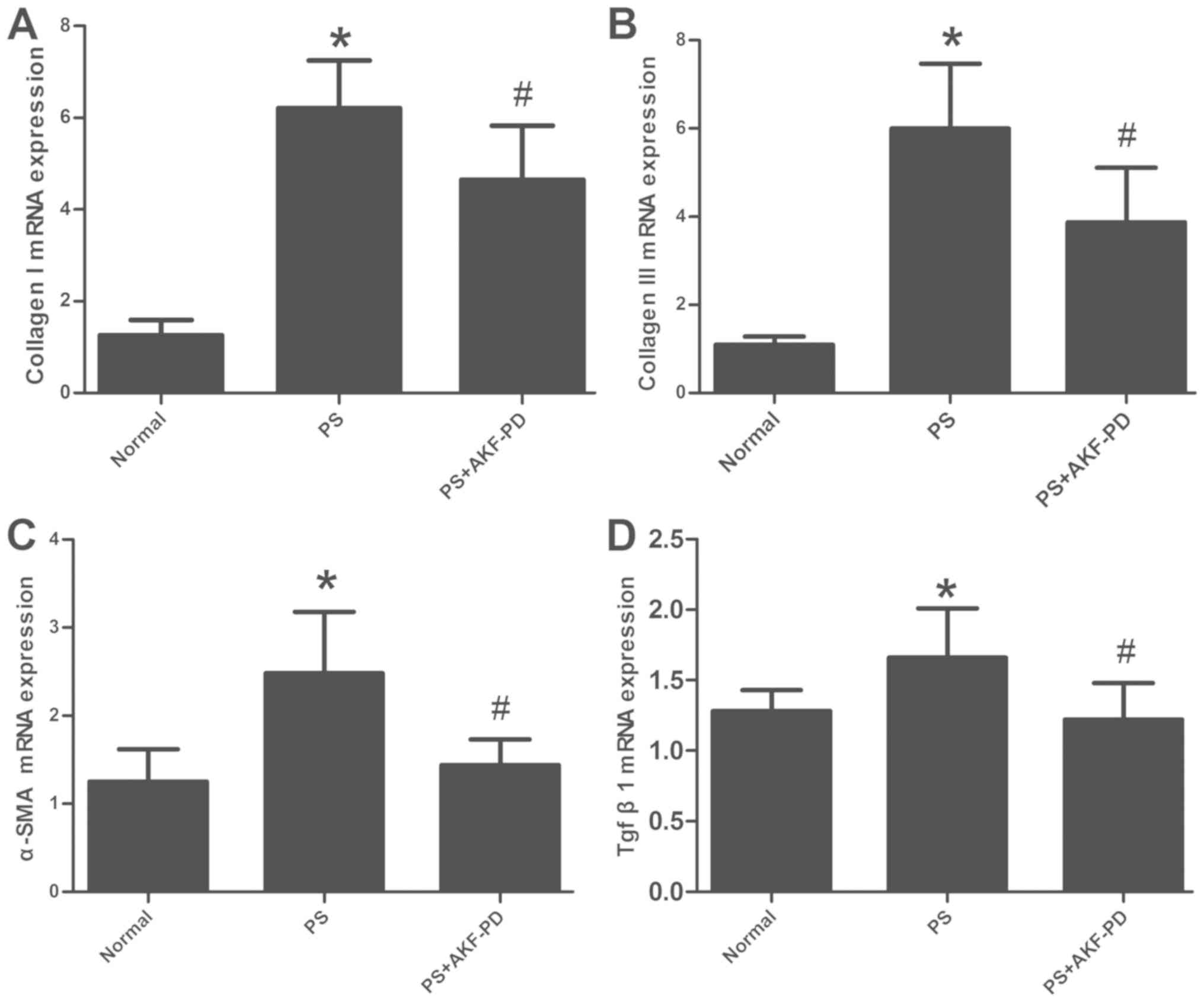
were confirmed by the RT-qPCR analysis of collagen type I, collagen
type III and α-SMA mRNA expression in liver tissue (P<0.05;
Fig. 3A-C).
