Introduction
Danon disease is a rare X-linked disorder clinically
characterized by a triad of cardiomyopathy, intellectual impairment
and skeletal myopathy (1). Danon
disease is caused by genetic defects in the lysosome-associated
membrane protein 2 (LAMP2) gene, which encodes the LAMP2 protein
(2). Cardiac involvement is
prominent in patients with Danon disease, while left ventricular
hypertrophy and dilation may be present with the former being more
prevalent in males (2,3). Cardiac symptoms usually occur in
adolescence and during the third decade of life, while most
patients die from heart failure. Electrical conduction
abnormalities are also common, presenting in >3/4 of males and
females with Danon disease. Specifically, ventricular
pre-excitation is the most common electrocardiographic pattern
(3).
Danon disease is an inherited disease that may be
passed on to the next generation. The present study reports on two
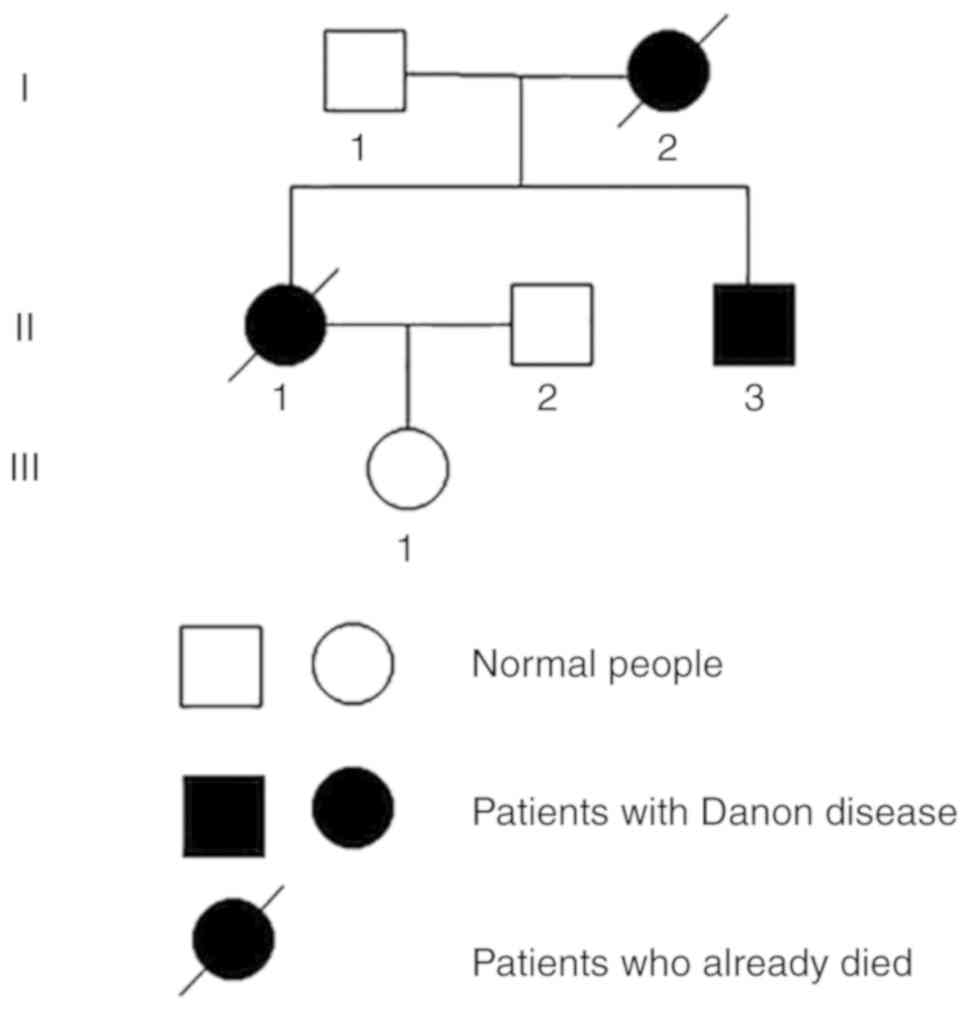
cases of Danon disease in a single family with distinctly different
phenotypes. In this pedigree, three generations involving 6 family
members were assessed, including one case of sudden death (Patient
I:2), and two cases of cardiomyopathy (Patients II.1 and II.3).
Patients II.1 and II.3 developed atrial fibrillation (AF) in the
later course of the disease, which is not common. In addition, a
literature review was performed by searching PubMed on the aspects
of clinical course, phenotype-genotype association, optimal
management and prognosis of Danon disease.
