Introduction
Traumatic brain injury (TBI) is of significant
public concern since it affects people in all demographics. TBI is
usually induced by the occurrence of mechanical forces to the head,
which can be followed by loss of consciousness. The severity,
pathogenesis, and treatment outcomes of brain trauma are
case-dependent. Brain trauma causes millions of deaths worldwide
with a tremendous life and economic cost each year (1). Current studies associate TBI with a
variety of chronic degenerative diseases, such as Alzheimer's and
Parkinson's disease (2). It is
necessary to understand the pathological mechanisms of TBI in order
to improve the clinical therapeutic strategies for the benefit of
patients.
Inflammation is one of the most prominent reactions
associated with TBI. When TBI occurs, inflammation is caused by
macrophages, microglia and other immune cells, such as monocytes
and T cells, in the central nervous system. In brief, macrophage
and microglia in the injured area quickly respond to the injury by
sending damage-associated molecular patterns leading to local
secretion of cytokines and chemokines (3). These cytokines and chemokines then
recruit immune cells such as neutrophils (4,5), which
infiltrate injured brain tissues and further promote the secretion
of cytokines and cause oxidative stress (6,7). The
exact mechanisms of TBI pathogenesis remain undetermined,
therefore, one area of focus is the study of the association
between neurodegeneration and inflammation, especially modulating
traumatic injury, through the adoption of immunosuppressants. The
inflammatory responses in TBI are required for healing and
defending the body against pathogens; however prolonged
inflammation in the injured brain can cause secondary damage.
Therefore anti-inflammatory drugs, including corticosteroids,
nonsteroidal anti-inflammatory drugs and cytokine inhibitors, have
been explored for treating TBI (8)
with a significant portion of these treatments able to reduce
symptoms and improve brain function.
Dexmedetomidine is an agonist that selectively
functions via targeting the 2-adrenergic receptor in the nervous
system. In a clinical setting, dexmedetomidine is widely employed
for sedating patients who need mechanical ventilation within 24 h
of incidence (9). Dexmedetomidine
can modulate the secretions of catecholamines in patients through
interacting with the 2-adrenergic receptor. Current studies have
also identified the multifaceted protective functions of
dexmedetomidine against inflammation (10–12).
In vitro studies have demonstrated that dexmedetomidine
significantly reduced lipopolysaccharide (LPS)-induced secretion of
pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α,
interleukin (IL)-6 and IL-8 (13).
In clinical practice, studies have illustrated similar
anti-inflammatory effects although inconsistencies between studies
exist (13–18). Dexmedetomidine also reduces patients'
need for anesthetic treatment (19),
and can reduce the blood pressure and heart rate of patients in a
dose-dependent manner (20).
Injection of dexmedetomidine prior to surgery can reduce the
consumption of oxygen in the intra-operative and post-operative
periods (21). In procedural
sedation, dexmedetomidine is used for the sedation of non-intubated
patients in procedures such as awake carotid endarterectomy and
vitreoretinal surgery (22–24). In pediatric practice, dexmedetomidine
is also reported to have different clinical sedation effects,
although the Food and Drug Administration has not approved it for
the pediatric population (25). For
example, one study demonstrated that dexmedetomidine could be
employed as a primary sedation agent during cardiac catheterization
in infants and children (26). In
addition, studies have indicated that dexmedetomidine can modulate
the immune system; for example, dexmedetomidine protected renal
functions in acute kidney injury patients by regulating
inflammatory cytokines, such as TNF-α (27), and also demonstrated the ability to
reduce the secretion of IL-6, IL-8 and TNF-α in critically septic
patients (28). Finally, it has been
determined that dexmedetomidine treatment reduced secretion of
TNF-α and IL-6 both in plasma and bronchoalveolar lavage fluid of
septic mice and inhibited the mRNA expression of toll-like receptor
4 and myeloid differentiation primary response 88 (29).
In our clinical practice, a decreased level of
inflammatory cytokines has been identified in TBI patients
following dexmedetomidine treatment, thus, it was hypothesized that
dexmedetomidine may regulate immune functions. To test this
hypothesis, the impact of dexmedetomidine administration on the
immune systems of mice was investigated, and specifically, the
effect on inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α. The
influence of dexmedetomidine on splenocytes was also analyzed, in
order to understand how dexmedetomidine affects different types of
immune cells. Finally, how dexmedetomidine regulates macrophage
activation and inflammatory function was investigated. The present
study may aid in the improved understanding of the regulatory
functions of dexmedetomidine in TBI.
Materials and methods
Animal model
Female C57BL/6J mice (4–7 weeks old, ~20 g weight; 8
mice per group) were purchased from Welitonghua Corporation
(Beijing, China). The animals were raised in a room with 12-h
light/dark cycle at 25°C with 45% humidity. The mice had free
access to food and water. The TBI model was established as
previously reported in the literature (30). In brief, the mice were anesthetized
using a mixture of 7.7 mg/ml ketamine and 12.3 mg/ml xylazine via
intraperitoneal injection. Following shaving the mice, a
petroleum-based jelly was put on their eyes to prevent drying. A
solution of 10% iodine and 70% ethanol was used for cleaning. The
head of each mouse was then fixed in a stereotactic frame using ear
bars and a bite plate. Scissors were then used to create a
longitudinal incision in the middle of each mouse's head and a
cotton-tipped applicator was used to expose the skull. Forceps were
employed to create pressure on the skull. Following the above
operation, an impact system was used to generate a TBI with the
actuator set at 3 m/sec achieving a 0.8 mm deformation depth.
Following impact, applicators with cotton tips were used to remove
any blood, and a warm pad was used to keep the mice warm. When
bleeding had ceased, the wound area was sutured and the mice were
returned into a clean cage with a warm pad. Mice were treated with
0.05–0.10 mg/kg buprenorphine via subcutaneous injection every 8 h
for 2 days.
The mice were divided into three groups each
containing 8 mice: Naive (mice with no treatment), trauma, and
trauma + DEX. Dexmedetomidine (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was used at a concentration of 20 µg/kg and was
employed to treat mice immediately following trauma induction and
was administered through and intravenous (i.v.) tail injection for
a single administration (25,31–33).
Yohimbine (YOH) was selected as the α-2-adrenergic receptor
antagonist (1 mg/kg; Sigma-Aldrich; Merck KGaA). All the animal
experiments were performed under the guidance of the Animal Care
and Use Committee at The First People's Hospital of Lianyungang
(Lianyungang, China) and approved by the Animal Care and Use
Committee. The animal study also followed the local and federal
laws and regulations on animal protection.
