Introduction
Autosomal dominant polycystic kidney disease (ADPKD)
is the most common hereditary kidney disorder found among all
ethnic groups worldwide, affecting 1 in 400 to 1 in 800 live births
among different populations (1,2). The
disease is characterized by the development and progressive
enlargement of renal cysts and is the fourth most common cause
leading to end-stage renal disease (ESRD) (2,3). The
etiology of this disease is associated with mutations in
PKD1 [Online Mendelian Inheritance in Man (OMIM) no. 601313;
16p13.3] and PKD2 (OMIM no. 173910; 4q21-22) genes, and
85–90% of ADPKD cases are caused primarily by mutations in the
PKD1 gene (4). PKD1
has 46 exons and encodes the transmembrane protein, polycystin-1
(PC1), which is composed of 4,303 amino acids. PKD2 is a
smaller gene with 15 exons. It encodes the transmembrane protein
polycystin-2 (PC2), which is composed of 968 amino acids (5,6). PC1
contains an N-terminal extracellular region, 11 membrane-spanning
domains and a cytoplasmic C-tail, whereas the PC2 protein possesses
a shorter N-terminal extracellular region and only 6
membrane-spanning domains (7,8).
The diagnosis of ADPKD is a challenge; renal
ultrasound, magnetic resonance imaging and computed tomography have
limited capability in terms of detecting ADPKD in patients
(9). With no hotspot mutation
information available for PKD1 and PKD2, clinical molecular
diagnostic techniques are difficult (10). To overcome these challenges, the
genotype variation of these two genes in patients with ADPKD has
been investigated using techniques including polymerase chain
reaction (PCR)-single-strand conformation polymorphism, denaturing
high-performance liquid chromatography, multiplex
ligation-dependent probe amplification and next-generation
sequencing (NGS) (11–14). Recent advancements in sequencing
technologies have enabled the rapid and cost-effective generation
of large quantities of data. By removing most of the throughput and
resource limitations of traditional methods, NGS enables
investigators to analyze PKD1 and PKD2 genes in a
single run (15–17).
In individuals diagnosed with ADPKD, PKD1
exhibits marked allelic heterogeneity, with a high level of gene
variation (18,19). To date, >2,300 germline mutations
of PKD1 have been identified; ~900 are likely to be neutral
polymorphisms and >150 are unclassified variants (20). In the current study, the
pathogenicity of an atypical splice variant in the PKD1
gene, rs201204878, was evaluated in an Iranian family with ADPKD.
In silico prediction-based models were used to evaluate its
pathogenicity.
Materials and methods
Ethical compliance
The study was approved by the ethics committee board
of Shahid Beheshti University of Medical Sciences (Tehran, Iran)
and all experiments were performed in adherence to the declaration
of Helsinki. Written informed consent was obtained from all
patients prior to their participation in the study.
Patient pedigree and subject data
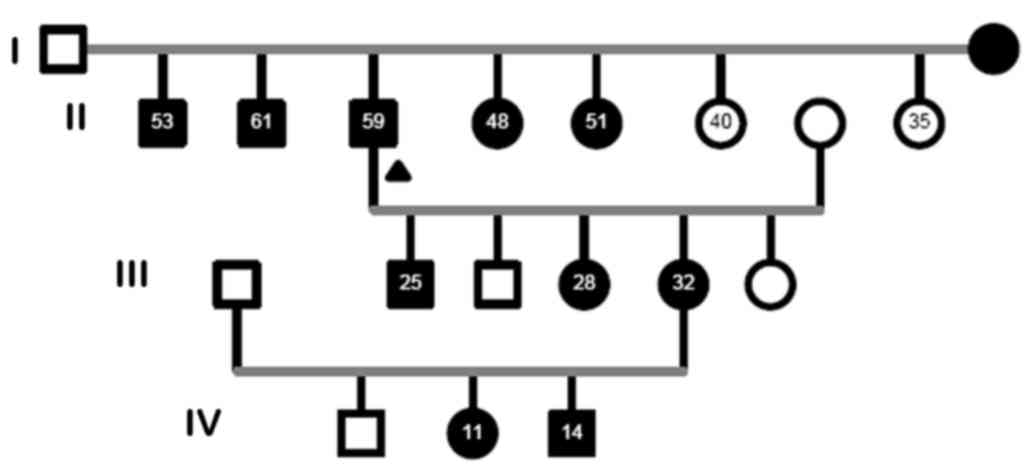
A large Iranian family of 19 members (10 males, nine
females; age, 40.8±15.7 years) with a history of ADPKD was
recruited for this study from dialysis centers and nephrology
clinics of Modarres Hospital (Tehran, Iran) in June 2015. In the
pedigree, 11 members were recruited; seven of them were diagnosed
with ADPKD by ultrasound examination, according to Ravine's
criteria (21). The proband selected
was diagnosed with ESRD, and had two affected brothers and two
affected sisters with early onset ESRD (age of onset, <50
years). A screening ultrasound was performed on 11 available
participants of the asymptomatic at-risk adult members in this
family. Details of the affected and non-affected members of the
family (Four generations, 19 members in total) are shown in
Fig. 1. Total genomic DNA from 11
available family members was extracted from blood samples using a
standard phenol-chloroform procedure as previously described
(22). The quality and concentration
of DNA samples was evaluated using spectrophotometry (260/280 nm)
and 1% agarose gel electrophoresis.