Case report
Patient II.1
Patient II.1 was the older child of the family, the
history of which comprised sudden cardiac death in the patient's
mother of 47 years old and hypertrophic cardiomyopathy (HCM) in the
patient's brother (Patient II.3). The pedigree chart is provided in
Fig. 1. Patient II.1, a 26-year-old
female, was initially diagnosed with dilated cardiomyopathy (DCM)
based on echocardiographic examination in a local hospital several
months previously and then gradually developed aggravated shortness
of breath and edema of the bilateral lower limbs. She was referred
to Department of Cardiology, Shijiazhuang Great Wall Hospital of
Integrated Traditional Chinese and Western Medicine in March 2016.
On physical examination, the patient was hemodynamically stable,
with positive hepatojugular reflex and edema in the lower limbs.
Pertinent laboratory parameters included elevated lactate
dehydrogenase (344.09 U/l; normal range, 120–250 U/l), elevated
α-hydroxybutyrate dehydrogenase (388.96 U; normal range, 72–182
U/l), elevated N-terminal pro-brain natriuretic peptide (4,212
pg/ml; normal range, 0–450 pg/ml), elevated aspartate transferase
(49.81 U/l; normal range, 13–35 U/l) and elevated
γ-glutamyltranspeptidase (135.14 U/l; normal range, 7–45 U/l). The
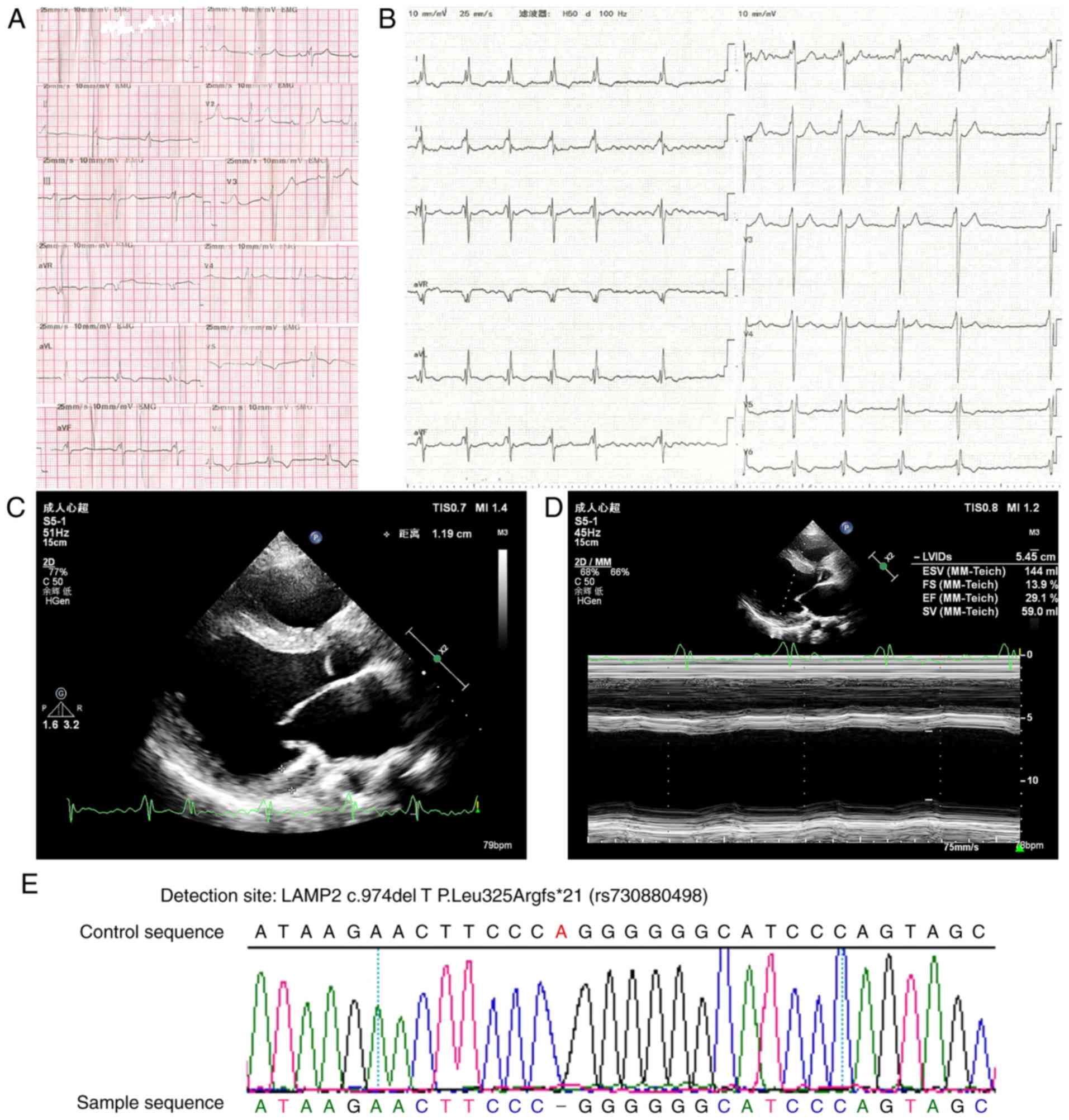
electrocardiogram (ECG) exhibited a ventricular pre-excitation
pattern at the first hospitalization (Fig. 2A). At nine months later, the patient
was admitted due to advanced heart failure with persistent AF
(Fig. 2B). The echocardiographic
examination revealed mild left ventricular hypertrophy
(interventricular septum, 13 mm; left ventricle anterior wall, 12
mm) and dilated left heart (atrium, 37 mm; left ventricle at end
diastole, 63 mm) (Fig. 2C). The left
ventricular systolic function was severely impaired with an
ejection fraction of 29% (Fig.