Patient samples
Patients' samples were collected from The First
People's Hospital of Lianyungang between 2012 to 2017. Among the
115 patients (Table I), blood
samples from 9 patients were used for cytokine-level tests. Male or
female patients aged between 18–75 were selected for the present
study. All patients did not have any other treatment at least one
month prior to enrollment and exhibited no major disease. Patients
who exhibited arrhythmia, renal, liver or heart failure were
excluded from the current study. All patients received
dexmedetomidine as a normal sedation procedure through i.v.
injection at a concentration of 0.2–0.3 µg/kg/h. The treatment was
performed immediately after patient admittance and lasted for ~100
min. Notably, these blood samples were not collected specifically
for the present study, instead the blood samples were originally
collected for various clinical examinations, and the present study
obtained permission to use these samples. The samples were not used
until written consent was obtained from each patient. The blood
samples were centrifuged at 15,000 × g for 5 min at 4°C to collect
the serum, then sealed in a 1.5 ml tube and stored at −80°C prior
to ELISA tests. Nine blood samples were also obtained from healthy
subjects who were doing blood tests for their annual physical exam.
The healthy subjects who participated in the tests were notified
about the study, and written permission was obtained. The present
study was approved by the Ethics Committee of The First People's
Hospital of Lianyungang and followed all the local and federal
laws.
 | Table I.Clinical characteristics of
patients. |
Table I.
Clinical characteristics of
patients.
| Clinical
characteristics | Group A (age ≤
50) | Group B (age >
50) | P-value |
|---|
| Gender
(male/female) | 13/45 | 10/47 | NS |
| Age | 43.2±8.2 | 61.3±5.1 | <0.001 |
| Systolic blood
pressure (mmHg) | 125±10.3 | 127±10.3 | NS |
| Diastolic blood
pressure (mmHg) | 73.9±7.5 | 74.9±6.7 | NS |
| Smoking | 37 | 35 | NS |
| Alcohol use | 7 | 5 | NS |
| Body mass index
(kg/m2) | 23.1±2.7 | 25.1±3.2 | NS |
| Blood urine
nitrogen (mg/dl) | 11.5±3.5 | 13.5±5.1 | NS |
| Total cholesterol
(mg/dl) | 179±37 | 191±29 | NS |
| High density
lipoprotein (mg/dl) | 51±12 | 50±11 | NS |
| Fasting blood
glucose (mg/dl) | 72.5±5.6 | 79.5±3.7 | NS |
ELISA analysis of mouse blood
samples
To assess the cytokine levels in mice, blood was
collected via retro-orbital collection. In brief, ~100 µl blood was
collected in heparinized tubes. The blood was then centrifuged at
15,000 × g for 5 min at 4°C to collect the serum and serum was then
stored at −80°C. The levels of cytokines IL-6 (cat. no. 550319),
IL-8 (cat. no. 555244), and TNF-α (cat. no. 558535) were determined
using ELISA kits (BD Bioscience, San Jose, CA, USA) according to
the manufacturer's instructions.
Isolation of splenocytes
Sham mice were anesthetized via gradual exposure to
carbon dioxide for 10–15 min. Cervical dislocation was used to
ensure a successful sacrifice. The spleen was collected from each
mouse, and the tissue forced through a cell strainer (40 µm) with a
syringe plunger to obtain a cell suspension. EDTA (10 mM) was added
to the cell suspension to prevent cells from aggregating. Following
PBS washes, the cells were cultured in RPMI culture medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 2%
fetal calf serum (Sigma-Aldrich; Merck KGaA).
Splenocyte viability assay
For viability tests, cells collected from the spleen
were cultured for 12 h. The cells were then washed with PBS twice
to remove any debris or free-floating dead cells. Cells were then
stained with trypan blue (1:1,000 in PBS) and viable cells were
counted using a cell counter.
ELISA analysis of mouse
splenocytes
ELISA was used to analyze the levels of cytokines in
the splenocyte culture medium. Cells were cultured for 60 h and 20
µl supernatant was collected and centrifuged at 15,000 × g for 5
min at 4°C to remove the debris. The levels of IL-6 (cat. no.
550319), IL-8 (cat. no. 555244), and TNF-α (cat. no. 558535) were
assessed using ELISA kits (BD Bioscience).
Macrophage viability, staining and
flow cytometry
Dexmedetomidine and YOH were used to treat
macrophages for viability tests. Macrophages were isolated from
mouse bone marrow and cultured in medium, as reported in the
literature (34). In brief,
macrophages were cultured under standard conditions (5%
CO2, 37°C) in a 96-well plate (2×105 cells
per well) and were treated with dexmedetomidine at different
concentrations (10, 30 and 100 µg/ml), YOH (10 µg/ml), or LPS (1
µg/ml). The cells were cultured for another 24 h, then collected by
centrifugation (500 × g for 5 min) and washed with PBS twice.
Trypan blue (1:1,000 dilution in PBS) was used to stain the cells
with the number of viable cells counted using a cell counter.
For flow cytometry assay, macrophages were treated
with LPS (1 µg/ml), dexmedetomidine (30 µg/ml), YOH (10 µg/ml),
dexmedetomidine (30 µg/ml) + LPS (1 µg/ml), and dexmedetomidine
(100 µg/ml) + LPS (1 µg/ml) + YOH (10 µg/ml), in 96-well plates
(1×105 cells per well). After 24 h, cells were washed
with PBS twice then Fc-block (1:200 in PBS plus 1% BSA) was added
to macrophages for 20 min at room temperature to prevent
non-specific binding. Cells were stained for cluster of
differentiation (CD)-80 and CD40 markers to assess the activation
levels. Fluorescein isothiocyanate-conjugated CD80 (cat. no.
561954; BD Biosciences) and allophycocyanin-conjugated CD40 (cat.
no. 124610; BioLegend, Inc., San Diego, CA, USA) were used diluted
at 1:200 in PBS+1% BSA for 25 min. Following staining, the cells
were washed with PBS twice. Cells were then added to PBS with DAPI
(1:1,000 dilution) at room temperature, and the levels of surface
markers were assessed by flow cytometry (FlowJo V7; FlowJo LLC,
Ashland, OR, USA).