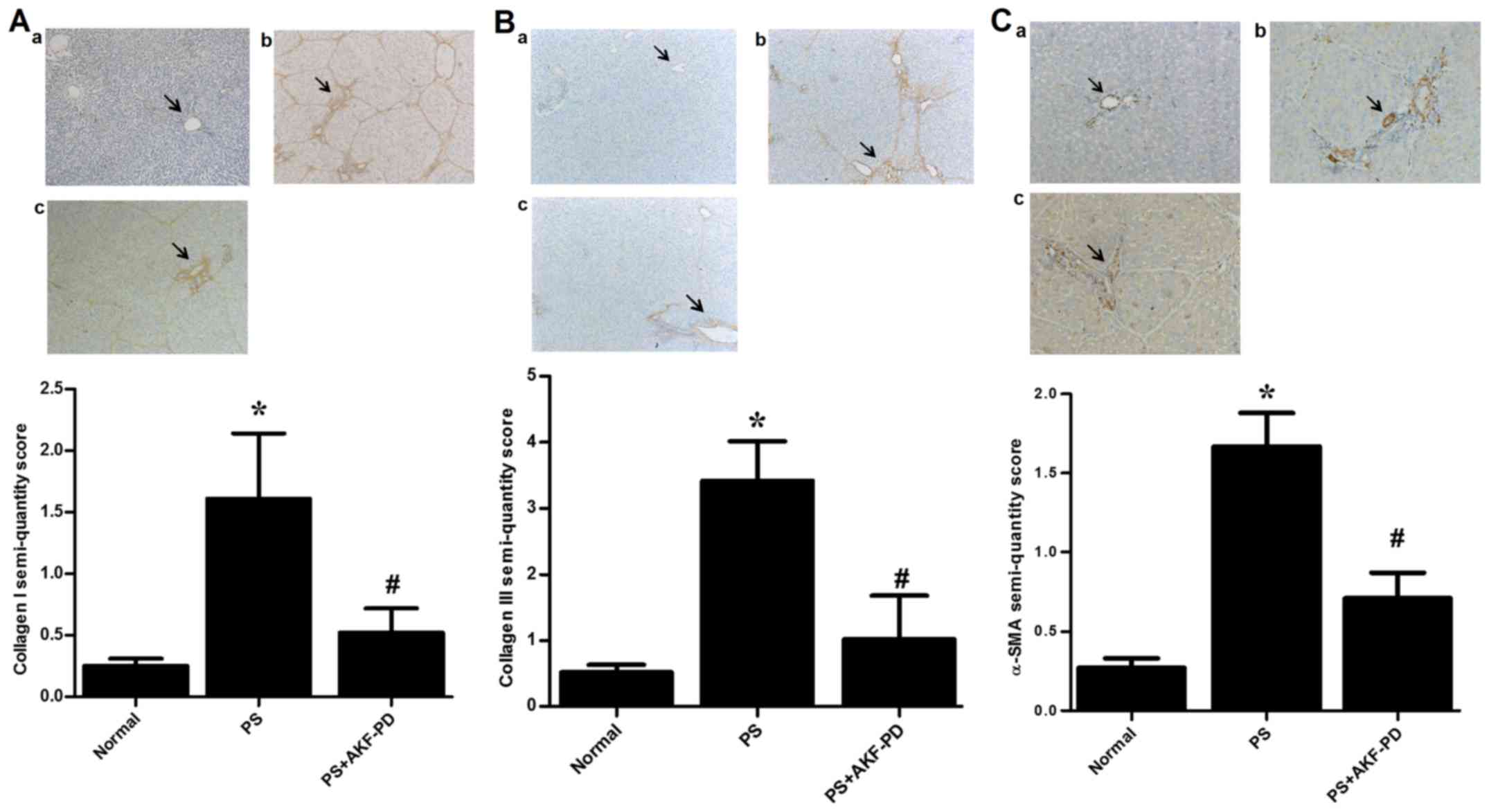 | Figure 2.AKF-PD attenuates ECM deposition and
HSC activation in rat fibrotic liver tissue. Immunohistochemical
staining for (A) collagen I, (B) collagen III and (C) α-SMA in
liver tissue in the (a) normal, (b) PS and (c) PS+AKF-PD groups are
presented (magnification, ×100). Arrows indicate the deposition of
collagen types I and III, the main ECM components of the fibrotic
liver, and α-SMA, a marker for HSC activation. Columns indicate the
semi-quantified scores for collagen I, collagen III and α-SMA
expression. Data were expressed as the mean ± standard deviation (6
rats/group). *P<0.05 vs. normal group; #P<0.05 vs.
PS group. AKF-PD, fluorofenidone; ECM, extracellular matrix; SMA,
smooth muscle actin; PS, pig serum; HSC, hepatic stellate cell. |
The expression of TGF-β1 is decreased
by AKF-PD in fibrotic liver tissue
TGF-β1 is a central mediator of liver fibrosis. The
upregulated expression of TGF-β1 has been detected in experimental
liver fibrosis models (20).