2D).
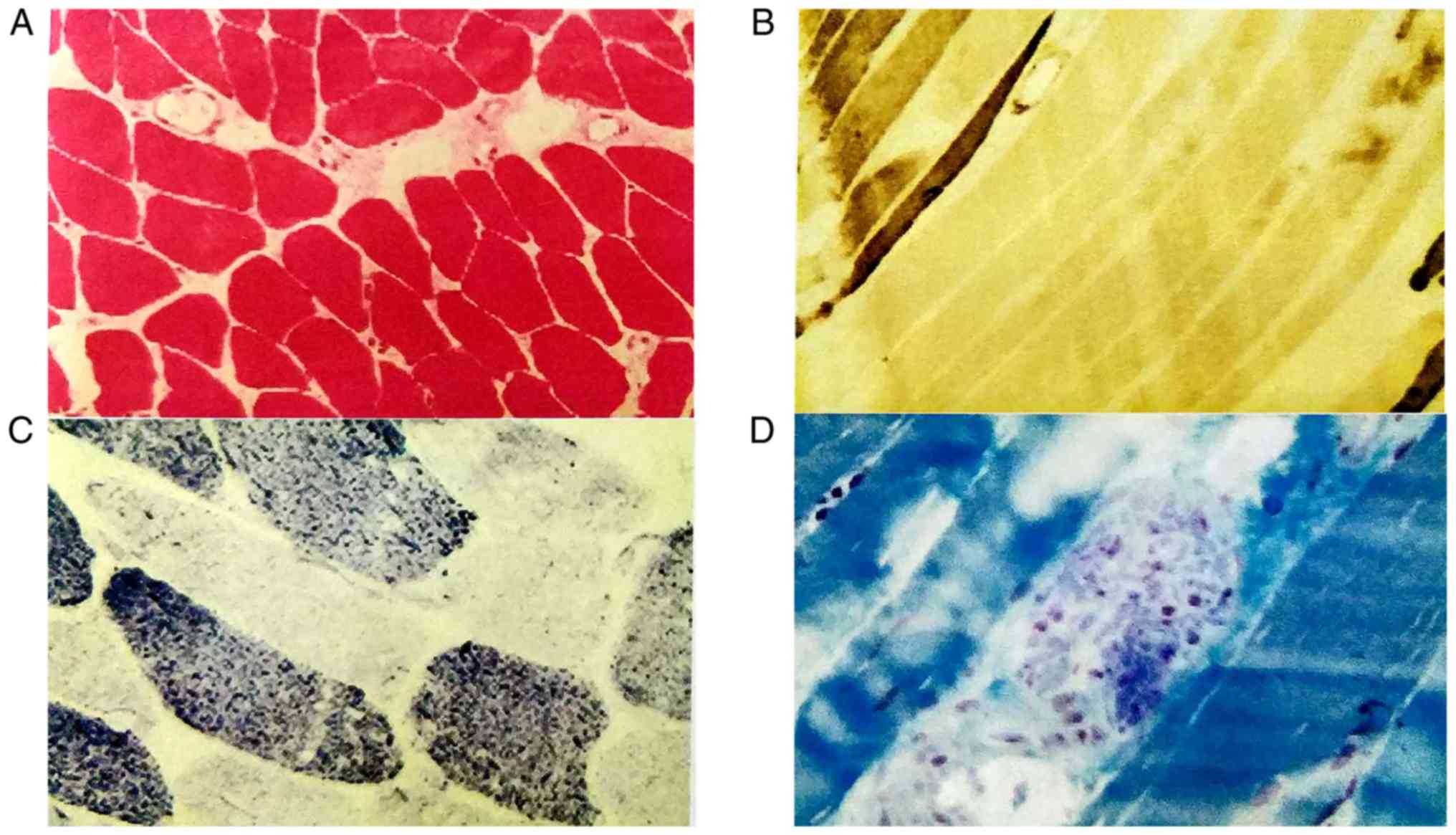
Biopsy specimens were taken from the patient's left
deltoid muscle. NADH enzyme staining indicated that small irregular
vacuoles were scattered in certain muscle fibers, and a small
amount of type I muscle fiber aggregated in edge particles
(Fig. 3C). ATPase staining revealed
clustering in some areas (Fig. 3B).