Statistical analysis
All data were expressed as average ± standard error
of the mean. The statistical differences between groups were
analyzed by unpaired t-tests using GraphPad Prism (v.6.02; GraphPad
Software, Inc., La Jolla, CA, USA). Multiple comparisons among
groups were analyzed using one-way ANOVA with a Tukey's post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
No significant difference in clinical
characteristics was observed in TBI patients
At our clinical practice, 115 patients who suffered
TBI from different causes (i.e., car accident, falling) had their
clinical characteristics recorded (Table
I). Patients were classified into two groups based on their age
for two key reasons: Firstly, aging might have a role in treatment;
and secondly, age is a mutually exclusive and a direct parameter
for grouping the patients. The following physical characteristics
were recorded: sex (male/female), age, systolic blood pressure,
diastolic blood pressure, smoker, alcohol use, body mass index,
blood urine nitrogen, total cholesterol (mg/dl), high-density
lipoprotein (mg/dl), and fasting blood glucose (mg/dl). No
statistical difference was observed between these two groups for
any parameter (Table I).
Patients with TBI have a higher level
of inflammatory cytokines in blood serum
Beyond recording basic clinical characteristics, the
aim of the present study was to analyze the levels of inflammatory
cytokines present in the blood serum of TBI patients. The levels of
IL-1β, IL-6, IL-8 and TNF-α were measured 24 h prior (Trauma group)
and 8 h following the administration of dexmedetomidine (Trauma +
Dex group). The cytokine level in healthy people was used as the
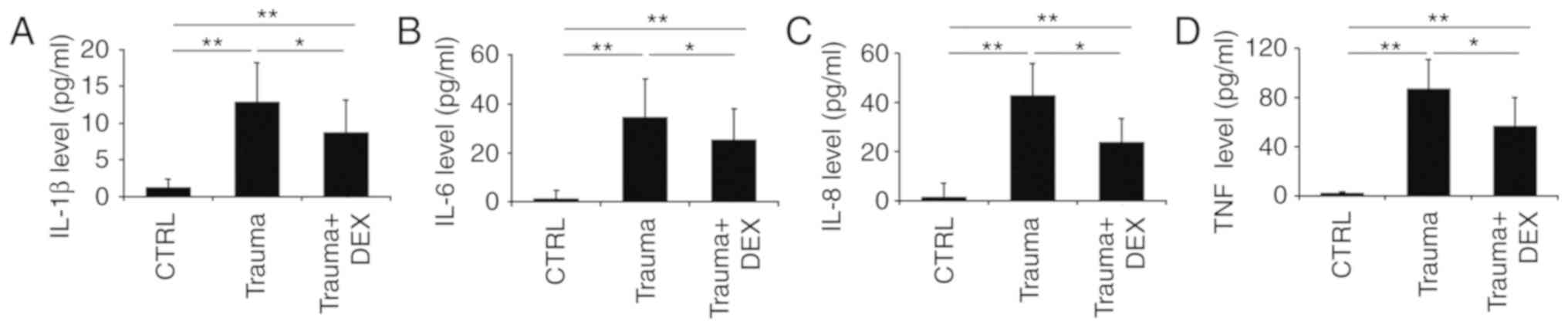
control (Fig. 1). Trauma patients
had significantly higher levels of IL-1β (Fig. 1A), IL-6 (Fig. 1B), IL-8 (Fig. 1C), and TNF-α (Fig. 1D) compared with the healthy control
group. However, following dexmedetomidine administration, the
levels of IL-1β (Fig. 1A), IL-6
(Fig. 1B), IL-8 (Fig. 1C), and TNF-α (Fig. 1D) were significantly decreased
compared with the trauma group. It is of note that the clinical
processing of patients with TBI requires complicated procedures,
where multiple therapeutic medicines or even surgery might be
required. This increased the complexity of the study, therefore the
effect of dexmedetomidine administration alone on levels of
inflammatory cytokines in these trauma patients could not be fully
confirmed. Thus, the effect of dexmedetomidine on TBI in a mouse
model was investigated.
Dexmedetomidine regulates the
secretion of inflammatory cytokines within 24 h in a mouse model of
TBI
The levels of IL-1β, IL-6, IL-8, and TNF-α cytokines
were monitored every 8 h over 24 h in three groups of mice: naive
mice, trauma mice, and trauma mice treated with dexmedetomidine.
Levels of IL-1β, IL-6, and IL-8 decreased over 24 h for trauma mice
treated with dexmedetomidine (Fig.
2A-C). It was also observed that the levels of IL-6 and IL-8 in
the mice with trauma did not vary significantly from 8 to 24 h,
indicating that the level of inflammation remained consistent over
this period (Fig. 2B and C). There
was no significant difference in TNF-α levels between the trauma
mice compared with the trauma mice with dexmedetomidine treatment,
which indicated that dexmedetomidine did not significantly affect
this cytokine in mice (Fig. 2D). The
levels of IL-1β, IL-6, IL-8 and TNF-α in the three groups of mice
were further analyzed at 24 h following treatment. There was a
significant difference (P<0.05) in the levels of IL-1β, IL-6,
and IL-8 between trauma mice and trauma mice with dexmedetomidine
treatment (Fig. 2E-G), indicating
that dexmedetomidine reduced inflammation in the mice. A somewhat
reduced level of TNF-α in the trauma mice with dexmedetomidine
treatment compared with trauma mice was observed; however, this
difference was not significant (Fig.
2H).