Mutational analysis by targeted
NGS
Targeted NGS experiments were performed by BGI Tech
Europe (Copenhagen, Denmark). The Genetic Sequencing Test was
performed using a custom-designed NimbleGen chip (Roche NimbleGen,
Inc., Madison, WI, USA) by BGI Tech Europe to capture the genes of
interest, which were then sequenced by NGS. In general, the test
platform examined >95% of the target gene with a sensitivity of
>99%. Point mutations, micro-insertions, deletions and
duplications (<20 bp) were detected simultaneously (14). SNPs and indels were identified via
the SOAP snp software (Release 1.03; http://soap.genomics.org.cn/soapsnp.html) and GATK
Indel Genotyper (Version 4.0.11; broadinstitute.org/gsa/wiki/index.php/).
Bioinformatics analysis and mutation
identification
Candidate variations were compared against
electronic database information and computational analysis was
performed. The detected sequence variations were compared against
the currently published list of PKD gene variants in the Human Gene
Mutation Database (23), the
Autosomal Dominant Polycystic Kidney Disease Mutation Database
version 3.1 (20) and the Iranian
Human Mutation Gene Bank (24). The
effects of variation were analyzed using web-based computational
pathogenicity prediction tools, including MutationTaster working on
current build of NCBI 137/Ensembl 69 (25), Sorting Intolerant from Tolerant based
on NCBI 137/Ensembl 66 (26) and
Polymorphism Phenotyping version 2 (27). Human Splice Finder (HSF) software
version 3.0 was also used to predict splicing (28).
Sanger sequencing
To validate the variants predicted to be associated
with ADPKD by computational analyses, the region of interest
surrounding the variant was amplified by PCR using Taq DNA
Polymerase Master Mix (Ampliqon A/S, Odense, Denmark). The variants
were sequenced using Sanger sequencing methods and appropriate
internal primers reported previously (Table I) (29).
 | Table I.Primers employed for Sanger
sequencing. |
Table I.
Primers employed for Sanger
sequencing.
| Exon | Primer sequence
(5′-3′) | PCR annealing
temperature (°C) | Cycle | (Refs.) |
|---|
| 41 |
F-CGGCCTCCTGACCAGCCTGGCTC | 64 | 30 | (31) |
|
|
R-TAGGCCAGCGGGGGCCGGAGGAGTG |
|
|
|
Segregation analysis
To confirm the association between mutation and the
pathogenicity of the disease, analysis of seven affected family
members in the pedigree was performed by direct sequencing (Sanger
method as previously described). The Iranian normal population
database contains data of the reported variants. Four unaffected
family members and four normal controls from the Iranian normal
population database were also checked for the mutation to confirm
the prediction.
Results
Mutational analysis
Mutational analysis of the PKD1 and
PKD2 genes was performed in a large Iranian family
consisting of 11 members (5 male and 6 females); 7 of them (3 male
and 4 female) diagnosed with ADPKD by renal ultrasound. Using
targeted NGS, three intronic variations and three synonymous exonic
variants were identified in the PKD2 gene, and two
non-synonymous exonic variants and eight intronic variants were
identified in the PKD1 gene (Table II). All variants except from three
intronic variants in PKD1 have been reported in the National
Centre for Biotechnology Information dbSNP database (build 151;
ncbi.nlm.nih.gov/SNP/) and are
considered to be known polymorphisms. The three novel mutations
were predicted to be deleterious polymorphisms by bioinformatics
analysis.
| Table II.Mutations identified in PKD1
and PKD2 genes in a large Iranian family with autosomal
dominant polycystic kidney disease. |
Table II.