The pathological diagnosis was myogenic fiber disorder myopathy
(Fig. 3).
For sequence analysis, DNA was extracted from blood.
A heterozygous frameshift missing mutation was identified in the
LAMP2 gene of the patient. The proband harbored an identical
c.974delTinsAA in exon 8 (Fig.
2E).
Collectively, the patient was admitted to the
hospital 6 times for heart failure decompensation within 2 years
and experienced sudden cardiac death during her last
hospitalization at the age of 28 years.
Patient II.3
Patient II.3, the younger brother of Patient II.1,
was a 23-year-old male presenting with a distinctly different
clinical course and initial manifestation. He was initially
referred to a local hospital due to an unexplained elevation of
cardiac enzyme in April 2016, one year before his referring to our
department, and dyspnea and weakness followed. Clinical examination
revealed hepatomegaly with no obvious edema. He reported mild
fatigue during physical activity. Abnormalities in laboratory
parameters included serum procarbamide transaminase (155 U/l;
normal range, 9–50 U/l), serum aspartate aminotransferase (215 U/l;
normal range, 15–40 U/l), serum creatine kinase (945 U/l; normal
range, 24–194 U/l), serum creatine kinase isoenzyme (45.40 U/l;
normal range, 0–24 U/l), serum α-hydroxybutyrate dehydrogenase (711
U/l; normal range, 24–194 U/l) and serum γ-glutamyl transpeptidase
(72 U/l; normal range, 10–60 U/l). The chest X-ray revealed mild
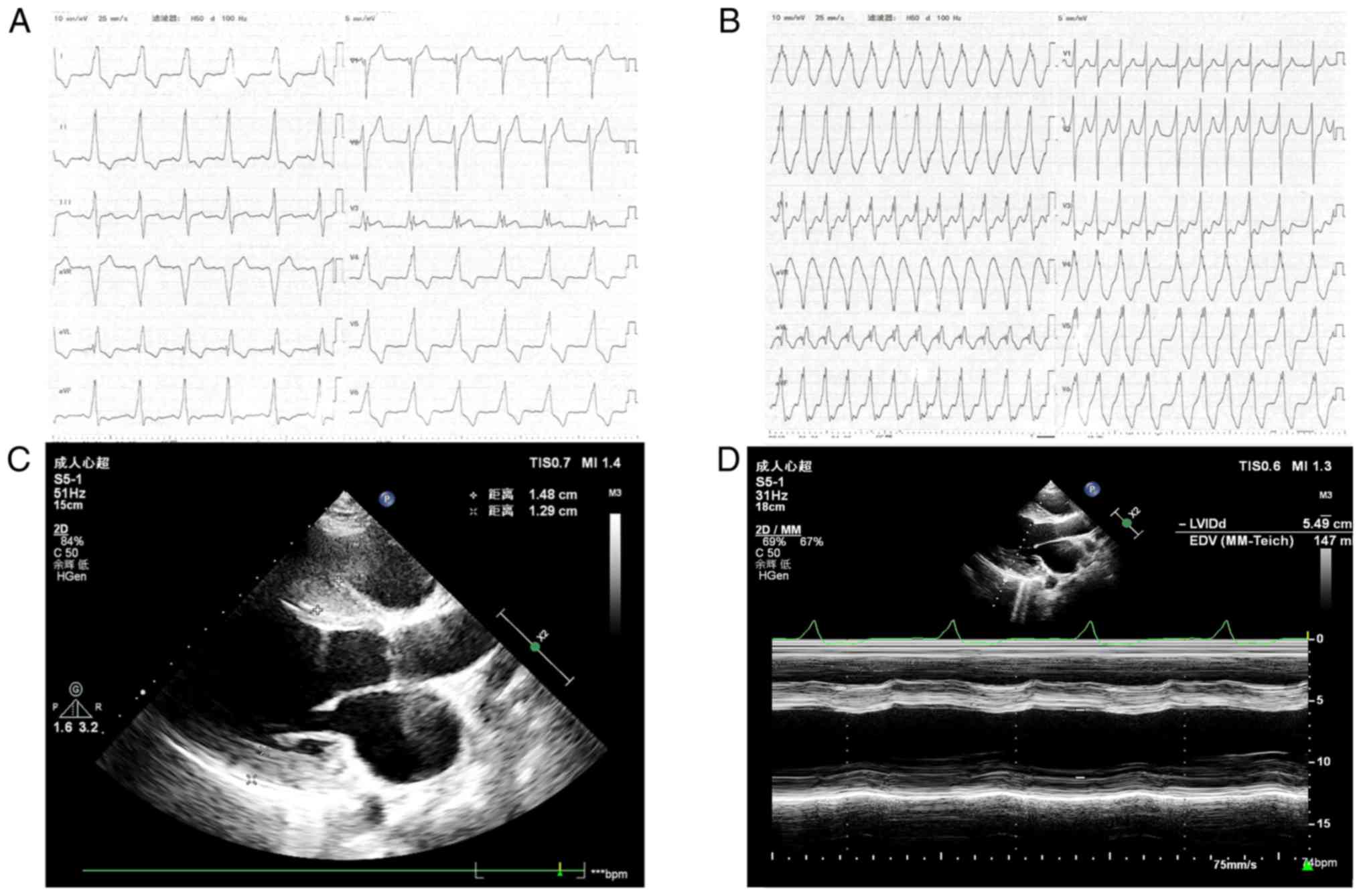
cardiomegaly. A ventricular pre-excitation pattern was evident in
the ECG (Fig. 4A). The ECG
examination revealed interventricular septum hypertrophy of 15 mm
and diffuse thickening of the left ventricular wall (Fig. 4C). The patient had a decreased left
ventricular systolic function with an ejection fraction of 37%
(Fig. 4D). Pathological diagnosis
was also myogenic fiber disorder myopathy, similar to that in
patient II.1 but with more obvious vacuoles in fibers.
DNA sequence analysis was also performed using a
sample extracted from an oral swab. An identical mutation to that
in patient II.1 was detected; the only difference was in the
homozygous presentation.
Over one year, patient II.3 was admitted to the
hospital 3 times for heart failure decompensation, while pertinent
laboratory results and cardiac function estimated by ECG did not
indicate any significant alterations. The patient developed AF
(Fig. 4B) during his second
hospitalization and ~1 year later his sinus rhythm was restored
following catheter radiofrequency ablation. At the time of writing