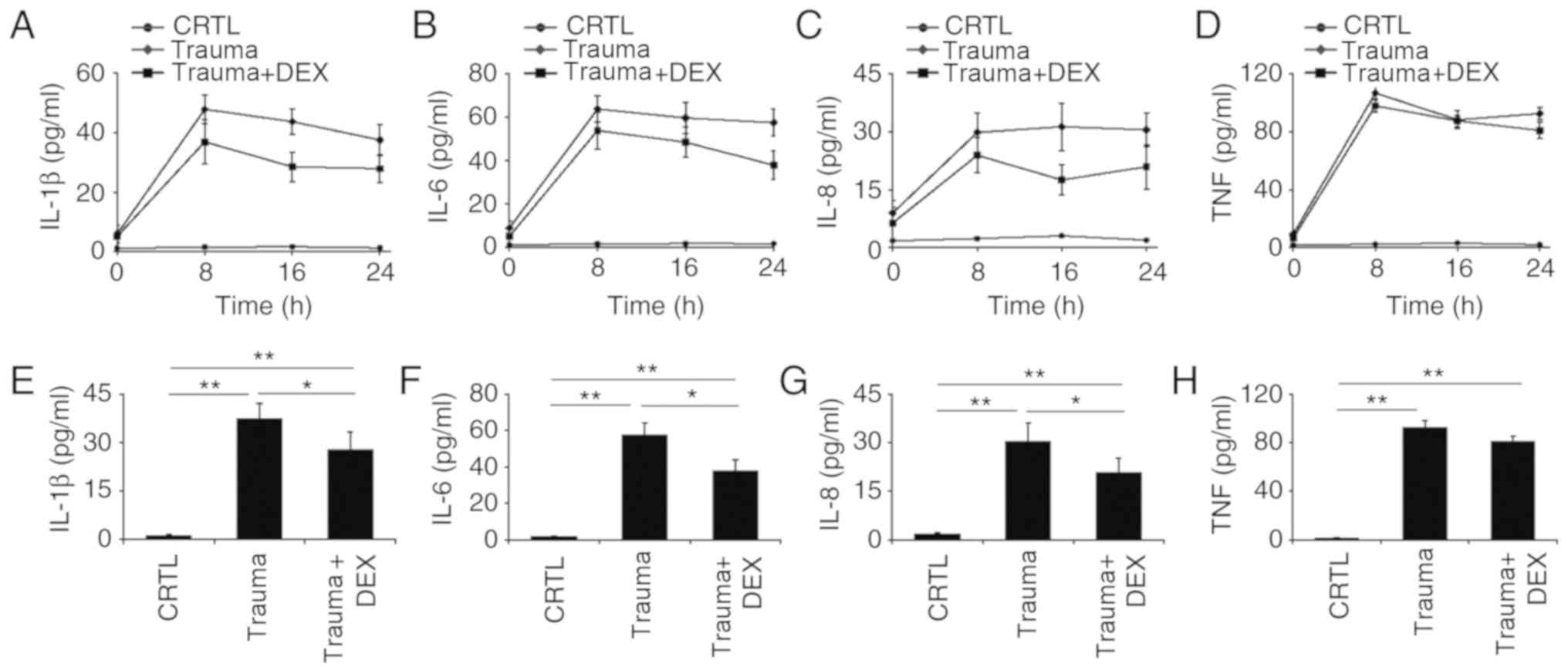 | Figure 2.Inflammatory cytokine levels over 24
h in naive mice (CRTL), mice with brain injury (Trauma), and
brain-injured mice with dexmedetomidine treatment (Trauma + Dex).
(A) The levels of IL-1β, (B) IL-6, (C) IL-8 and (D) TNF-α cytokines
were measured with ELISA at 0, 8, 16 and 24 h post-injury. (E)
Statistical analyses of the levels of IL-1β, (F) IL-6, (G) IL-8 and
(H) TNF-α cytokines at 24 h post-injury. *P<0.05 and **P<0.01
with comparisons indicated by lines. CTRL, control; DEX,
dexmedetomidine; IL, interleukin; TNF, tumor necrosis factor. |
Dexmedetomidine reduces inflammatory
cytokine secretion in splenocytes without affecting their
viability
Splenocytes were selected for experimentation, as
the spleen is a major secondary immune organ and contains different
types of immune cells. The spleen has similar structure to a lymph
node and has vital roles functioning as part of the mononuclear
phagocyte system, metabolizing haemoglobin from red blood cells
(35). In addition, the spleen can
produce antibodies and remove bacteria or old blood cells. The
spleen contains at least half of the monocytes in the body which
are capable of differentiating into dendritic cells and
macrophages, both of which are vital immune cells (35).
Splenocytes were collected from the spleens of naive
mice and processed into a single-cell suspension. It was identified
that dexmedetomidine at various concentrations (10, 30 and 100
µg/ml) and YOH treatment did not affect the viability of
splenocytes (Fig. 3A). Next, the
levels of inflammatory cytokines in the different treatment groups
were analyzed at 48 h post-treatment (Fig. 3B-E). Following LPS treatment,
increased levels of IL-1β, IL-6, IL-8 and TNF-α were observed