Therefore, it was considered whether AKF-PD diminished the
expression of TGF-β1. It was revealed that the expression of TGF-β1
was notably increased following PS treatment, and that AKF-PD
significantly attenuated the expression of TGF-β1 (P<0.05;
Fig. 3D).
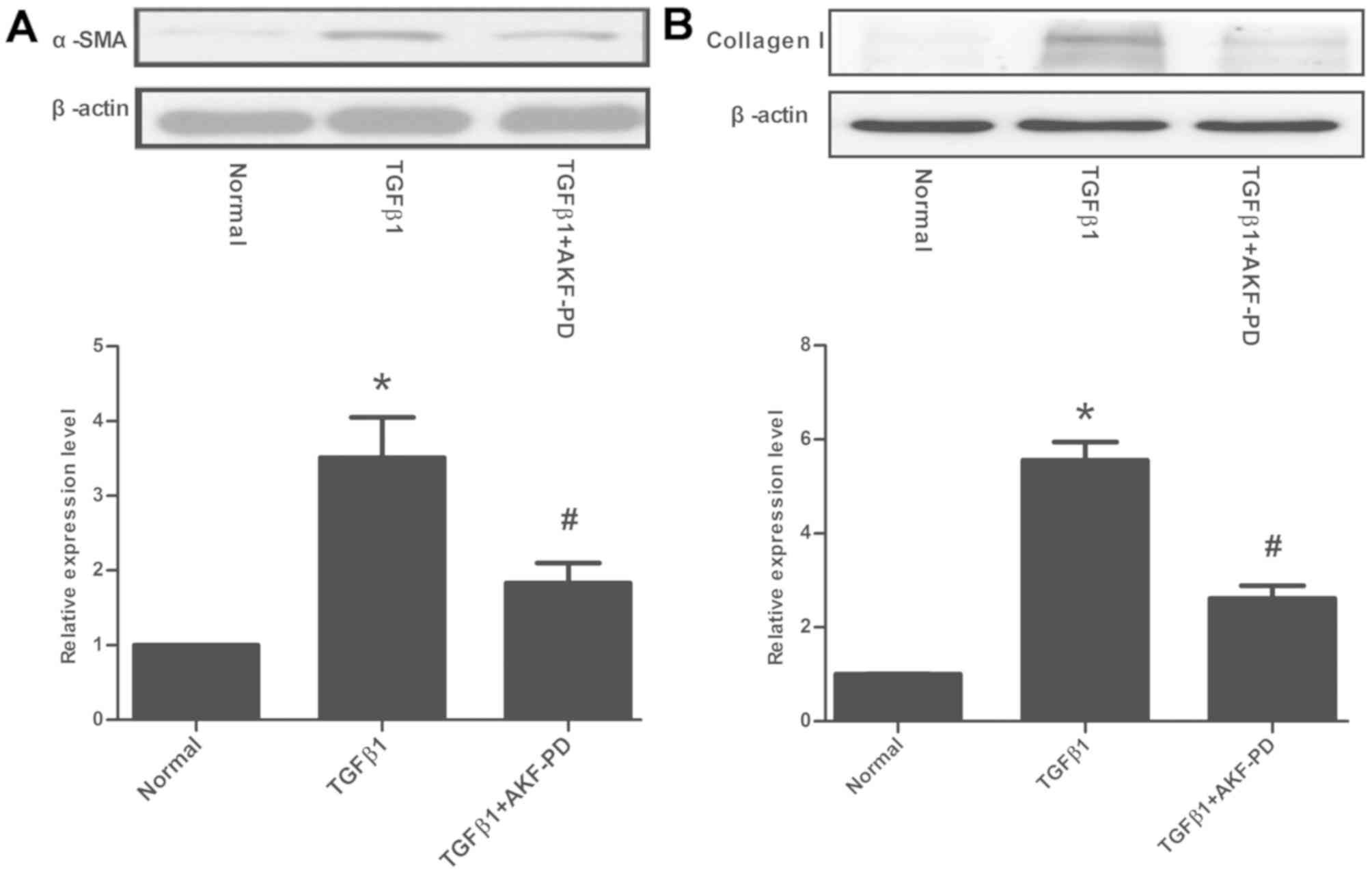
AKF-PD alleviates TGF-β1-induced α-SMA
and collagen I expression in HSCs
Having demonstrated that AKF-PD can effectively
attenuate liver fibrosis in experimental animals, the cellular
mechanism by which AKF-PD influences HSCs, the central cell type
associated with liver fibrosis, was considered in the LX-2 cell
line. Following the incubation of LX-2 cells with AKF-PD, the
expression of α-SMA initiated by TGF-β1 was significantly decreased
(P<0.05; Fig. 4A). The protein
expression of the fibrosis marker collagen I was also detected. As
indicated in Fig. 4B, a 2.6-fold
increase in collagen I expression was induced by TGF-β1, which was
suppressed by AKF-PD to a variable extent (P<0.05).
