Introduction
Ischemia/reperfusion (I/R) injury is caused by the
restoration of blood flow following ischemia. I/R injury is
associated with the generation of reactive oxygen species (ROS),
calcium overload, release and activation of inflammatory cytokines
and alexins, fibrinolysis imbalance, blood coagulation and
infiltration of inflammatory cells (1), which result in serious damage in
various diseases, including myocardial infarction (1–4).
However, the intracellular mechanisms of I/R injury remain largely
unclear.
Autophagy is an important cellular mechanism that
degrades large structures, such as organelles and protein
aggregates (5). During I/R injury,
tissue is deprived of oxygen and nutrients, which are then
restored, triggering changes in autophagy. Previous studies have
mainly focused on the effects of autophagy in myocardial I/R
injury, yet the role of autophagy is under debate. Elevated
autophagy during ischemia significantly reduces myocardiocyte death
(6–9), whereas the role of autophagy during
reperfusion remains elusive. Several studies have demonstrated that
an increase in autophagy during reperfusion reduces the death of
cardiomyocytes (10–12), whereas other studies have reported
the opposite effect (9). The
association between autophagy and I/R injury in the brain and
kidneys has also been reported recently, with autophagy shown to
have a protective role in kidney I/R injury, whereas its effect in
brain I/R injury was unclear (13,14).
The status and roles of autophagy vary among
different organs and tissues, and the exact function of autophagy
in hindlimb I/R injury has not been established. Thus, the current
study aimed to explore the role of autophagy, and the findings
indicated that enhanced autophagy protected cells from hindlimb I/R
injury.
Materials and methods
Cell culture and hypoxia/reoxygenation
(H/R) cell model
Mouse C2C12 myoblast cells (Shanghai Cellular
Institute of China Scientific Academy) were cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific
Inc.) supplemented with 10% fetal bovine serum (Thermo Fisher
Scientific Inc.) in 5% CO2 at 37°C. In order to
establish the H/R cell model, when cells reached 80% confluence,
hypoxia was induced by changing the air content to 0.5%
O2, 93% N2 and 5% CO2 gas mixture
in a tri-gas CO2 incubator (Thermo Fisher Scientific
Inc.) and replacing the media with fresh deoxygenated serum-free
high glucose DMEM for 6 h. Subsequently, the medium was replaced
with the normal media, and incubated in 95% air and 5%
CO2 for 6 h for reoxygenation. The normal group was
cultured in normal conditions for the same period of time,
respectively. TUNEL staining and western blot, chloroquine (40 nM)
was added to media prior to the hypoxia and H/R phase for the
regulation of autophagy flux.
Animals
Approval of all animal experiments was obtained from
the Institutional Review Board of The First Affiliated Hospital of
Sun Yat-sen University (Guangzhou, China). Male C57BL6 mice (age,
10–12 weeks; weight, ~20 g) were used to establish the I/R injury
model. Mice (n=45) were randomly assigned to the control, ischemia,
I/R, rapamycin + ischemia and rapamycin + I/R groups (n=9 per
group). The rapamycin (3 mg/kg body weight, dissolved in DMSO) was
intraperitoneally administered three times a week for two
consecutive weeks prior to I/R modeling, and equal volume saline
was intraperitoneally administered at the same frequency for the
control, ischemia and I/R groups (15).
I/R injury model
Animals were anesthetized with isoflurane and then
placed on a heating pad to maintain the body temperature at 37°C.
Blood flow was blocked in the left hindlimbs by placing orthodontic
rubber bands (0.3 cm; 128 g) high up on the thigh. The rubber bands
were immediately removed after placement in the control group. Mice
in the ischemia group were euthanized immediately after 3 h of
ischemia, while mice in the I/R group were euthanized 4 h after the
bands were removed following 3 h of ischemia. Gastrocnemius muscles
were immediately harvested following euthanasia and stored at −80°C
for subsequent protein extraction, paraffin-embedded and glutaric
dialdehyde fixated at 4°C for 4 h for TUNEL staining and electron
microscopy, respectively.
Laser Doppler imaging
A laser Doppler imager (Blood Perfusion Imager;
Perimed AB) was used to assess limb perfusion at the baseline (10
min after anesthesia), ischemia (10 min after ligation) and the
reperfusion phase (10 min after band removal). Fur was completely
removed from the two hindlimbs using a depilatory cream subsequent
to the induction of anesthesia. The laser Doppler detector was
placed 10 cm above the heating pad when the mice were restrained on
it. The laser beam (780 nm), which was reflected by moving red
blood cells in capillaries, arterioles and venules, was detected.
Next, a computerized, color-coded image was produced. Mean flux
values representing tissue perfusion were calculated from the
relative flux (in U/cm2) in the areas corresponding to
the plantar aspect of the hindlimb or tail, with the application of
image analysis software (PIMsoft 2.0.3; Perimed AB).
Triphenyl tetrazolium chloride assay
(TTCA)
TTCA was applied to measure the infarct size induced
by I/R in skeletal muscle (16). At
the end of the I/R procedure (3-h ischemia, followed by 4-h
reperfusion), gastrocnemius muscles from the I/R and control
animals were immediately harvested and washed with 0.9% normal
saline. Subsequent to the removal of the adherent fascias, fat and
tendons, the muscles were cut into 1.5-mm transverse slices and
washed with cold normal saline to eliminate any blood. The slices
were then blotted dry with paper towels and incubated in 1% TTC
(cat. no. 298964; Sigma-Aldrich; Merck KGaA) solution for staining
at room temperature for 1 h. The muscles were separated into viable
skeletal muscles with a natural dark purple-red color and infarcted
muscles with a pale brown color stained using TTC according to the
aforementioned conditions. Infarct size in each slice was
calculated using Adobe Photoshop CS3 (Photoshop Extended; Adobe
Systems, Inc.), and the ratio (%) of the infarcted muscle to total
gastrocnemius muscles (viability plus infarcted muscles) was
recorded.