of the manuscript, one year after the intervention, the patient
remained in a relatively stable state.
Other family members
Patients I.1 and III.1 had normal psychomotor
development and no exercise intolerance. The LAMP2 gene mutation
was not identified in these two family members. Patient I.2, the
mother of Patient II.1, suffered a sudden cardiac death without an
accurate diagnosis. Patient II.2 was normal with no sign of mental
or physical disorders.
Discussion
The present study described 2 cases of Danon disease
in a single family, which clearly differed in terms of severity of
the underlying cardiomyopathy. In previous cases, AF has not been
widely described to develop after progression of the disease, as in
the two patients of the present study. In the subsequent section, a
review of the literature is provided and the epidemiology,
manifestations, cardiac electrical abnormalities, imaging
examinations and genetics of Danon disease are discussed.
The literature search was performed using PubMed to
identify articles and case reports without any time limitation to
identify articles and case reports on Danon disease. The search
terms were a combination of ‘Danon disease’, ‘lysosomal glycogen
storage disease without acid maltase deficiency’ and ‘glycogen
storage disease IIb’. First, the epidemiology of Danon disease was
reviewed. Of note, the majority of published data on Danon disease
originate from case reports and the prevalence of this disease is
undetermined. A recent study reported a prevalence of 1.5% in a
cohort with unexplained left ventricular hypertrophy, rising to 30%
in patients with unexplained left ventricular hypertrophy and
pre-excitation on ECG (4). Almost
all patients had the manifestation of cardiomyopathy, with males
predominantly presenting with a HCM phenotype, while females
presented with HCM or DCM equally (3). It is noteworthy that patients with
idiopathic HCM most commonly exhibit preserved systolic function
and better prognosis (5). However,
patients with Danon disease presenting with an HCM pattern and
manifesting with preserved systolic function early in the course of
disease may later progress with a worse prognosis. One study
identified LAMP2 mutations in 4 of 64 patients with HCM, while no
LAMP2 mutation was detected in 72 patients with DCM (6). In another study of an affected family,
contrary to common sense, DCM was predominant in male patients
(7). Of note, the prevalence of
diagnosed cases of Danon disease may be rising due to increased
detection based on the expanding knowledge of this rare
disease.