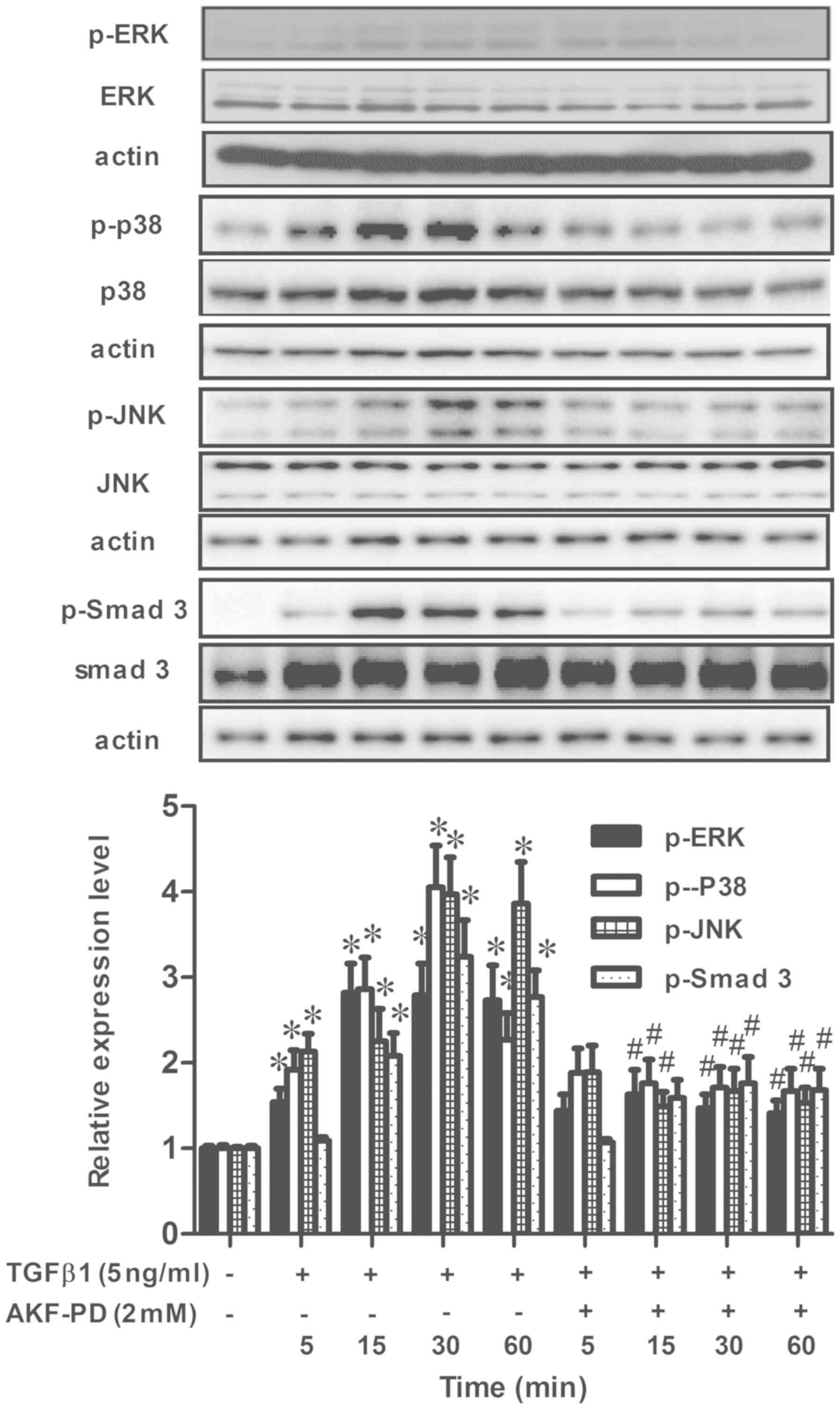
AKF-PD inhibits the TGF-β1/Smad and
MAPK pathways in HSCs
To further identify the molecular mechanisms
responsible for the inhibitory effects of AKF-PD on TGF-β1-induced
HSC activation, the effects of AKF-PD on TGF-β1-induced TGF-β1/Smad
and MAPK pathways in LX-2 cells were analyzed. The time course of
the activation of Smad-3, ERK, p38 and JNK in reaction to TGF-β1
was detected in LX-2 cells. Significant increases in p-Smad-3,
p-ERK, p-p38 and p-JNK were observed as early as 15 min following
TGF-β1 treatment (Fig. 5). The
levels of p-Smad3, p-ERK, p-p38 and p-JNK were markedly decreased
by co-treatment with AKF-PD. Therefore, these data suggest that
AKF-PD inhibits TGF-β1-induced HSCs activation by targeting the
TGF-β1/Smad and MAPK signaling pathways.