Terminal deoxynucleotidyl transferase
dUTP nick end-labelling (TUNEL) staining
In vitro, apoptotic cells were identified by
TUNEL staining according to the manufacturer's protocol using a
commercially available kit (One Step TUNEL Apoptosis Assay kit;
Beyotime Institute of Biotechnology). Mouse C2C12 myoblast cells
were fixed with 4% paraformaldehyde at room temperature for 30 min.
As a positive control, untreated cells were pre-incubated with
DNase I recombinant (5 µg/ml) for 10 min at room temperature.
Subsequent to rinsing with phosphate-buffered saline (PBS), cells
were incubated with TUNEL reaction mixture for 60 min at 37°C in
the dark. The grey intensity of TUNEL-positive nuclei was
quantified in five random fields in each group with ImageJ software
(National Institutes of Health) under a fluorescence microscope
(Leica DMI8; Leica Microsystems GmbH). In vivo, apoptotic
cells were identified according to the manufacturer's protocol
using a commercially available kit (In situ Cell Death
Detection Kit; Roche). The tissue sections of skeletal muscle were
dewaxed and rehydrated according to a standard protocol. Next,
slides were incubated with Proteinase K working solution for 15 min
at room temperature. After rinsing with PBS, slides were incubated
with TUNEL reaction mixture for 60 min at 37°C in the dark. Next,
Converter-POD antibody (1:500) from aforementioned TUNEL kit was
used for further reaction with 30 min at 37°C, followed by the
addition of DAB substrate for 10 min at room temperature.
Subsequently, the slides were mounted with PBS/glycerol. The number
of TUNEL-positive nuclei (brown) was normalized to total nuclei
(blue) from 10 random fields of each tissue cross-section under a
light microscope (Zeiss Axio Observer Z1; Carl Zeiss). All TUNEL
assay images were obtained under identical magnification and
microscopy conditions.
Electron microscopy
Gastrocnemius muscles were cut into small sections
(1 mm3), and fixed with 2% glutaraldehyde and 2%
paraformaldehyde in sodium phosphate buffer (pH 7.4) overnight at
4°C. The tissue samples were post-fixed with 1% OsO4 for
1 h at room temperature, dehydrated in a series of aqueous alcohol
solutions and finally 100% alcohol, and then embedded in epoxy
resin. Ultrathin sections cut using a Leica ultramicrotome were
stained with lead citrate and uranyl acetate, and examined using a
JEM-1400/JEM-1400 PLUS electron microscope (Jeol USA, Inc.) at 100
kV.
Western blotting
Cultured cell and muscle tissues were homogenized in
lysis buffer (containing 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1
mM EGTA, 1% Triton X-100, 1 mM PMSF, pH 7.4 with protease
inhibitors; Cell Signaling Technology, Inc.), and the homogenate
was centrifuged at 11,800 × g for 30 min at 4°C. Next, the
supernatant was collected and its protein content was measured. An
aliquot of the supernatant (20 µg/ml protein) was subsequently
subjected to SDS-PAGE using 10% gels, and the proteins were
transferred onto PVDF membranes (EMD Millipore). The membranes were
incubated with polyclonal antibodies [1:1,000 dilution in TBS/Tween
20 (TBST)] against P62 (cat. no. P0067; Sigma-Aldrich; Merck KGaA),
microtubule associated protein 1 light chain 3 (LC3; cat. no.
L8918; Sigma-Aldrich; Merck KGaA) and cleaved caspase-3 (cat. no.
9661; Cell Signaling Technology, Inc.) overnight at 4°C. Membranes
were then incubated with horseradish peroxidase (HRP)-conjugated
goat anti-rabbit IgG secondary antibodies (1:1,000 dilution in
TBST; cat. no. 7074; Cell Signaling Technology, Inc.) for 1 h at
room temperature, and the bound antibody was detected using
Chemiluminescence Reagent Plus (PerkinElmer, Inc.). The intensity
of each band was quantified using a densitometer. GAPDH, serving as
the internal control, was detected using a rabbit anti-GAPDH
primary antibody (1:3,000; cat. no. G9545; Sigma-Aldrich; Merck
KGaA) and HRP-conjugated goat anti-rabbit IgG secondary antibody
(1:1,000).
Statistical analysis
Data were analyzed using SPSS version 24.0 software
(IBM Corp.). One-way and two-way analysis of variance (ANOVA) was
performed for the comparison of multiple groups. Bonferroni
post-hoc testing was used following ANOVA for analyzing all
pairwise comparisons between groups. Subsequent contrast analysis
was also performed when necessary. P<0.05 was considered to
indicate a statistically significant difference.