compared with control untreated cells (Fig. 3B-E). Dexmedetomidine reduced the
levels of IL-1β, IL-6, IL-8 and TNF-α, with dexmedetomidine at a
higher concentration (100 µg/ml) more effective in reducing
inflammation compared with dexmedetomidine at a low concentration
(30 µg/ml; Fig. 3B-E). Combination
treatment with YOH, dexmedetomidine and LPS resulted in increased
secretion of cytokines compared with the samples treated with
dexmedetomidine and LPS alone (Fig.
3B-E), suggesting that YOH compromised the anti-inflammatory
effects of dexmedetomidine.
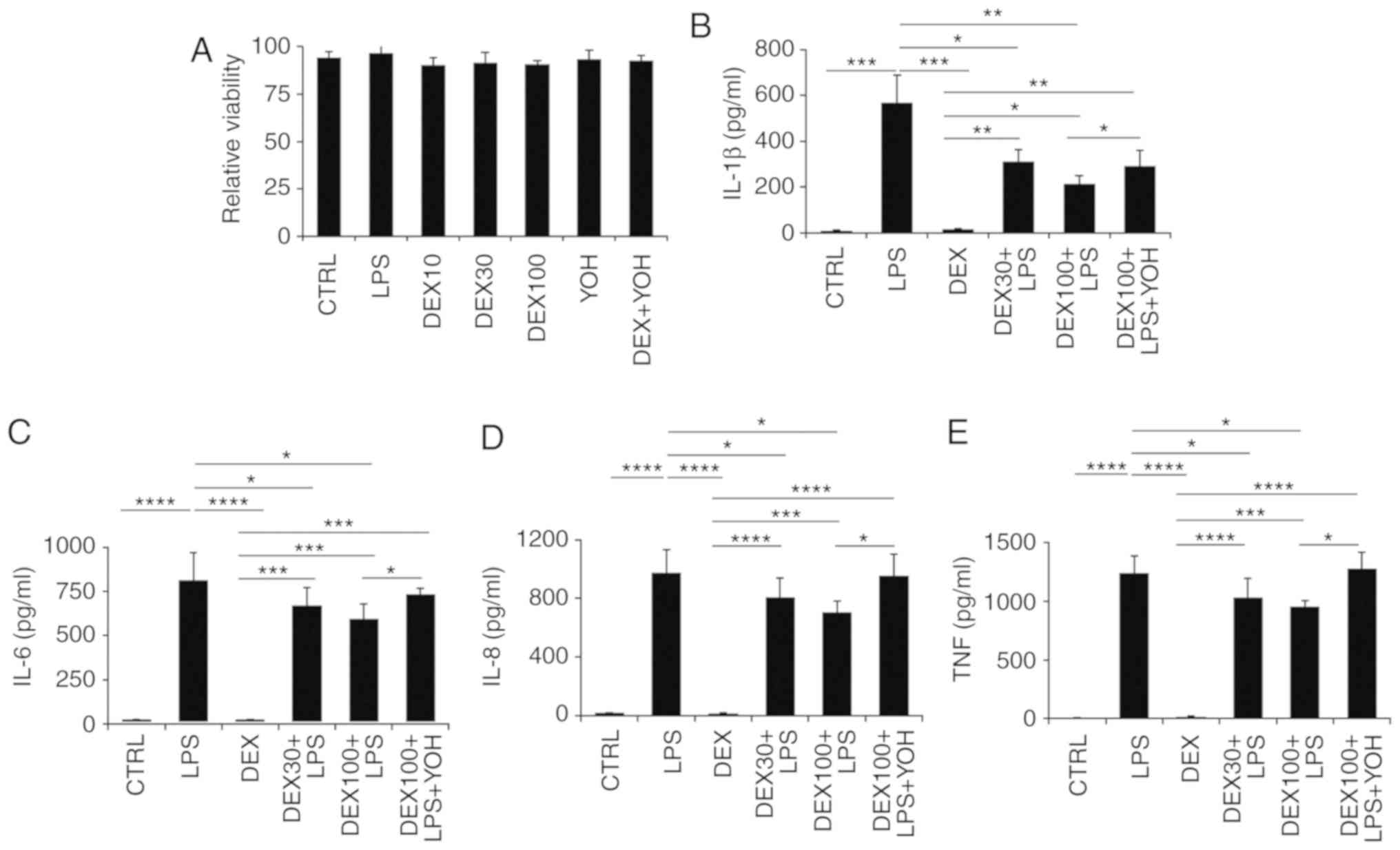 | Figure 3.Effect of dexmedetomidine on
splenocyte inflammatory cytokine expression. (A) The impact of
dexmedetomidine (10, 30, and 100 µg/ml,) and YOH on the viability
of splenocytes. (B) The impact of dexmedetomidine (30 and 100
µg/ml) on the secretion of IL-1β, (C) IL-6, (D) IL-8, and (E) TNF-α
in splenocytes. LPS was used to stimulate the splenocytes and YOH
was used to compromise the function of dexmedetomidine. *P<0.05,
**P<0.01, ***P<0.001 and ****P<0.0001 with comparisons
indicated by lines. YOH, yohimbine; IL, interleukin; TNF, tumor
necrosis factor; LPS, lipopolysaccharide; CTRL, control; DEX,
dexmedetomidine. |
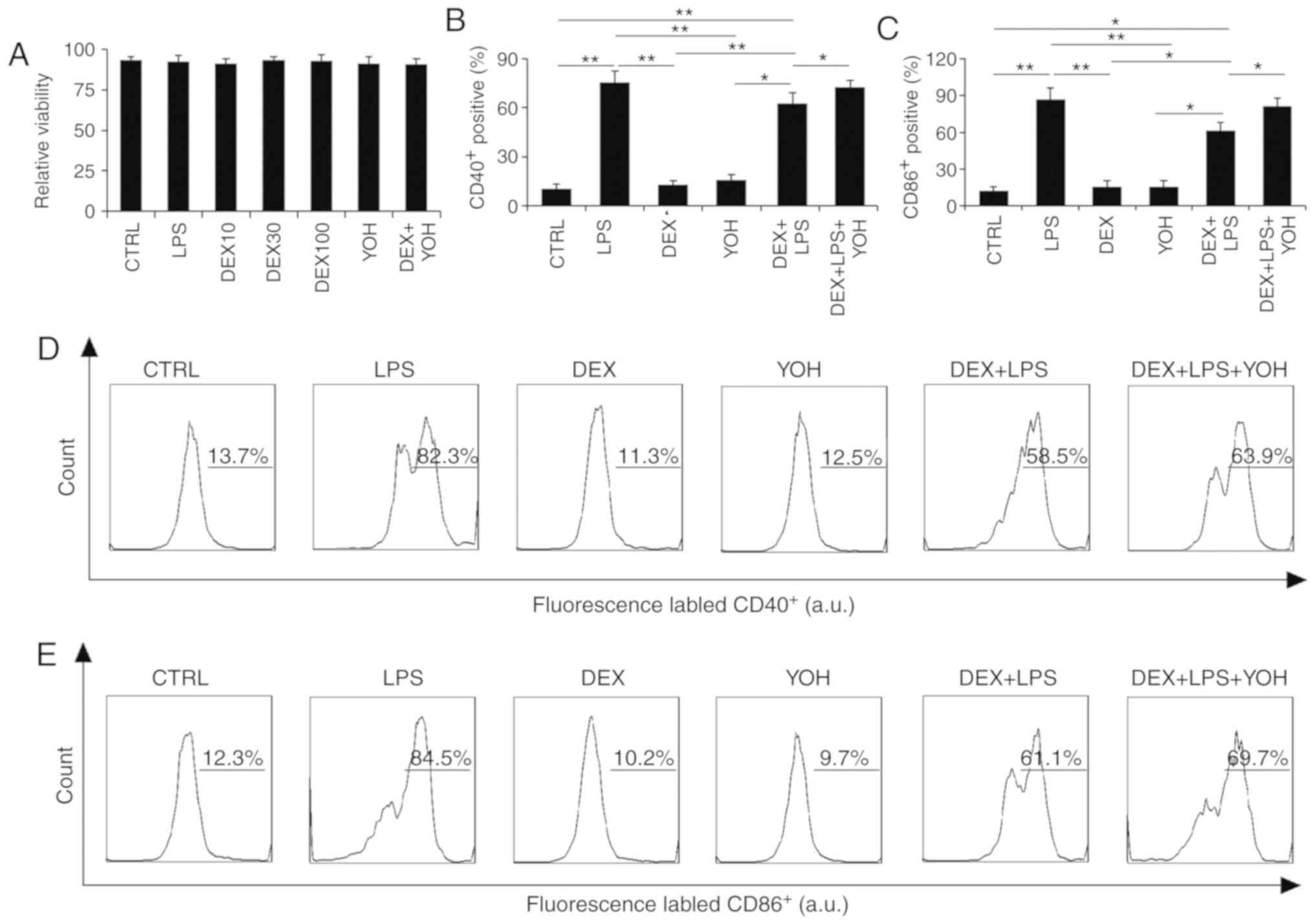
Dexmedetomidine regulates the
expression of CD40 and CD86 surface markers on macrophages
Macrophages are one of the most important cells that
regulate the immune system, therefore the impact of dexmedetomidine
on macrophage function was investigated. Results demonstrated that
viability was not affected by the various treatments tested in the
present study (LPS, dexmedetomidine, dexmedetomidine + LPS,
dexmedetomidine + LPS + YOH; Fig.