The clinical features of Danon disease were then
reviewed. Danon disease has been increasingly reported, mostly as
case reports, during the past few years. To the best of our
knowledge, female patients with Danon disease mostly present with
mild symptoms and a later onset (13.3±8.0 years of age for male
patients and 28.9±14.2 years for female patients; P=0.0008)
(3). Of note, the female patient of
the present study manifested with an early onset as a female and
had an unfavorable prognosis. Indeed, the phenotypes in the
different genders are distinctly different given that males are
homozygous and females are heterozygous, presumably due to the gene
dosage difference with haploinsufficiency and skewed X-chromosome
inactivation (8–10). The clinical course in female patients
with Danon disease may have a broad spectrum. The youngest female
patient reported had an onset at the age of 12 years (11). In addition, a 38-year-old female,
whose son was diagnosed with Danon disease based on genetic
analysis, harbored the same mutation as her son and was
asymptomatic with no specific abnormalities in the ECG, physical
examination and blood biochemistry (12). Regarding the variety and severity of
cardiomyopathy symptoms, Hedberg Oldfors et al (13) reported that an uneven distribution of
LAMP2 protein causing deleterious effects in the myocardium may
have a more significant role than an overall moderate reduction of
LAMP2 protein. Cardiomyopathy is present in almost all patients
with Danon disease. Of note, the two probands of the present study
did not exhibit any clinical indication of skeletal myopathy or
intellectual disability. Due to lack or decrease of LAMP2 protein
in all body parts of the patients, certain less prevalent symptoms
may also occur. In the eye, LAMP2 is expressed only in the retinal
pigment epithelium (14), and
retinal abnormalities are estimated to occur in 64–69% of patients
(3). Hepatic disease has also been
described in patients with Danon disease (2,15).
Autism associated with Danon disease was reported in a 16-month-old
male with normal presentation on cardiac evaluation (16). All available evidence indicates that
Danon disease is a multi-system disease and therefore, attention
should be paid to symptoms of systems other than cardiovascular
system in order to avoid a missed diagnosis of this lethal disease.
Regarding cardiac electrical abnormalities, a ventricular
pre-excitation pattern was evident in the patients, but no
electrophysiological evaluation was performed. Ventricular
pre-excitation is the most frequent abnormality on ECG in this
setting and it has been demonstrated in 68% of males and 27% of
females (3). However, in a recent
study, pre-excitation was identified in only 11 out of 27 patients
with Danon disease (17). Of note,
in a study that performed an electrophysiologic evaluation of 4
male patients with Danon disease and initial QRS complex mimicking
ventricular pre-excitation on ECG, no accessory pathway was
identified (18). On the other hand,
a fasciculoventricular pathway was discovered on electrophysiology
examination in a 13-year-old male with Danon disease from Spain in
2008 (19). These results were
consistent with those of a further case (20). In addition, a recently published
study demonstrated that a fasciculoventricular pathway was present
in 2 out of 3 patients with Danon disease, with no
electrophysiology results available in the one remaining patient
(4). According to the aforementioned
studies focusing on pre-excitation, a fasciculoventricular pathway
may represent a hallmark of Danon disease.
The present study provided electrocardiographic
evidence of AF in the Patient II.1 and Patient II.3 during the
progression of the disease. At present, available data regarding AF
in Danon disease are limited. D'Souza et al (20) reported on a 14-year-old male
diagnosed with Danon disease who presented with persistent
palpitations at the age of 8 years. He had Wolff-Parkinson-White
syndrome, brief runs of atrial tachycardia and pre-excited AF, and
experienced sudden cardiac death at the age of 14. Konrad et
al (18) observed AF in 5 out of
7 patients with Danon disease from 3 families and hypothesized that
AF may be associated with thromboembolic events in young
individuals, which is supported by another three cases reported
previously (21). In the patients of
the present study, AF developed and aggravated the heart failure.
There are only few reports of AF in Danon disease, and it may
indicate a worse prognosis of this disease. Therefore, it is
important to pay attention to the development of AF in patients
when the diagnosis is already certain. However, whether aggressive
treatment of AF in this setting favorably affects the evolution of
heart failure remains elusive. Of note, AF ablation in Patient II.3
of the present study effectively restored the sinus rhythm and
improved his general condition.
The genetics associated with Danon disease were then
reviewed. The LAMP2 c.974delT mutation identified in the cases of
the present study is a frameshift mutation in exon 8 of the LAMP2
gene that was first reported by Nishino et al (1). Genotype-phenotype associations appear
to have a significant role in age of onset of Danon disease.
Nonsense, frameshift and large deletion/duplication mutations were
associated with the earliest age of onset (22). In the family described in the present
study, the mother of Patient II.1 suffered sudden death at the age
of 47, respectively, much later than Patient II.1, and not in
accordance with the early-onset pattern of a frameshift mutation.
Bertini et al (23) reported
that the same mutation of the LAMP2 gene resulted in various
phenotypes. Although the mutation type significantly affected the
phenotype, there are other factors contributing to the final
phenotype, which may explain for the heterogeneity of phenotypes.