Results
H/R model reveals that downregulation
of autophagy increases apoptosis in C2C12 cells
To study autophagic flux and its function in I/R
injury, mouse C2C12 myoblast cells were subjected to H/R, serving
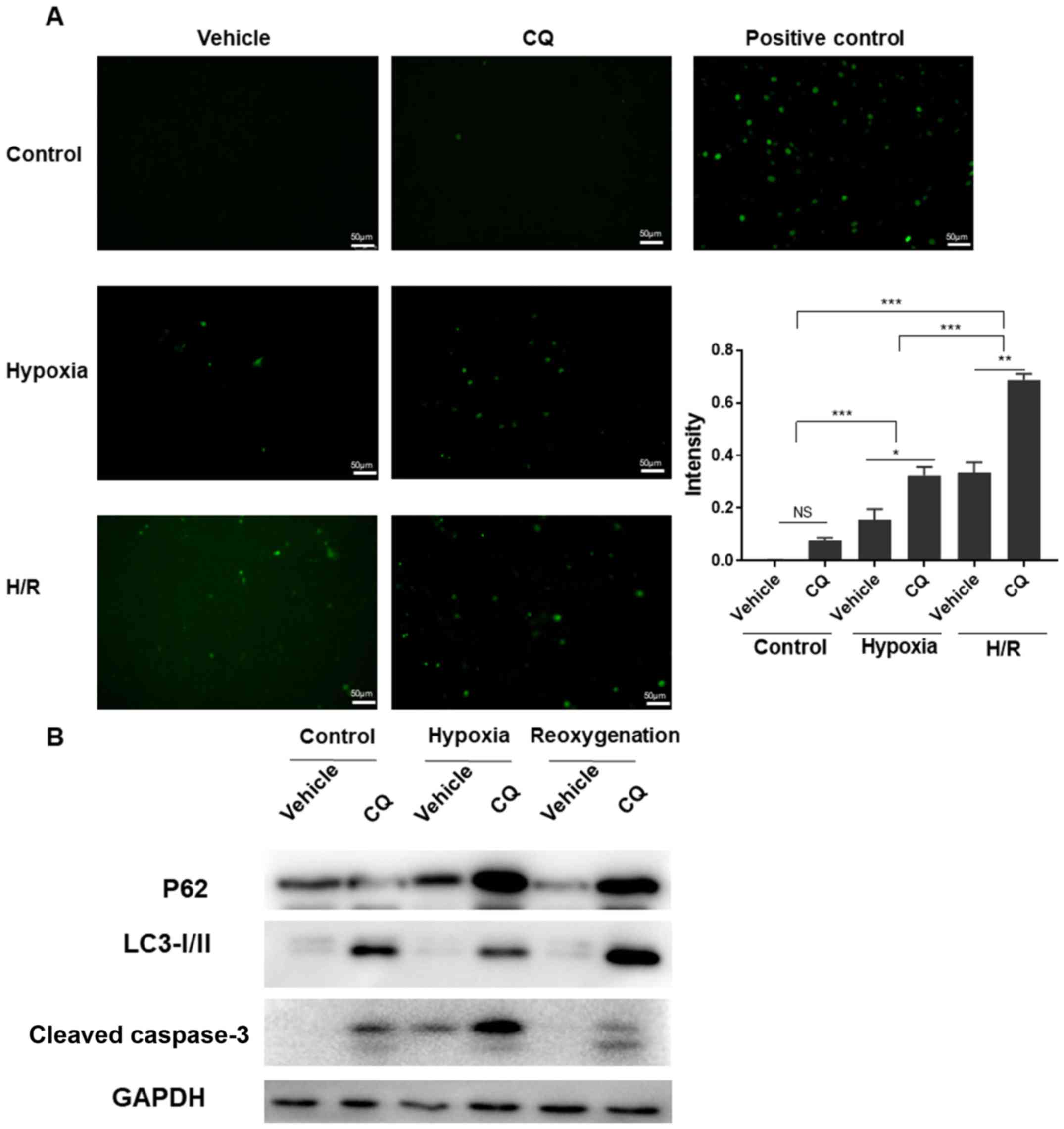
as a model of in vivo I/R conditions. Firstly, TUNEL
staining (Fig. 1A; P<0.05 vehicle
vs. chloroquine in the hypoxia group; P<0.01 vehicle vs.
chloroquine in the H/R group) demonstrated that downregulation of
autophagic flux resulted in more severe apoptosis during H/R
injury. Subsequently, as shown in Fig.
1B, western blot analysis demonstrated that chloroquine
downregulated autophagic flux, while detection of cleaved caspase-3
illustrated that apoptosis was increased under hypoxia and H/R.
Taken together, these results revealed that inhibition of
autophagic flux with chloroquine exacerbated cell apoptosis during
H/R. These in vitro data suggested that upregulation of
autophagic flux may augment I/R injury in mice in order to reduce
the injury severity.