4A).
The activation of macrophage surface markers was
then analyzed by flow cytometry. LPS stimulated the activation of
CD40 and CD86 surface markers on macrophages (Fig. 4B-D). By contrast, dexmedetomidine and
YOH treatments alone did not stimulate activation of CD40 or CD86
surface markers compared with control, instead displaying similar
levels of expression to the control, indicating that these drugs
did not promote macrophage activation (Fig. 4D). The use of dexmedetomidine reduced
the expression of CD40 and CD86 markers compared with cells treated
with LPS alone, which indicated that dexmedetomidine can offset the
stimulatory functions of LPS (Fig.
4B-D). Compared with cells treated with LPS + dexmedetomidine,
the cells treated with LPS + dexmedetomidin + YOH had increased
expression of these surface markers, indicating that YOH can
inhibit dexmedetomidine function (Fig.
4B and C). The percentage of cells expressing CD40 on their
cell surface in the CTRL, LPS, dexmedetomidine, YOH,
dexmedetomidine + LPS, and dexmedetomidine + LPS + YOH groups was
13.7, 82.3, 11.3, 12.5, 58.5, and 63.9%, respectively (Fig. 4D). The percentage of cells expressing
CD86 on their cell surface in the CTRL, LPS, dexmedetomidine, YOH,
dexmedetomidine + LPS, and dexmedetomidine + LPS + YOH groups was
12.3, 84.5, 10.2, 9.7, 61.1 and 69.7%, respectively. Taken
together, these findings indicated that dexmedetomidine reduced the
inflammatory responses of macrophages.
Discussion
TBI is typically caused by external forces, such as
blast waves, rapid acceleration or deceleration, which leads to
temporary or long-term damage to cognitive function. Worldwide, TBI
caused >57 million hospitalizations and >10 million deaths
each year (36,37). Clinically, TBI is complicated and
usually includes immediate mechanical damage to brain tissues,
causing injury to blood vessels and stretching of neurons. Beyond
the primary direct damage, TBI also involves secondary damage from
the cascades of metabolic and molecular mechanisms, causing
neuronal death and tissue damage. In clinical practice, patients
may receive different types of treatments to prevent further trauma
damage, which may potentially influence the functions of
dexmedetomidine. For example, intravenous anesthesia, including
ketamine, propofol or etomidate, might be utilized (38,39)
which typically will not interact with dexmedetomidine; however
detailed interactions require future study. For the animal model of
the present study, this factor was not considered since this would
make the study excessively complicated. However, to understand the
impact of dexmedetomidine comprehensively, future studies need to
illustrate the potential influence of other accepted clinical
treatments to dexmedetomidine functions.
The present study selected IL-1β, IL-6, IL-8, and
TNF-α as the target cytokines to analyze the roles of
dexmedetomidine in modulating inflammation. IL-1β is a key mediator
of inflammation in both the peripheral and central nervous systems.
The association between IL-1β and TBI has been comprehensively
studied in both the focal and diffuse injury models (40,41). In
healthy individuals, the levels of IL-1β are barely detectable. By
contrast, in TBI patients, the levels of IL-1β increase due to the
inflammation (42). Current studies
have also identified IL-1β concentrations in post-mortem tissues of
TBI patients within several hours of injury occurrence (43). TNF is a multifunctional
pro-inflammatory cytokine. Studies using TNF and TNF receptor
knockout mice identified that TNF knockout mice with TBI have
higher mortality rates and longer recovery times (44,45).
IL-6 has been extensively investigated in different physiological
and pathophysiological processes, confirming the regulatory roles
of IL-6 in inflammation, immunity, and neural development (46). It is of note that IL-6 is suspected
to have vital roles in other diseases, such as autoimmune disease,
Alzheimer's disease and TBI, where it is upregulated (47,48).
Similarly, studies have demonstrated that levels of the
inflammatory cytokine IL-8 peaks following TBI (49). Therefore, the present study
investigated these cytokines in both patients and mice with TBI.
The current study demonstrated that patients with TBI had a higher
level of inflammatory cytokines. The level of inflammatory
cytokines decreased over 24 h following dexmedetomidine treatment.
These results are consistent with existing studies that have
revealed that the level of inflammatory cytokines including IL-1β
and IL-6 increased in patients with TBI (42,46).
The roles of macrophages have been previously
studied in central nervous system injury models, such as ischemic
stroke and TBI. Necrotic debris clearing and wound repair are vital
functions of macrophages (50–52).
Studies using different models, such as experimental autoimmune
encephalomyelitis, demonstrated that macrophages also regulate the
production of IL-10 and transforming growth factor cytokines,
indicating that macrophages have different roles in brain injury
(53,54). Therefore, the present study selected
macrophages as the candidate cell for investigating the roles of
dexmedetomidine in the TBI mouse model. The immune system is very
complicated with a variety of immune cells therefore the present
study focused on investigating the roles of splenocytes and
macrophages. These two large types of cells were selected because
the spleen is a key secondary immune organ and macrophages have a
vital role in regulating inflammation. The present study revealed
an enhanced level of inflammatory cytokines in mice stimulated with
LPS. These results indicated that the use of dexmedetomidine
reduced the level of inflammatory cytokines. Treatment with YOH
offset the function of dexmedetomidine and caused an increase in
the level of inflammatory response. These results indicated that
dexmedetomidine reduced inflammation in patients with TBI, thus
providing protection from secondary damage. Additionally, one of
the major functions of dexmedetomidine is clinical sedation. The
sedative and anti-inflammatory functions indicate that
dexmedetomidine is a valuable choice for the treatment of patients
with TBI.
Future work will involve analyzing the impact of
dexmedetomidine on different types of immune cells, such as
regulatory T cells, cytotoxic T cells and dendritic cells, in the
disease mouse model. In brief, investigation into whether
dexmedetomidine has a role in balancing the relative ratios of
different types of T cells and also whether it can impact
antigen-presenting functions of dendritic cells is planned. In
addition, the present study focused on the impact of
dexmedetomidine on the secretion of inflammatory cytokines over 24
h in the mouse model, since TBI requires fast treatment to the
patients; however, the long-term effects of dexmedetomidine also
need to be investigated.