Mutations identified in PKD1
and PKD2 genes in a large Iranian family with autosomal
dominant polycystic kidney disease.
| Gene | Genomic
position | RefSeq | Nucleic acid
alteration | Mutation
location | SNPID |
|---|
| PKD2 | 88,957,562 | NM_000297 | T/C | Intronic | rs17786456 |
| PKD2 | 88,959,381 | NM_000297 | G/A | Intronic | rs2725221 |
| PKD2 | 88,959,745 | NM_000297 | A/G | Intronic | rs17013735 |
| PKD1 |
140,294 | NM_001009944 | C/T | Exonic | rs148478410 |
| PKD1 |
141,714 | NM_001009944 | G/A | ncRNA_
intronic | Novel |
| PKD1 |
141,776 | NM_001009944 | -/CCC | ncRNA_
intronic | rs201204878 |
| PKD1 |
142,113 | NM_001009944 | G/A | Exonic | rs145955373 |
| PKD1 |
152,574 | NM_001009944 | C/T | Exonic | rs374619113 |
| PKD1 |
154,478 | NM_001009944 | A/G | Intronic | rs4786209 |
| PKD1 |
156,369 | NM_001009944 | G/A | Intronic | rs142761413 |
| PKD1 |
158,176 | NM_001009944 | T/A | Intronic | rs200363107 |
| PKD1 |
163,115 | NM_001009944 | G/C | ncRNA_
intronic | Novel |
| PKD1 |
163,579 | NM_001009944 | G/C | Intronic | rs56326527 |
| PKD1 |
164,808 | NM_001009944 | C/T | Exonic | rs40433 |
| PKD1 |
166,769 | NM_001009944 | G/A | Intronic | Novel |
| PKD1 |
167,874 | NM_001009944 | G/A | Exonic | rs35842 |
Analysis of splice site mutations
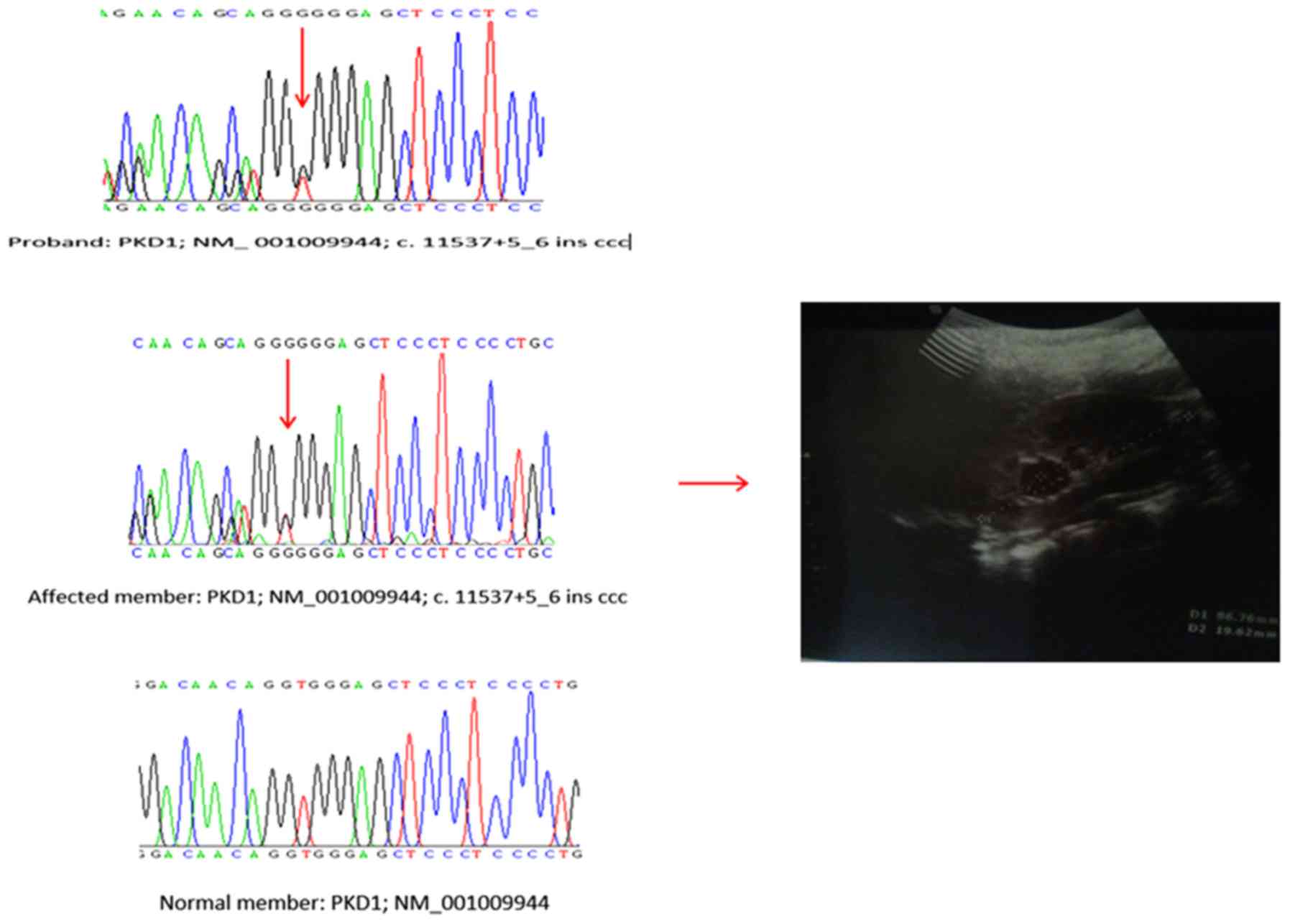
Among the three reported non-coding RNA (ncRNA)
intronic variations, one (rs201204878) was identified as a splice
region variant (c. 11537+5_6 ins ccc). The pathogenic relevance of
putative splice site mutations was evaluated using HSF software.
The HSF results indicated that this mutation occurred in the late
exonic positions and generated a new exonic splice site that
potentially altered splicing. Therefore this mutation was
classified as a highly likely pathogenic mutation (30). This variant was assessed for its
co-segregation with the disease in three affected and two
unaffected family members The mutation was identified in all
affected family members, and was absent in unaffected family
members, as well as in 400 normal subjects in the Iranian
population (Fig. 2).
Segregation analysis
In the present study, the allele frequency of the c.
11537+5_6 ins ccc variation in world population groups from the
1,000 Genomes Project was analyzed using the Ensembl database
(internationalgenome.org/category/ensembl/). The
frequency of this mutation in sub-populations from America, Africa