Hindlimb I/R injury is associated with
augmented damage compared with simple ischemic injury
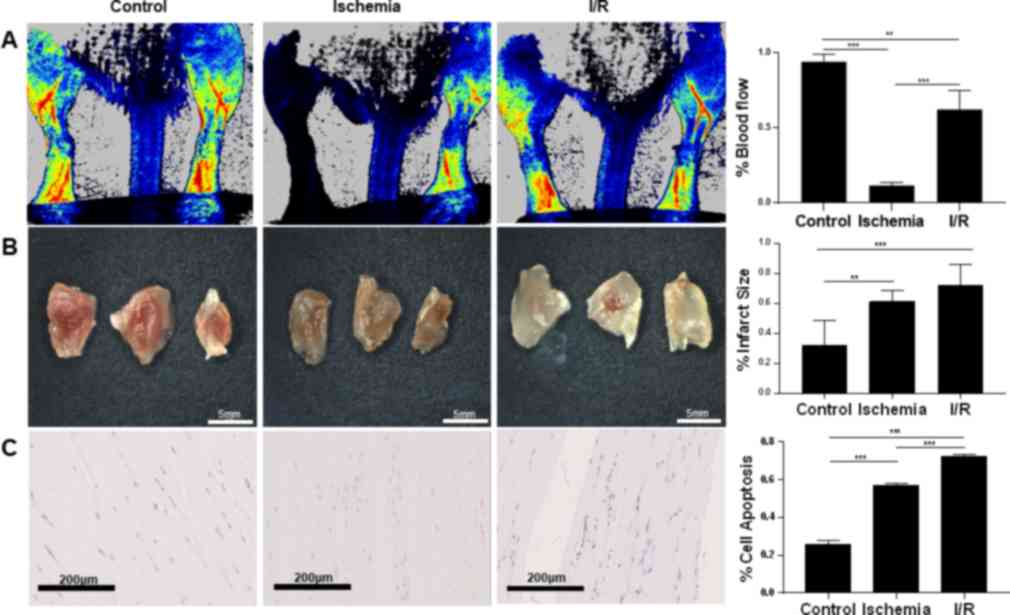
Ischemia and I/R injury murine models were
established according to the aforementioned protocol. Laser Doppler
imaging confirmed that blood flow was reduced by 82% after the
ligation and was restored to 62% of the baseline after the ligation
was removed (P<0.001 control vs. ischemia; P<0.01 control vs.
I/R; P<0.001 ischemia vs. I/R; n=3; Fig. 2A). TTCA demonstrated that murine
gastrocnemius muscles suffered more infarct in the ischemia
(61.16%; P<0.01) and I/R groups (77.44%; P<0.001), compared
with the control group (30.55%; n=3; Fig. 2B). The infarct size ratio was
consistently higher in the I/R group compared with the ischemia
condition, although it did not reach statistical significance.
TUNEL staining demonstrated that the percentage of apoptotic cells
was significantly higher in the ischemia group (57.1%) in
comparison with the control group (25.4%). However, the I/R group
(72.3%) exhibited more apoptotic cells than the ischemia group
(P<0.001 control vs. ischemia; P<0.001 control vs. I/R;
P<0.001 ischemia vs. I/R; n=3; Fig.
2C). Taken together, these results suggested that murine
gastrocnemius muscle in the ischemia group was associated with cell
damage, which was exacerbated in the I/R injury group.
Autophagy is upregulated by ischemia
and downregulated by I/R injury
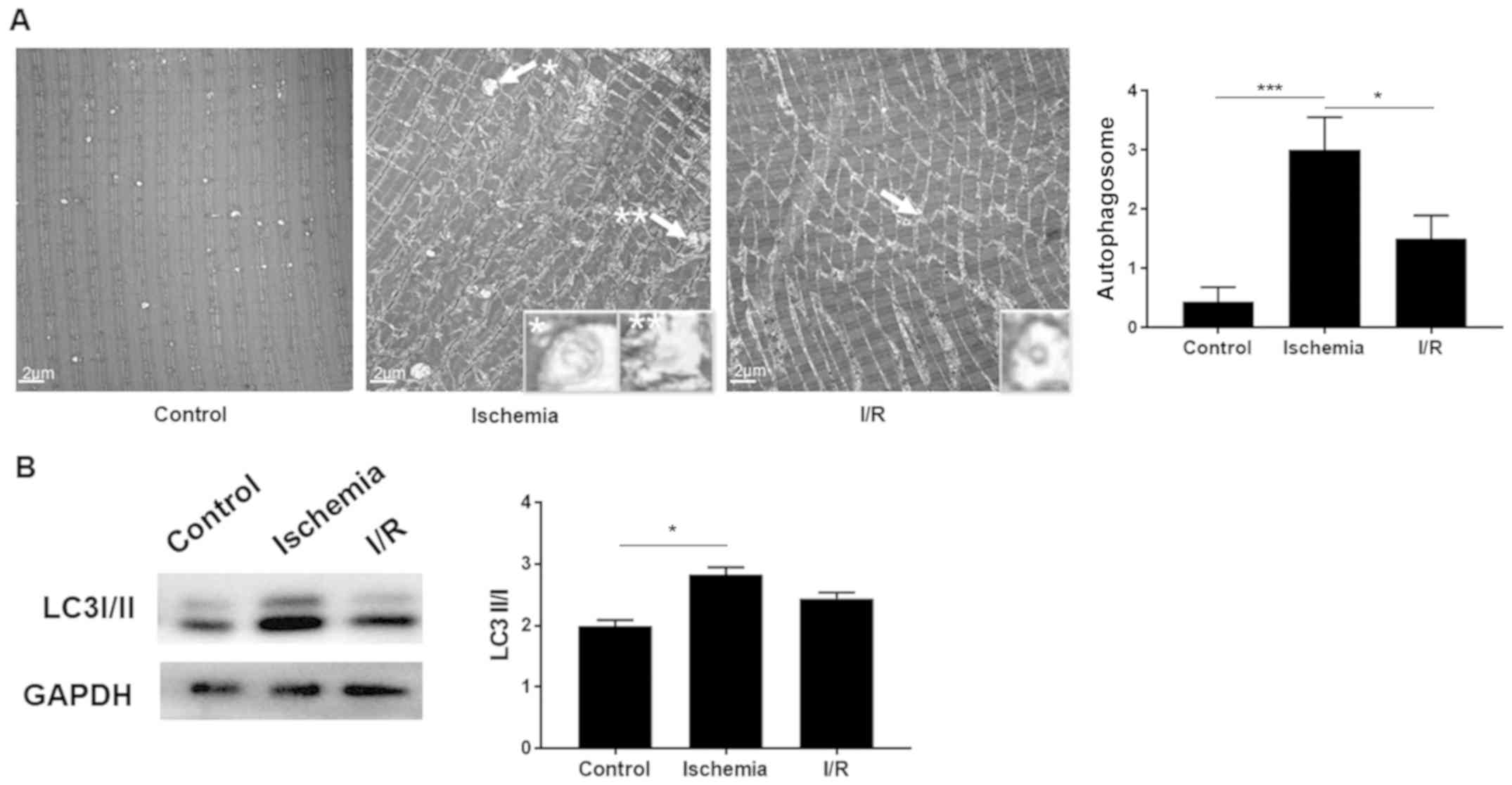
In order to compare the changes in autophagy,