In summary, the present study observed decreased
levels of inflammatory cytokines following administration of
dexmedetomidine in patients with TBI. It was hypothesized that
dexmedetomidine could modulate immune functions in TBI. To test
this hypothesis, the impact of dexmedetomidine on the production of
inflammatory cytokines in mice with TBI was investigated. In
addition, the influence of dexmedetomidine on splenocytes was
determined to understand how dexmedetomidine affects different
types of immune cells. It was demonstrated that dexmedetomidine
could downregulate inflammatory cytokines. Finally, how
dexmedetomidine mediates macrophage activation and inflammatory
cytokine production was determined. The present study provided
evidence aiming to better understand the regulatory functions of
dexmedetomidine in TBI.
Acknowledgements
Not applicable.
Funding
The present study was supported by the First
People's Hospital of Lianyungang (grant no. LYG201605).
Availability of data and materials
All relevant data and its supporting information
files are within the present study.
Authors' contribution
MD and YW designed the experiments and wrote the
majority of the paper. MD performed a majority of the experiments.
YC, HL and ZZ carried out ELISA experiments and contributed to part
of the paper.
Ethics approval and consent to
participate
The patients that participated in the study signed a
consent form, allowing the use of their blood samples. The Research
Ethics Committee of The First Hospital of Lianyungang approved the
study. Protocols involving the use of animals were approved by the
Animal Care and Use Committee of The First People's Hospital of
Lianyungang.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
References
|
1
|
Woodcock T and Morganti-Kossmann MC: The
role of markers of inflammation in traumatic brain injury. Front
Neurol. 4:182013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith DH, Johnson VE and Stewart W:
Chronic neuropathologies of single and repetitive TBI: Substrates
of dementia? Nat Rev Neurol. 9:211–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Russo MV and McGavern DB: Inflammatory
neuroprotection following traumatic brain injury. Science.
353:783–785. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bye N, Habgood MD, Callaway JK, Malakooti
N, Potter A, Kossmann T and Morganti-Kossmann MC: Transient
neuroprotection by minocycline following traumatic brain injury is
associated with attenuated microglial activation but no changes in
cell apoptosis or neutrophil infiltration. Exp Neurol. 204:220–233.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clark RS, Schiding JK, Kaczorowski SL,
Marion DW and Kochanek PM: Neutrophil accumulation after traumatic
brain injury in rats: Comparison of weight drop and controlled
cortical impact models. J Neurotrauma. 11:499–506. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dinkel K, Dhabhar FS and Sapolsky RM:
Neurotoxic effects of polymorphonuclear granulocytes on hippocampal
primary cultures. Proc Natl Acad Sci USA. 101:331–336. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen HX, O'Barr TJ and Anderson AJ:
Polymorphonuclear leukocytes promote neurotoxicity through release
of matrix metalloproteinases, reactive oxygen species, and
TNF-alpha. J Neurochem. 102:900–912. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bergold PJ: Treatment of traumatic brain
injury with anti-inflammatory drugs. Exp Neurol. 275:367–380. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martin E, Ramsay G, Mantz J and Sum-Ping
ST: The role of the alpha2-adrenoceptor agonist dexmedetomidine in
postsurgical sedation in the intensive care unit. J Intensive Care
Med. 18:29–41. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sukegawa S, Higuchi H, Inoue M, Nagatsuka
H, Maeda S and Miyawaki T: Locally injected dexmedetomidine
inhibits carrageenin-induced inflammatory responses in the injected
region. Anesth Analg. 118:473–480. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Taniguchi T, Kidani Y, Kanakura H,
Takemoto Y and Yamamoto K: Effects of dexmedetomidine on mortality
rate and inflammatory responses to endotoxin-induced shock in rats.
Crit Care Med. 32:1322–1326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yagmurdur H, Ozcan N, Dokumaci F, Kilinc
K, Yilmaz F and Basar H: Dexmedetomidine reduces the
ischemia-reperfusion injury markers during upper extremity surgery
with tourniquet. J Hand Surg Am. 33:941–947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kawasaki T, Kawasaki C, Ueki M, Hamada K,
Habe K and Sata T: Dexmedetomidine suppresses proinflammatory
mediator production in human whole blood in vitro. J Trauma Acute
Care Surg. 74:1370–1375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim Y, Kang SH, Hong TH, Cho ML, Han HJ,
Kwon SJ and Lee J: Effects of dexmedetomidine on the ratio of T
helper 1 to T helper 2 cytokines in patients undergoing
laparoscopic cholecystectomy. J Clin Anesth. 26:281–285. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Naguib AN, Tobias JD, Hall MW, Cismowski
MJ, Miao Y, Barry N, Preston T, Galantowicz M and Hoffman TM: The
role of different anesthetic techniques in altering the stress
response during cardiac surgery in children: A prospective,
double-blinded, and randomized study. Pediatr Crit Care Med.
14:481–490. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ueki M, Kawasaki T, Habe K, Hamada K,
Kawasaki C and Sata T: The effects of dexmedetomidine on
inflammatory mediators after cardiopulmonary bypass. Anaesthesia.
69:693–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia R, Yin H, Xia ZY, Mao QJ, Chen GD and
Xu W: Effect of intravenous infusion of dexmedetomidine combined
with inhalation of isoflurane on arterial oxygenation and
intrapulmonary shunt during single-lung ventilation. Cell Biochem
Biophys. 67:1547–1550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xin J, Zhang YB, Zhou L, Liu F, Zhou X,
Liu B and Li Q: Effect of dexmedetomidine infusion for intravenous
patient-controlled analgesia on the quality of recovery after
laparotomy surgery. Oncotarget. 8:100371–100383. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aho M, Erkola O, Kallio A, Scheinin H and
Korttila K: Dexmedetomidine infusion for maintenance of anesthesia
in patients undergoing abdominal hysterectomy. Anesth Analg.