Danon disease is caused by mutation of the LAMP2 gene; however, in
a previous study, a patient with normal DNA sequencing results
presented with symptoms of Danon disease, and a LAMP2
microduplication was confirmed by PCR and RT-PCR analyses (24). From the above, it is apparent that
genotype-phenotype associations summed up previously may not apply
in numerous cases.
Regarding imaging examination and treatment, ECG,
particularly three-dimensional speckle-tracking imaging (25), and cardiac magnetic resonance
(26) have a significant role in the
diagnosis and assessment of Danon disease, as reported in numerous
cases (25,27). The final treatment of Danon
cardiomyopathy is heart transplantation, while applying a left
ventricular assist device at the appropriate time is a crucial
management method prior to heart transplantation (28). Implantation of a cardioverter
defibrillator is also a useful treatment, and subcutaneous
implantable cardioverter defibrillators have the advantage of
terminating ventricular tachycardia or fibrillation much better
than transvenous implantable cardioverter defibrillators (29).
In conclusion, the present study reported on a
frameshift mutation in the LAMP2 gene in 2 siblings with
cardiomyopathy. For patients with DCM or HCM, elevated serum
myocardial enzyme and serum liver enzyme, a pre-excitation in the
ECG, fibrosis or dynamic abnormities on ECG examination or cardiac
magnetic resonance imaging, retinopathy myopathy and intellectual
impairment, particularly with an X-linked family history, point to
the diagnosis of Danon disease. Muscle biopsy and genetic mutation
analysis are necessary to make a definite diagnosis. The
development of AF may represent a significant feature of Danon
disease. The prognostic significance of AF and its impact on the
management of patients with Danon disease requires further
study.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TL, FH, SG and LZ contributed to the writing and
preparation of the manuscript. RW, ZL, HX and BH collected the
clinical and imaging data. SG, LZ, RW and PK performed the
literature review. SG, LZ, FH and TL were major contributors in
writing and revising the manuscript. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patients provided written informed consent;
however, the authors made efforts to remove identifying information
to protect the privacy of the patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nishino I, Fu J, Tanji K, Yamada T,
Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al: Primary
LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and
myopathy (Danon disease). Nature. 406:906–910. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sugie K, Yamamoto A, Murayama K, Oh SJ,
Takahashi M, Mora M, Riggs JE, Colomer J, Iturriaga C, Meloni A, et
al: Clinicopathological features of genetically confirmed danon
disease. Neurology. 58:1773–1778. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boucek D, Jirikowic J and Taylor M:
Natural history of Danon disease. Genet Med. 13:563–568. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y, Wang F, Chen X, Liang Y, Deng H,
Liao H, Rao F, Wei W, Zhang Q, Zhang B, et al: Fasciculoventricular
pathways responsible for ventricular preexcitation in patients with
danon disease. Circ Arrhythm Electrophysiol. 11:e0067042018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rowin EJ, Maron BJ, Kiernan MS, Casey SA,
Feldman DS, Hryniewicz KM, Chan RH, Harris KM, Udelson JE, DeNofrio
D, et al: Advanced heart failure with preserved systolic function
in nonobstructive hypertrophic cardiomyopathy: Under-recognized
subset of candidates for heart transplant. Circ Heart Fail.
7:967–975. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fu L, Luo S, Cai S, Hong W, Guo Y, Wu J,
Liu T, Zhao C, Li F, Huang H, et al: Identification of LAMP2
mutations in early-onset danon disease with hypertrophic
cardiomyopathy by targeted next-generation sequencing. Am J
Cardiol. 118:888–894. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Taylor MR, Ku L, Slavov D, Cavanaugh J,
Boucek M, Zhu X, Graw S, Carniel E, Barnes C, Quan D, et al: Danon
disease presenting with dilated cardiomyopathy and a complex
phenotype. J Hum Genet. 52:830–835. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dougu N, Joho S, Shan L, Shida T, Matsuki
A, Uese K, Hirono K, Ichida F, Tanaka K, Nishino I and Inoue H:
Novel LAMP-2 mutation in a family with Danon disease presenting
with hypertrophic cardiomyopathy. Circ J. 73:376–380. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arad M, Maron BJ, Gorham JM, Johnson WH,
Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman
CE and Seidman JG: Glycogen storage diseases presenting as
hypertrophic cardiomyopathy. N Engl J Med. 352:362–372. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Majer F, Vlaskova H, Krol L, Kalina T,
Kubanek M, Stolnaya L, Dvorakova L, Elleder M and Sikora J: Danon
disease: A focus on processing of the novel LAMP2 mutation and
comments on the beneficial use of peripheral white blood cells in
the diagnosis of LAMP2 deficiency. Gene. 498:183–195. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sugie K, Yoshizawa H, Onoue K, Nakanishi
Y, Eura N, Ogawa M, Nakano T, Sakaguchi Y, Hayashi YK, Kishimoto T,
et al: Early onset of cardiomyopathy and intellectual disability in
a girl with Danon disease associated with a de novo novel mutation
of the LAMP2 gene. Neuropathology. 36:561–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen XL, Zhao Y, Ke HP, Liu WT, Du ZF and
Zhang XN: Detection of somatic and germline mosaicism for the LAMP2
gene mutation c.808dupG in a Chinese family with danon disease.