electron microscopy was applied to observe the subcellular
structures. The average number of autophagosomes in the control,
ischemia and I/R groups was 0.4, 3.00 and 1.5 per field,
respectively (P<0.001 control vs. ischemia; P<0.05 ischemia
vs. I/R; n=3; Fig. 3). The amount of
autophagosomes was significantly increased in the ischemia group as
compared with that in the control, whereas it declined in the I/R
group. Subsequently, western blot analysis was used to further
confirm this result. The LC3II/I ratio was significantly increased
in the ischemia group compared with the control (P<0.05),
whereas a decreased ratio was observed in the I/R group in
comparison with the ischemia group, although the difference was not
statistically significant (n=3; Fig.
3B). These results suggest that autophagy was upregulated in
the ischemia group, but subsequently downregulated by I/R injury,
indicating that autophagy was undamaged in skeletal muscle under
I/R conditions.
Rapamycin protects skeletal muscle
during ischemia by autophagy upregulation
Rapamycin is an autophagy inducer that inhibits the
autophagy suppressor mTOR. Considering that long-term
administration of rapamycin would enhance autophagic flux under
ischemic conditions, ischemia alone and rapamycin + ischemia groups
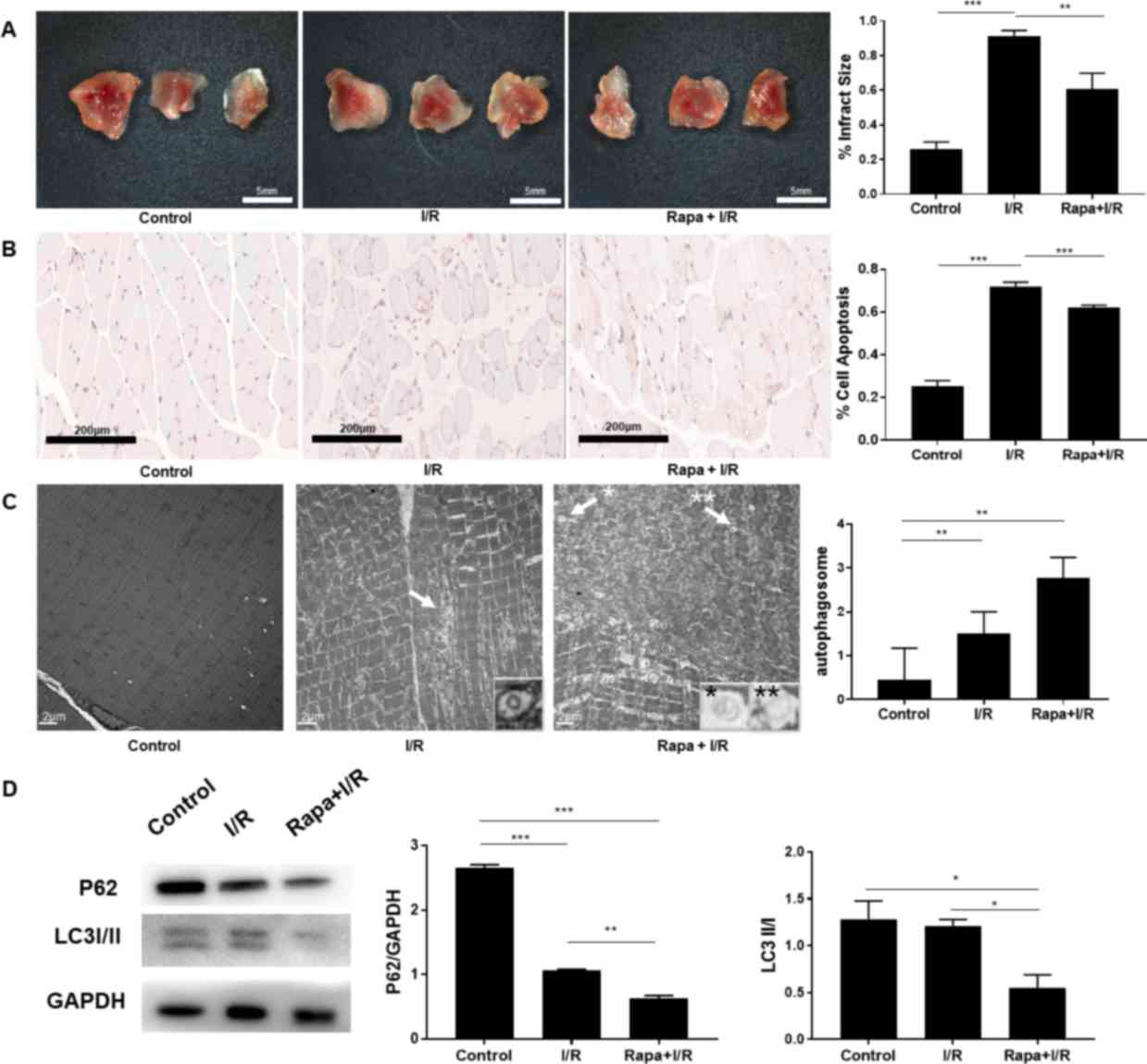
were examined in the animal model. TTCA demonstrated that rapamycin
significantly reduced the infarct area percentage from 80.28 to
52.67% (P<0.01; n=9; Fig. 4A).