and East Asia was 0%. The frequency of this mutation in European
and South Asian sub-populations was ~2 and ~3%, respectively.
However, the homozygous genotype frequency in these sub-populations
was 0%. This appears to be consistent with the experimental results
of the current study, and may be associated with the truncation
effect of this variant and the subsequent abortion of embryos
carrying the homozygous mutation in mice models (31). The co-segregation analysis and HSF of
rs201204878 showed no co-relation with the severity of ADPKD in the
affected family members.
Discussion
According to the genotype-phenotype correlation, the
ADPKD phenotype exhibits both genetic and allelic dependency.
PKD1 mutations are correlated with disease severity and the
onset of ESRD in ADPKD patients (32). The c. 11537+5_6 ins ccc splice
mutation identified in the present study is a type of truncating
mutation, which has been associated with a more severe ADPKD
phenotype when compared with non-truncating PDK1 mutations
(4). To the best of our knowledge,
the present study is the first to report the rs201204878 pathogenic
variant in the Iranian population, as this variation was not
observed in 400 subjects from the Iranian normal population
database. The clinical severity of ADPKD in affected family members
was consistent with the obtained splicing mutation results from HSF
and co-segregation analyses. Hence, follow-up in other young
affected family members becomes necessary to verify the reported
association. The average age of ESDR onset varies with the type of
gene involved, and it was reported to be 54.3 years in patients
with PKD1 mutations and 74 years in patients with PKD2 mutations
(33).
In the present study, the heterozygous 11537+5_6 ins
ccc mutation (rs201204878) was identified in intron 41 of the
PKD1 gene, and no other truncation mutations were identified
by targeted NGS. This was demonstrated to be a pathogenic mutation,
which extends the associated phenotypic and genotypic spectrums for
ADPKD. The rs201204878 mutation has been previously reported to be
benign (33), but in the present
study it was predicted that the splice mutation may lead to
abnormal splicing of PKD1. This is expected to affect PC1
function, which is consistent with a previous report demonstrating
that the expression of the truncated protein is correlated with the
early onset of ESRD (34).