Discussion
The inordinate accumulation of ECM proteins such as
collagen leads to liver fibrosis, which is associated with the
majority of chronic liver diseases. Advanced stages of liver
dysfunction and cirrhosis result in liver fibrosis (21). However, despite the advancements in
knowledge regarding the molecular and cellular mechanisms of liver
fibrosis and cirrhosis, the available anti-fibrotic treatment
options are insufficient. In certain cases, causes including
alcohol-induced liver fibrosis may further the progression of liver
fibrosis, even following the withdrawl of the causative agent
(alcohol) (22).
In the present study, the anti-fibrotic effect of
AKF-PD and its potential mechanisms were identified: AKF-PD was
revealed to reduce the number of activated HSCs, as demonstrated by
the decrease in α-SMA (a myofibroblast marker) and ECM protein
expression, which was mediated at least partly by attenuating the
TGF-β1-induced upregulation of the TGF-β/Smad and MAPK
pathways.
The progression of liver fibrosis is a dynamic
process comprising several types of cells in the hepatic sinusoids.
It is characterized by disturbed hepatic architecture and the
accumulation of ECM proteins. The activation of HSCs in response to
hepatic injury is a key process in hepatic fibrogenesis, comprising
the transformation of quiescent vitamin A-rich cells into
proliferative, fibrogenic and contractile myofibroblasts (23). α-SMA is a marker for the detection of
activated HSCs during fibrogenesis (24). The CCl4- and
dimethylnitrosamine-induced liver fibrosis models are chemically
induced injuries, whereas PS immunologically induced injury is more
likely to simulate hepatic fibrosis, caused by hepatitis B
(13). In the present study, the
oral administration of AKF-PD to rats hindered PS-induced α-SMA
expression and collagen type I and III deposition, as evaluated by
immunohistochemistry, a result that was also identified at the mRNA
level. These data implied that PS-induced liver fibrosis can be
attenuated by AKF-PD.
Hepatic fibrosis is characterized by the inordinate
yield and deposition of ECM proteins, resulting in the devastation
of the ordinary hepatic parenchyma and interruption of the liver
structure (25,26). Collagen types I and III and other ECM
proteins can be excessively deposited, thereby causing organ
malfunction and failure (27). The
regulation of collagen I and III expression has been extensively
studied to determine the mechanism of fibrosis (28). TGF-β1 is a central mediator of HSC
activation and ECM protein accumulation, as it gives rise to
fibrosis. The secretion and activation of TGF-β1 stimulates the
synthesis of various ECM components, including collagen I and III
(29). TGF-β1 expression is markedly
augmented in cirrhotic liver tissue; it is a potent inducer of
stellate collagen production and cell proliferation (30). In the present study, TGF-β1
expression was reduced by AKF-PD in PS-induced liver fibrosis,
suggesting that the anti-fibrotic effects of AKF-PD may be mediated
through the suppression of TGF-β1.
HSCs are activated by a variety of cytokines,
including PDGF and TGF-β1, which are produced by endothelial cells,
hepatocytes and Kupffer cells (29).
PDGF is the most potent proliferative cytokine in HSCs, whereas
TGF-β1 is the strongest pro-fibrotic factor (31,32). The
transformation of HSCs into myofibroblasts can be promoted by
TGF-β1; the synthesis and degradation of the ECM can also be
stimulated and inhibited by TGF-β1 (33). In the present study, AKF-PD
attenuated the TGF-β1-induced expression of collagen I and α-SMA
in vitro, suggesting that AKF-PD decreases the extent of
liver fibrosis by suppressing TGF-β1.