The percentage of apoptotic cells, as indicated by TUNEL staining,
was significantly reduced in the rapamycin + ischemia group
(47.77%) compared with that in the ischemia group (58.31%;
P<0.001; n=9; Fig. 4B).
Additionally, electron microscopy and western blot analysis were
performed to study changes in autophagic flux during ischemia. The
average number of autophagosomes was increased from 3.0 to 7.2
subsequent to rapamycin intervention in the ischemia group
(P<0.05; n=9; Fig. 4C).
Consistent with the electron microscopy data, the expression levels
of P62 and LC3, which are two commonly used markers of autophagic
protein turnover, demonstrated that autophagy was significantly
enhanced by rapamycin during ischemia (P<0.01; n=9; Fig. 4D). These results indicated that
rapamycin upregulated autophagy in the ischemic phase and
consequently protected skeletal muscle from injury.
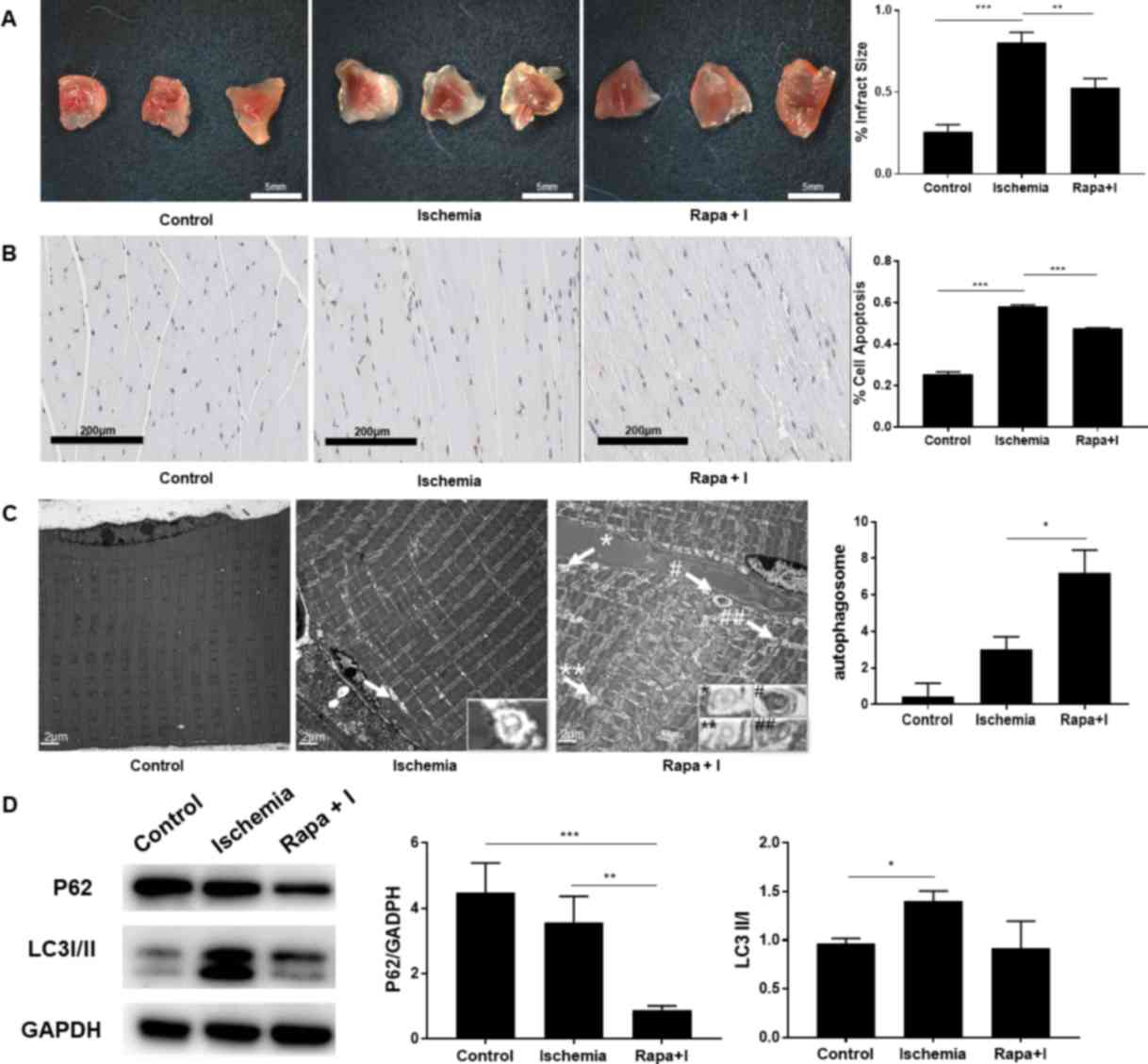 | Figure 4.(A) The ratio of infarct area to gross
area in the Rapa + I group was lower compared with that in the
ischemia group. (n=9 each) (B) The status of apoptosis was improved
in the Rapa + I group, as compared with the ischemia alone group
(n=9 each; magnification, ×100). (C) Electron microscopy revealed
that the autophagosomes (indicated by arrows and symbols) were
increased in the Rapa + I group compared with the ischemia alone
group (n=9 each; magnification, ×10,000). (D) P62 protein was
degraded following the administration of rapamycin in the ischemic
phase, as the formation of autophagosomes was increased, indicated
by LC3II/I expression (n=6 each). *P<0.05, **P<0.01 and
***P<0.001. I, ischemia; Rapa, rapamycin; LC3, microtubule
associated protein 1 light chain 3. |
Rapamycin alleviates skeletal muscle
injury during I/R
To further explore the specific role of autophagy
during I/R, autophagic flux was altered using rapamycin treatment
under I/R conditions. Following the administration of rapamycin,
TTCA demonstrated that the ratio of the infarct size was
significantly decreased (60.89%), compared with the I/R group
(91.29%; P<0.01; n=9; Fig. 5A).