In the current study, targeted NGS of the
PKD1 and PKD2 genes produced high-coverage sequencing
data with high sensitivity and specificity. The results were
confirmed with Sanger sequencing, which is the standard approach
for clinical genetic testing in ADPKD. The results suggest that
targeted NGS may potentially replace Sanger sequencing for clinical
genetic testing in ADPKD as the former is a faster and more
accurate procedure In silico and co-segregation analyses
concluded that the rs201204878 variant may be considered as a
potential functional mutation. However, further mini gene analysis
may provide further insight into the pathogenicity of this
mutation. In addition, as the Iranian population is not included in
the 1,000 Genomes Project (35),
performing genetic variation studies will add to the plethora of
data available on mutations; such studies on ethnic populations may
aid in the diagnosis, prognosis and management of this disease. In
conclusion, the polymorphisms identified in the present study may
contribute to improving the diagnosis, genetic counseling and
treatment of patients with ADPKD.
Acknowledgements
The authors want to thank the Urology and Nephrology
Research Center (UNRC) of Shahid Beheshti University of Medical
Sciences for their support.
Funding
This trial was supported by a grant from Royan
Institute and the Royan Charity Association for Health Research
(grant no. 93/566).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AB conceived of the present study. FR and AT
performed the experiments. AB, NA and RM analysed the results. FR
and AB wrote the manuscript. NA and RM edited the manuscript.
Ethics approval and consent to
participate
The study was approved by the ethics committee board
of Shahid Beheshti University of Medical Sciences (Tehran, Iran)
and all experiments were performed in adherence to the declaration
of Helsinki. Written informed consent was obtained from all
patients prior to their participation in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Trujillano D, Bullich G, Ossowski S,
Ballarín J, Torra R, Estivill X and Ars E: Diagnosis of autosomal
dominant polycystic kidney disease using efficient PKD1 and PKD2
targeted next-generation sequencing. Mol Genet Genomic Med.
2:412–421. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wilson PD: Polycystic kidney disease. N
Engl J Med. 350:151–164. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Srivastava A and Patel N: Autosomal
dominant polycystic kidney disease. Am Fam Physician. 90:303–307.
2014.PubMed/NCBI
|
|
4
|
Kurashige M, Hanaoka K, Imamura M, Udagawa
T, Kawaguchi Y, Hasegawa T, Hosoya T, Yokoo T and Maeda S: A
comprehensive search for mutations in the PKD1 and PKD2 in Japanese
subjects with autosomal dominant polycystic kidney disease. Clin
Genet. 87:266–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Germino GG, Weinstat-Saslow D, Himmelbauer
H, Gillespie GA, Somlo S, Wirth B, Barton N, Harris KL, Frischauf
AM and Reeders ST: The gene for autosomal dominant polycystic
kidney disease lies in a 750-kb CpG-rich region. Genomics.
13:144–151. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cornec-Le Gall E, Audrézet MP, Le Meur Y,
Chen JM and Férec C: Genetics and pathogenesis of autosomal
dominant polycystic kidney disease: 20 years on. Hum Mutat.
35:1393–1406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hughes J, Ward CJ, Peral B, Aspinwall R,
Clark K, San Millán JL, Gamble V and Harris PC: The polycystic
kidney disease 1 (PKD1) gene encodes a novel protein with multiple
cell recognition domains. Nat Genet. 10:151–160. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nims N, Vassmer D and Maser RL:
Transmembrane domain analysis of polycystin-1, the product of the
polycystic kidney disease-1 (PKD1) gene: Evidence for 11
membrane-spanning domains. Biochemistry. 42:13035–13048. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pei Y, Obaji J, Dupuis A, Paterson AD,
Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, et al:
Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc
Nephrol. 20:205–212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tan AY, Michaeel A, Liu G, Elemento O,
Blumenfeld J, Donahue S, Parker T, Levine D and Rennert H:
Molecular diagnosis of autosomal dominant polycystic kidney disease
using next-generation sequencing. J Mol Diagn. 16:216–228. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ding L and Zhang S, Qiu W, Xiao C, Wu S,
Zhang G, Cheng L and Zhang S: Novel mutations of PKD1 gene in
Chinese patients with autosomal dominant polycystic kidney disease.
Nephrol Dial Transplant. 17:75–80. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu C, Yang Y, Zou L, Hu Z, Li J, Liu Y, Ma
Y, Ma M, Su D and Zhang S: Identification of novel mutations in
Chinese Hans with autosomal dominant polycystic kidney disease. BMC
Med Genet. 12:1642011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu G, Tan AY, Michaeel A, Blumenfeld J,
Donahue S, Bobb W, Parker T, Levine D and Rennert H: Development
and validation of a whole genome amplification long-range PCR
sequencing method for ADPKD genotyping of low-level DNA samples.