TGF-β1 signaling is transduced through the
sequential activation of its two serine/threonine kinase receptors
(TGF-β type I receptor and TGF-β type II receptor), which in turn
phosphorylate Smad-3 or Smad-2 to induce nuclear translocation
(34). Smad-3 regulates the
transcription of fibrogenic genes, including α-SMA and pro-collagen
type I, which are important for the production of ECM proteins to
repair damaged tissues (35).
Therefore, the inhibition of TGF-β1 signaling in HSCs is a
therapeutic target for inhibiting the initiation and progression of
hepatic fibrosis. TGF-β1 is the primary isoform associated with
liver fibrosis. The results of the present study revealed that
treatment with 5 ng/ml TGF-β1 activated HSCs, as confirmed by the
increase in α-SMA and collagen I protein expression. Furthermore,
the signaling pathway activated by TGF-β1 affects the type I
receptor-mediated phosphorylation of Smad-3 (36), which was also suppressed by AKF-PD.
These data suggest that AKF-PD blocks fibrogenesis via an effect on
the TGF-β/Smad signaling pathway.
Smad-3 was demonstrated to be an effector of
TGF-β1-induced fibrosis (37).
Additionally, a number of non-Smad signaling molecules, including
ERK, p38, JNK, and phosphatidylinositol-3-kinase have previously
been identified as mediators of TGF-β1-induced fibrosis (38). Smads and the canonical Smad
transcription factor pathway have been the focus of previous
research (39). The linker domains
of Smad2/3 can act as sensors of Smad independent or non-Smad
signal transduction cascades. There are several possible ERK
phosphorylation sites within the linker region domains of Smad2 and
Smad3 (40). In addition to the
Smad-mediated canonical TGF-β1 signaling pathway, the ERK, p38 MAPK
and JNK signaling pathways can also be activated by TGF-β1
(41). Furthermore, TGF-β1 can
augment collagen type I expression through the MAPK and Akt
pathways (42), and the signaling
pathway activated by TGF-β1 comprises non-Smad signaling molecules,
including ERK, p38 and JNK, which were also suppressed by AKF-PD.
These data indicated that AKF-PD blocks fibrogenesis through the
MAPK signaling pathway.
Considering the ability of AKF-PD to ameliorate
liver fibrosis, the present study investigated its effect on
TGF-β1-mediated HSC activation in LX-2 cells and on the
fibrogenesis induced by PS in rats. The present study demonstrated
that AKF-PD inhibits PS-stimulated liver fibrosis and suppresses
TGF-β1-mediated HSCs activation. The impacts of AKF-PD on HSCs
activation through the TGF-β1/Smad and MAPK pathways, which serve
crucial roles in fibrogenesis, were also identified. The results of
the present study may inform a novel therapeutic strategy for
preventing liver fibrosis and other liver diseases.
Acknowledgements
The authors would like to thank Professor Scott L.
Friedman (Icahn School of Medicine at Mount Sinai, New York, NY,
USA) for donating a sample of the LX-2 cell line.
Funding
The present study was supported by the grants from
National Natural Science Foundation of China (grant nos. 81400642,
81370547 and 81673499).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YP, LT and HY conceived and designed the current
study. YP, LL, XZ, MX, CY and ST performed the experiments. YP
analyzed the data. YP, HS and GH interpreted the results of
experimentation. YP prepared the figures and drafted the
manuscript. YP and HS edited and revised manuscript. YP, HY and LT
approved the final version of manuscript. All authors read and
approved the manuscript.
Ethics approval and consent to
participate
The present protocol was approved by the Ethics
Review Committee for Animal Experimentation of Central South
University (Changsha, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman SL and Bansal MB: Reversal of
hepatic fibrosis e fact or fantasy? Hepatology 43(2 Suppl 1).
S82–S88. 2006. View Article : Google Scholar
|
|
3
|
Parola M and Robino G: Oxidative
stress-related molecules and liver fibrosis. J Hepatol. 35:297–306.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Balta C, Herman H, Boldura OM, Gasca I,
Rosu M, Ardelean A and Hermenean A: Chrysin attenuates liver
fibrosis and hepatic stellate cell activation through TGF-b/Smad
signaling pathway. Chem Biol Interact. 240:94–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Williams E and Iredale J: Hepatic
regeneration and TGF-beta: Growing to a prosperous perfection. Gut.