The percentage of apoptotic cells, as indicated by TUNEL staining,
demonstrated that apoptosis was reduced in the rapamycin + I/R
group (62.21%) as compared with the I/R alone group (71.99%;
P<0.001; n=9; Fig. 5B). These
results indicated that rapamycin alleviated I/R injury. Further
western blot analysis revealed the upregulation of autophagy, as
increased turnover of P62 and autophagosome degradation, as
indicated by the changes in LC3II, were observed. Consistently, the
number of autophagosomes was increased by ~2-fold following
administration of rapamycin; however, this change was not
statistically significant (n=9; Fig. 5C
and D).
Discussion
During the ischemic phase, depletion of oxygen and
nutrients leads to upregulation of autophagy (17); however, the status of autophagic flux
in the reperfusion phase remains unclear. Furthermore, the specific
role of autophagy during I/R is controversial. In the current
study, a murine hindlimb I/R injury model was used to demonstrate
that autophagy was enhanced during ischemia, but declined during
reperfusion. Notably, rapamycin treatment upregulated autophagy and
significantly alleviated cell death under I/R conditions,
indicating the protective role of autophagy during I/R injury,
which was likely initiated in the ischemic phase and persisted
through the reperfusion phase.
Recent studies have illustrated that autophagy has a
critical role in I/R injury (3,4,18,19).
However, it raises an important question as to whether autophagy
has a protective or deleterious function during I/R in skeletal
muscle. To address this, in the present study, chloroquine was used
to manipulate autophagic flux in C2C12 myoblast cells under H/R
conditions. Downregulation of autophagy aggravated the apoptosis of
C2C12 cells, while upregulation of autophagy had no significant
effect. The in vitro results indicated that autophagy is
directly associated with apoptosis in I/R conditions. To further
investigate this, rapamycin, a classical autophagy inducer
(20,21), was applied to manipulate the status
of autophagy in mice. As expected, rapamycin intervention
significantly upregulated autophagic flux and alleviated cell death
in the ischemia group, suggesting that enhancement of autophagy has
a protective role in the ischemic phase. Furthermore, rapamycin
treatment significantly alleviated cell death in the I/R group.
Consistently, the number of autophagosomes was increased by ~2-fold
following rapamycin intervention. Taken together, the data
suggested that autophagy is activated during I/R injury in skeletal
muscle, and upregulated autophagy counteracts the induction of
apoptosis, thus protecting skeletal muscle from damage.
Consistent with previous findings (22), the results of the current study
revealed that autophagosomes were increased in the ischemic phase,
as a stress response to nutrient depletion and hypoxia. According
to previous research, during ischemia, damaged mitochondria with
highly reductive conditions increase electron transfer to
O2 in the mitochondrial electron transport chain
(23,24), which consequently increases the
formation of ROS. Hydrogen peroxide, one of the most well-studied
ROS, was reported to regulate autophagosome formation by
inactivating Atg4. In brief, an essential cysteine residue of Atg4
is oxidized by hydrogen peroxide under hypoxic conditions, leading
to accumulation of LC3-phosphatidylethanolamine on the phagophore
membrane and the formation of autophagosomes (24,25). In
return, upregulated autophagy eliminates damaged mitochondria
(mitophagy), which prevents further release of apoptosis-promoting
factors from mitochondria, in order to alleviate cell apoptosis.
According to these aforementioned observations, the findings of the
present study suggest that mitophagy was induced by rapamycin,
which increases the scavenging of damaged mitochondria and ROS, and
consequently alleviates cell apoptosis under I/R conditions. This
Atg4-ROS-mitophagy mechanism may be an important regulatory pathway
during I/R injury, and more experiments are required to investigate
this further.
Growing evidence has illustrated that I/R injury
impairs autophagosome clearance, which leads to accumulation of
autophagosomes and increased cardiomyocyte death in the reperfusion
phase (9,19). Notably, the current study data
demonstrated a decreased number of autophagosomes during the
reperfusion phase in skeletal muscle cells, indicating that
autophagic flux was likely undamaged. This controversial
observation may be due to the multifaceted role of autophagy in
different cell types.