Gene. 507:174–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hedberg Oldfors C, Mathe G, Thomson K,
Tulinius M, Karason K, Östman-Smith I and Oldfors A: Early onset
cardiomyopathy in females with Danon disease. Neuromuscul Disord.
25:493–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schorderet DF, Cottet S, Lobrinus JA,
Borruat FX, Balmer A and Munier FL: Retinopathy in Danon disease.
Arch Ophthalmol. 125:231–236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Danon MJ, Oh SJ, DiMauro S, Manaligod JR,
Eastwood A, Naidu S and Schliselfeld LH: Lysosomal glycogen storage
disease with normal acid maltase. Neurology. 31:51–57. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Burusnukul P, de Los Reyes EC, Yinger J
and Boué DR: Danon disease: An unusual presentation of autism.
Pediatr Neurol. 39:52–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
López-Sainz A, Salazar-Mendiguchía J,
Garcia-Alvarez A, Campuzano Larrea O, Lopez-Garrido MÁ,
Garcia-Guereta L, Fuentes Cañamero ME, Climent Payá V, Pena-Peña
ML, Zorio-Grima E, et al: Clinical findings and prognosis of danon
disease. An analysis of the Spanish multicenter danon Registry. Rev
Esp Cardiol (English, Spanish). 72:479–486. 2018. View Article : Google Scholar
|
|
18
|
Konrad T, Sonnenschein S, Schmidt FP,
Mollnau H, Bock K, Ocete BQ, Münzel T, Theis C and Rostock T:
Cardiac arrhythmias in patients with danon disease. Europace.
19:1204–1210. 2017.PubMed/NCBI
|
|
19
|
García Seara FJ, Martínez Sande JL, Cid
Alvarez B and González Juanatey JR: Wolff-Parkinson-White's
syndrome and danon's disease. Med Clin (Barc) (Spanish).
130:2772013. View
Article : Google Scholar
|
|
20
|
D'Souza RS, Mestroni L and Taylor MRG:
Danon disease for the cardiologist: Case report and review of the
literature. J Community Hosp Intern Med Perspect. 7:107–114. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Spinazzi M, Fanin M, Melacini P,
Nascimbeni AC and Angelini C: Cardioembolic stroke in danon
disease. Clin Genet. 73:388–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
D'Souza RS, Levandowski C, Slavov D, Graw
SL, Allen LA, Adler E, Mestroni L and Taylor MR: Danon disease:
Clinical features, evaluation, and management. Circ Heart Fail.
7:843–849. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bertini E, Donati MA, Broda P, Cassandrini
D, Petrini S, Dionisi-Vici C, Ballerini L, Boldrini R, D'Amico A,
Pasquini E, et al: Phenotypic heterogeneity in two unrelated Danon
patients associated with the same LAMP-2 gene mutation.
Neuropediatrics. 36:309–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lines MA, Hewson S, Halliday W, Sabatini
PJ, Stockley T, Dipch AI, Bowdin S and Siriwardena K: Danon disease
due to a novel LAMP2 microduplication. JIMD Rep. 14:11–16. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miani D, Nucifora G, Piccoli G, Proclemer
A and Badano LP: Incremental value of three-dimensional strain
imaging in danon disease. Eur Heart J Cardiovasc Imaging.
13:8042012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piotrowska-Kownacka D, Kownacki L, Kuch M,
Walczak E, Kosieradzka A, Fidzianska A and Krolicki L:
Cardiovascular magnetic resonance findings in a case of danon
disease. J Cardiovasc Magn Reson. 11:122009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Le DD, Alvarez P, Barrios R, Arriaga M,
Cherry M, Shah D and Lin CH: Hypertrophic cardiomyopathy with
unusual extensive scarring pattern: Danon disease. Methodist
DeBakey Cardiovasc J. 12:227–229. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kitahara H, Nawata K, Kinoshita O, Itoda
Y, Shintani Y, Fukayama M and Ono M: Implantation of a left
ventricular assist device for danon cardiomyopathy. Ann Thorac
Surg. 103:e39–e41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zaki A, Zaidi A, Newman WG and Garratt CJ:
Advantages of a subcutaneous implantable cardioverter-defibrillator
in LAMP2 hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol.
24:1051–1053. 2013. View Article : Google Scholar : PubMed/NCBI
|