46:593–594. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng Y, Yang H, Wang N, Ouyang Y, Yi Y,
Liao L, Shen H, Hu G, Wang Z and Tao L: Fluorofenidone attenuates
hepatic fibrosis by suppressing the proliferation and activation of
hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol.
306:253–263. 2014. View Article : Google Scholar
|
|
8
|
Peng Y, Yang H, Zhu T, Zhao M, Deng Y, Liu
B, Shen H, Hu G, Wang Z and Tao L: The antihepatic fibrotic effects
of fluorofenidone via MAPK signalling pathways. Eur J Clin Invest.
43:358–368. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang Y, Zhang F, Huang L, Yuan Q, Qin J,
Li B, Wang N, Xie Y, Wang L, Wang W, et al: The protective
mechanism of fluorofenidone in renal interstitial inflammation and
fibrosis. Am J Med Sci. 350:195–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song C, He L, Zhang J, Ma H, Yuan X, Hu G,
Tao L, Zhang J and Meng J: Fluorofenidone attenuates pulmonary
inflammation and fibrosis via inhibiting the activation of NALP3
inflammasome and IL-1β/IL-1R1/MyD88/NF-κB pathway. J Cell Mol Med.
20:2064–2077. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen LX, Yang K, Sun M, Chen Q, Wang ZH,
Hu GY and Tao LJ: Fluorofenidone inhibits transforming growth
factor-beta1-induced cardiac myofibroblast differentiation.
Pharmazie. 67:452–456. 2012.PubMed/NCBI
|
|
12
|
Xu L, Hui AY, Albanis E, Arthur MJ,
O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL and Eng FJ: Human
hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis
of hepatic fibrosis. Gut. 54:142–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Paronetto F and Popper H: Chronic liver
injury induced by immunologic reactions Cirrhosis following
immunization with heterologous sera. Am J Pathol. 49:1087–1101.
1966.PubMed/NCBI
|
|
14
|
Benedetti A, Di Sario A, Casini A, Ridolfi
F, Bendia E, Pigini P, Tonnini C, D'Ambrosio L, Feliciangeli G,
Macarri G and Svegliati-Baroni G: Inhibition of the NA(+)/H(+)
exchanger reduces rat hepatic stellate cell activity and liver
fibrosis: An in vitro and in vivo study. Gastroenterology.
120:545–556. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Di Sario A, Bendia E, Taffetani S,
Marzioni M, Candelaresi C, Pigini P, Schindler U, Kleemann HW,
Trozzi L, Macarri G and Benedetti A: Selective Na+/H+ exchange
inhibition by cariporide reduces liver fibrosis in the rat.
Hepatology. 37:256–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chevallier M, Guerret S, Chossegros P,
Gerard F and Grimaud JA: A histological semi-quantitative scoring
system for evaluation of hepatic fibrosis in needle liver biopsy
specimens: Comparison with morphometric studies. Hepatology.
20:349–355. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pei H, Zhu H, Zeng S, Li Y, Yang H, Shen
L, Chen J, Zeng L, Fan J, Li X, et al: Proteome analysis and tissue
microarray for profiling protein markers associated with lymph node
metastasis in colorectal cancer. J Proteome Res. 6:2495–2501. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Broekema M, Harmsen MC, van Luyn MJ,
Koerts JA, Petersen AH, van Kooten TG, van Goor H, Navis G and Popa
ER: Bone marrow-derived myofibroblasts contribute to the renal
interstitial myofibroblast population and produce procollagen I
after ischemia/reperfusion in rats. J Am Soc Nephrol. 18:165–175.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Winer J, Jung CK, Shackel I and Williams
PM: Development and validation of real-time quantitative reverse
transcriptase-polymerase chain reaction for monitoring gene
expression in cardiacmyocytes in vitro. Anal Biochem. 270:41–49.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsui HT, Wea LL, Zi HC, Lee YJ, Shie MS,
Lee KF, Shen CH and Kuo HC: Moniliformediquinone as a potential
therapeutic agent, inactivation of hepatic stellate cell and
inhibition of liver fibrosis in vivo. J Transl Med. 14:2632016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferrell LD and Kakar S: Liver Pathology.
Demos Medical. (New York, NY). 2011.
|
|
22
|
Saber S, Goda R, El-Tanbouly GS and Ezzat
D: Lisinopril inhibits nuclear transcription factor kappa B and
augments sensitivity to silymarin in experimental liver fibrosis.