In conclusion, the findings of the present study
illustrated that enhanced autophagy in the ischemic phase protected
murine hindlimb from I/R injury, suggesting that upregulation of
the autophagosome response with rapamycin may be a potential
therapeutic strategy for the treatment of hindlimb I/R injury.
Acknowledgements
The authors would like to thank Dr Zhang Jian from
Zhongshan Ophthalmic Center of SYSU for assistance with statistical
analysis, as well as Dr Zhou Yi and Dr Yating Wang from the First
Affiliated Hospital of SYSU for technical help.
Funding
This study was supported by grants from the Young
Scientists Fund of the National Natural Science Foundation of China
(no. 81300237), the National Natural Science Foundation of China
(no. 81670439), the Doctoral Fund of Ministry of Education of China
(no. 20130171120079) and the Science and Technology Program of
Guangzhou, China (grant nos. 201710010056 and 20158020116). The
funders had no role in study design, data collection and analysis,
decision to publish or preparation of the manuscript.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
All authors have read and approved the submitted
manuscript. CL performed the TUNEL staining and TTCA, wrote the
paper and performed data analysis. MP performed the electron
microscopy studies, I/R modeling and data analysis. LZ performed
the laser Doppler imaging studies, I/R modeling, animal surgery and
data analysis. YZ performed the I/R modeling, animal surgery and
data analysis. RW designed the study concept and performed animal
surgery. QS monitored the animal welfare, and selected the animals
and experimental models. SC and ZL participated in designing the
study concept and experimental design, and wrote the
manuscript.
Ethics approval and consent to
participate
Approval of all animal experiments was obtained from
the Institutional Review Board of the First Affiliated Hospital of
Sun Yat-sen University, in accordance with the National Institutes
of Health Guide for the Care and Use of Laboratory Animals [NIH
Publications no. 8023, revised 1978; permit number, 2017 (35)].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
I/R
|
ischemia/reperfusion
|
|
TTCA
|
triphenyl tetrazolium chloride
assay
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end-labelling
|
References
|
1
|
Dai S, Xu Q, Liu S, Yu B, Liu J and Tang
J: Role of autophagy and its signaling pathways in
ischemia/reperfusion injury. Am J Transl Res. 9:4470–4480.
2017.PubMed/NCBI
|
|
2
|
Tran TP, Tu H, Pipinos II, Muelleman RL,
Albadawi H and Li YL: Tourniquet-induced acute ischemia-reperfusion
injury in mouse skeletal muscles: Involvement of superoxide. Eur J
Pharmacol. 650:328–334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gustafsson AB and Gottlieb RA: Autophagy
in ischemic heart disease. Circ Res. 104:150–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vignaud A, Hourde C, Medja F, Agbulut O,
Butler-Browne G and Ferry A: Impaired skeletal muscle repair after
ischemia-reperfusion injury in mice. J Biomed Biotechnol.
2010:7249142010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
May D, Gilon D, Djonov V, Itin A, Lazarus
A, Gordon O, Rosenberger C and Keshet E: Transgenic system for
conditional induction and rescue of chronic myocardial hibernation
provides insights into genomic programs of hibernation. Proc Natl
Acad Sci USA. 105:282–287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matsui Y, Kyoi S, Takagi H, Hsu CP,
Hariharan N, Ago T, Vatner SF and Sadoshima J: Molecular mechanisms
and physiological significance of autophagy during myocardial
ischemia and reperfusion. Autophagy. 4:409–415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sengupta A, Molkentin JD, Paik JH, DePinho
RA and Yutzey KE: FoxO transcription factors promote cardiomyocyte
survival upon induction of oxidative stress. J Biol Chem.
286:7468–7478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McCormick J, Suleman N, Scarabelli TM,
Knight RA, Latchman DS and Stephanou A: STAT1 deficiency in the
heart protects against myocardial infarction by enhancing
autophagy. J Cell Mol Med. 16:386–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Fang YQ, Xue ZF, He YP, Fang RM and
Li L: Beta-asarone attenuates ischemia-reperfusion-induced
autophagy in rat brains via modulating JNK, p-JNK, Bcl-2 and Beclin
1. Eur J Pharmacol. 680:34–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu S, Hartleben B, Kretz O, Wiech T,
Igarashi P, Mizushima N, Walz G and Huber TB: Autophagy plays a
critical role in kidney tubule maintenance, aging and
ischemia-reperfusion injury. Autophagy. 8:826–837. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J,
et al: FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab. 6:458–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lie JT, Holley KE, Kampa WR and Titus JL:
New histochemical method for morphologic diagnosis of early stages
of myocardial ischemia. Mayo Clin Proc. 46:319–327. 1971.PubMed/NCBI
|
|
17
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sadoshima J: The role of autophagy during
ischemia/reperfusion. Autophagy. 4:402–403. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Kim SG and Blenis J: Rapamycin: One
drug, many effects. Cell Metab. 19:373–379. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ryter SW, Lee SJ, Smith A and Choi AM:
Autophagy in vascular disease. Proc Am Thorac Soc. 7:40–47. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nohl H and Jordan W: The mitochondrial
site of superoxide formation. Biochem Biophys Res Commun.
138:533–539. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Becker LB, vanden Hoek TL, Shao ZH, Li CQ
and Schumacker PT: Generation of superoxide in cardiomyocytes
during ischemia before reperfusion. Am J Physiol. 277:H2240–H2246.
1999.PubMed/NCBI
|
|
25
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|