Gene. 550:131–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang T, Meng Y, Wei X, Shen J, Zhang M, Qi
C, Wang C, Liu J, Ma M and Huang S: Identification of novel
mutations of PKD1 gene in Chinese patients with autosomal dominant
polycystic kidney disease by targeted next-generation sequencing.
Clin Chim Acta. 433:12–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rossetti S, Hopp K, Sikkink RA, Sundsbak
JL, Lee YK, Kubly V, Eckloff BW, Ward CJ, Winearls CG, Torres VE
and Harris PC: Identification of gene mutations in autosomal
dominant polycystic kidney disease through targeted resequencing. J
Am Soc Nephrol. 23:915–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eisenberger T, Decker C, Hiersche M,
Hamann RC, Decker E, Neuber S, Frank V, Bolz HJ, Fehrenbach H, Pape
L, et al: An efficient and comprehensive strategy for genetic
diagnostics of polycystic kidney disease. PloS One.
10:e01166802015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Edrees BM, Athar M, Al-Allaf FA, Taher MM,
Khan W, Bouazzaoui A, Al-Harbi N, Safar R, Al-Edressi H, Alansary
K, et al: Next-generation sequencing for molecular diagnosis of
autosomal recessive polycystic kidney disease. Gene. 591:214–226.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harris PC and Hopp K: The mutation, a key
determinant of phenotype in ADPKD. J Am Soc Nephrol. 24:868–870.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Yu C, Tao Y, Yang Y, Hu Z and Zhang
S: Putative mutation of PKD1 gene responsible for autosomal
dominant polycystic kidney disease in a Chinese family. Int J Urol.
18:240–242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gout AM, Martin NC, Brown AF and Ravine D:
PKDB: Polycystic kidney disease mutation database-a gene variant
database for autosomal dominant polycystic kidney disease. Hum
Mutat. 28:654–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ravine D, Gibson RN, Walker RG, Sheffield
LJ, Kincaid-Smith P and Danks DM: Evaluation of ultrasonographic
diagnostic criteria for autosomal dominant polycystic kidney
disease 1. Lancet. 343:824–827. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sambrook J and Russell DW: Purification of
nucleic acids by extraction with phenol: Chloroform. CSH Protoc.
2006.pdb.prot44552006.PubMed/NCBI
|
|
23
|
Stenson PD, Mort M, Ball EV, Howells K,
Phillips AD, Thomas NS and Cooper DN: The human gene mutation
database: 2008 update. Genome Med. 1:132009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human Splicing Finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rossetti S, Chauveau D, Walker D,
Saggar-Malik A, Winearls CG, Torres VE and Harris PC: A complete
mutation screen of the ADPKD genes by DHPLC. Kidney Int.
61:1588–1599. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kinoshita M, Higashihara E, Kawano H,
Higashiyama R, Koga D, Fukui T, Gondo N, Oka T, Kawahara K, Rigo K,
et al: Technical evaluation: Identification of pathogenic mutations
in PKD1 and PKD2 in patients with autosomal dominant polycystic
kidney disease by next-generation sequencing and use of a
comprehensive new classification system. PloS One. 11:e01662882016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Flaherty L, Bryda EC, Collins D, Rudofsky
U and Montgomery JC: New mouse model for polycystic kidney disease
with both recessive and dominant gene effects. Kidney Int.
47:552–558. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cornec-Le Gall E, Audrézet MP, Chen JM,
Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau
E, Jousset P, et al: Type of PKD1 mutation influences renal outcome
in ADPKD. J Am Soc Nephrol. 24:1006–1013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heyer CM, Sundsbak JL, Abebe KZ, Chapman
AB, Torres VE, Grantham JJ, Bae KT, Schrier RW, Perrone RD, Braun
WE, et al: Predicted mutation strength of nontruncating PKD1
mutations aids genotype-phenotype correlations in autosomal
dominant polycystic kidney disease. J Am Soc Nephrol. 27:2872–2884.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gainullin VG, Hopp K, Ward CJ, Hommerding
CJ and Harris PC: Polycystin-1 maturation requires polycystin-2 in
a dose-dependent manner. J Clin Invest. 125:607–620. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Siva N: 1000 Genomes project. Nat
Biotechnol. 26:2562008. View Article : Google Scholar : PubMed/NCBI
|