Int Immunopharmacol. 64:340–349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Domitrović R and Jakovac H: Effects of
standardized bilberry fruit extract (Mirtoselect®) on
resolution of CCl4-induced liver fibrosis in mice. Food Chem
Toxicol. 49:848–854. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsai JH, Liu JY, Wu TT, Ho PC, Huang CY,
Shyu JC, Hsieh YS, Tsai CC and Liu YC: Effects of silymarin on the
resolution of liver fibrosis induced by carbon tetrachloride in
rats. J Viral Hepat. 15:508–514. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhan L, Huang C, Meng XM, Song Y, Wu XQ,
Yang Y and Li J: Hypoxia-inducible factor-1alpha in hepatic
fibrosis: A promising therapeutic target. Biochimie. 108:1–7. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li J, Li X, Xu W, Wang S, Hu Z, Zhang Q,
Deng X, Wang J, Zhang J and Guo C: Antifibrotic effects of luteolin
on hepatic stellate cells and liver fibrosis by targeting
AKT/mTOR/p70S6K and TGFb/Smad signalling pathways. Liver Int.
35:1222–1233. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park JH, Yoon J, Lee KY and Park B:
Effects of geniposide on hepatocytes undergoing
epitheliale-mesenchymal transition in hepatic fibrosis by targeting
TGFb/Smad and ERK-MAPK signaling pathways. Biochimie. 113:26–34.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang JH, Kim SC, Kim KM, Jang CH, Cho SS,
Kim SJ, Ku SK, Cho IJ and Ki SH: Isorhamnetin attenuates liver
fibrosis by inhibiting TGF-β/Smad signaling and relieving oxidative
stress. Eur J Pharmacol. 783:92–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Friedman SL: Molecular regulation of
hepatic fibrosis, an integrated cellular response to tissue injury.
J Biol Chem. 275:2247–2250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Borkham-Kamphorst E, Herrmann J, Stoll D,
Treptau J, Gressner AM and Weiskirchen R: Dominant-negative soluble
PDGF-beta receptor inhibits hepatic stellate cell activation and
attenuates liver fibrosis. Lab Invest. 84:766–777. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tahashi Y, Matsuzaki K, Date M, Yoshida K,
Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y and Inoue
K: Differential regulation of TGF-beta signal in hepatic stellate
cells between acute and chronic rat liver injury. Hepatology.
35:49–61. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marra F, Galastri S, Aleffi S and Pinzani
M: Stellate cells. Dufour JF and Clavien PA: Signaling pathways in
liver diseases. (Springer-Verlag, Berlin, Heidelberg).
2010.41–68
|
|
34
|
Friedman SL: Mechanisms of disease:
Mechanismsofhepatic fibrosis and therapeutic implications. Nat Clin
Pract Gastroenterol Hepatol. 1:98–105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gauldie J, Bonniaud P, Sime P, Ask K and
Kolb M: TGF-beta, Smad3 and the process of progressive fibrosis.
Biochem Soc Trans. 35:661–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Parsons CJ, Takashima M and Rippe RA:
Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol
Hepatol. 22 (Suppl 1):S79–S84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cho HJ, Baek KE, Saika S, Jeong MJ and Yoo
J: Snail is required for transforming growth factor-beta-induced
epithelial-mesenchymal transition by activating PI3 kinase/Akt
signal pathway. Biochem Biophys Res Commun. 353:337–343. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Burch ML, Zheng W and Little PJ: Smad
linker region phosphorylation in the regulation of extracellular
matrix synthesis. Cell Mol Life Sci. 68:97–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matsuura I, Wang G, He D and Liu F:
Identification and characterization of ERK MAP kinase
phosphorylation sites in Smad3. Biochemistry. 44:12546–12553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim YS, Kim J, Kim KM, Jung DH, Choi S,
Kim CS and Kim JS: Myricetin inhibits advanced glycation end
product (AGE)-induced migration of retinal pericytes through
phosphorylation of ERK1/2, FAK-1, and paxillin in vitro and in
vivo. Biochem Pharmacol. 93:496–505. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lechuga CG, Hernández-Nazara ZH, Domínguez
Rosales JA, Morris ER, Rincón AR, Rivas-Estilla AM, Esteban-Gamboa
A and Rojkind M: TGF-beta1 modulates matrix metalloproteinase-13
expression in hepatic stellate cells by complex mechanisms
involving p38MAPK, PI3-kinase, AKT, and p70S6k. Am J Physiol
Gastrointest Liver Physiol. 287:G974–G987. 2004. View Article : Google Scholar : PubMed/NCBI
|