75:940–946. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Talke P, Richardson CA, Scheinin M and
Fisher DM: Postoperative pharmacokinetics and sympatholytic effects
of dexmedetomidine. Anesth Analg. 85:1136–1142. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taittonen MT, Kirrvelä OA, Aantaa R and
Kanto JH: Effect of clonidine and dexmedetomidine premedication on
perioperative oxygen consumption and haemodynamic state. Br J
Anaesth. 78:400–406. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bekker A, Gold M, Ahmed R, Kim J, Rockman
C, Jacobovitz G, Riles T and Fisch G: Dexmedetomidine does not
increase the incidence of intracarotid shunting in patients
undergoing awake carotid endarterectomy. Anesth Analg. 103:955–958.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bekker AY, Basile J, Gold M, Riles T,
Adelman M, Cuff G, Mathew JP and Goldberg JD: Dexmedetomidine for
awake carotid endarterectomy: Efficacy, hemodynamic profile, and
side effects. J Neurosurg Anesthesiol. 16:126–135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaygusuz K, Gokce G, Gursoy S, Ayan S,
Mimaroglu C and Gultekin Y: A comparison of sedation with
dexmedetomidine or propofol during shockwave lithotripsy: A
randomized controlled trial. Anesth Analg. 106:114–119, table of
contents. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Naaz S and Ozair E: Dexmedetomidine in
current anaesthesia practice-a review. J Clin Diagn Res.
8:GE01–GE04. 2014.PubMed/NCBI
|
|
26
|
Munro HM, Tirotta CF, Felix DE, Lagueruela
RG, Madril DR, Zahn EM and Nykanen DG: Initial experience with
dexmedetomidine for diagnostic and interventional cardiac
catheterization in children. Paediatr Anaesth. 17:109–112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tasdogan M, Memis D, Sut N and Yuksel M:
Results of a pilot study on the effects of propofol and
dexmedetomidine on inflammatory responses and intraabdominal
pressure in severe sepsis. J Clin Anesth. 21:394–400. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Memiş D, Hekimoğlu S, Vatan I, Yandim T,
Yüksel M and Süt N: Effects of midazolam and dexmedetomidine on
inflammatory responses and gastric intramucosal pH to sepsis, in
critically ill patients. Br J Anaesth. 98:550–552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang J, Wang Z, Wang Y, Zhou G and Li H:
The effect of dexmedetomidine on inflammatory response of septic
rats. BMC Anesthesiol. 15:682015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Romine J, Gao X and Chen J: Controlled
cortical impact model for traumatic brain injury. J Vis Exp.
e517812014.PubMed/NCBI
|
|
31
|
Inada T, Sumi C, Hirota K, Shingu K,
Okamoto A, Matsuo Y and Kamibayashi T: Mitigation of inflammation
using the intravenous anesthetic dexmedetomidine in the mouse air
pouch model. Immunopharm Immunot. 39:225–232. 2017. View Article : Google Scholar
|
|
32
|
Cagle LA, Franzi LM, Epstein SE, Kass PH,
Last JA and Kenyon NJ: Injectable anesthesia for mice: Combined
effects of dexmedetomidine, tiletamine-zolazepam, and butorphanol.
Anesthesiol Res Pract. 2017:91610402017.PubMed/NCBI
|
|
33
|
Pan W, Hua X, Wang Y, Guo R, Chen J and Mo
L: Dose response of dexmedetomidine-induced resistance to hypoxia
in mice. Mol Med Rep. 14:3237–3242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weischenfeldt J and Porse B: Bone
marrow-derived macrophages (BMM): Isolation and applications. CSH
Protoc 2008: pdb prot5080. 2008.
|
|
35
|
Swirski FK, Nahrendorf M, Etzrodt M,
Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler
RH, Chudnovskiy A, Waterman P, Aikawa E, et al: Identification of
splenic reservoir monocytes and their deployment to inflammatory
sites. Science. 325:612–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maas AI, Stocchetti N and Bullock R:
Moderate and severe traumatic brain injury in adults. Lancet
Neurol. 7:728–741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Langlois JA, Rutland-Brown W and Wald MM:
The epidemiology and impact of traumatic brain injury: A brief
overview. J Head Trauma Rehabil. 21:375–378. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Behrens V, Dudaryk R, Nedeff N, Tobin JM
and Varon AJ: The ryder cognitive aid checklist for trauma
anesthesia. Anesth Analg. 122:1484–1487. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tobin JM, Grabinsky A, McCunn M, Pittet
JF, Smith CE, Murray MJ and Varon AJ: A checklist for trauma and
emergency anesthesia. Anesth Analg. 117:1178–1184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brough D, Tyrrell PJ and Allan SM:
Regulation of interleukin-1 in acute brain injury. Trends Pharmacol
Sci. 32:617–622. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fan L, Young PR, Barone FC, Feuerstein GZ,
Smith DH and McIntosh TK: Experimental brain injury induces
expression of interleukin-1 beta mRNA in the rat brain. Brain Res
Mol Brain Res. 30:125–130. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Singhal A, Baker AJ, Hare GMT, Reinders
FX, Schlichter LC and Moulton RJ: Association between cerebrospinal
fluid interleukin-6 concentrations and outcome after severe human
traumatic brain injury. J Neurotrauma. 19:929–937. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Frugier T, Morganti-Kossmann MC, O'Reilly
D and McLean CA: In situ detection of inflammatory mediators in
post mortem human brain tissue after traumatic injury. J
Neurotrauma. 27:497–507. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Scherbel U, Raghupathi R, Nakamura M,
Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW and McIntosh
TK: Differential acute and chronic responses of tumor necrosis
factor-deficient mice to experimental brain injury. Proc Natl Acad
Sci USA. 96:8721–8726. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stahel PF, Shohami E, Younis FM, Kariya K,
Otto VI, Lenzlinger PM, Grosjean MB, Eugster HP, Trentz O, Kossmann
T, et al: Experimental closed head injury: Analysis of neurological
outcome, blood-brain barrier dysfunction, intracranial neutrophil
infiltration, and neuronal cell death in mice deficient in genes
for pro-inflammatory cytokines. J Cereb Blood Flow Metab.
20:369–380. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Romano M, Sironi M, Toniatti C,
Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van
Hinsbergh V, Sozzani S, et al: Role of IL-6 and its soluble
receptor in induction of chemokines and leukocyte recruitment.
Immunity. 6:315–325. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Banks WA: Blood-brain barrier transport of
cytokines: A mechanism for neuropathology. Curr Pharm Des.
11:973–984. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morganti-Kossmann MC, Kossmann T and Wahl
SM: Cytokines and neuropathology. Trends Pharmacol Sci. 13:286–291.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kushi H, Saito T, Makino K and Hayashi N:
IL-8 is a key mediator of neuroinflammation after traumatic brain
injury. Crit Care Med. 30:A822002. View Article : Google Scholar
|
|
50
|
Dimitrijevic OB, Stamatovic SM, Keep RF
and Andjelkovic AV: Absence of the chemokine receptor CCR2 protects
against cerebral Ischemia/reperfusion injury in mice. Stroke.
38:1345–1353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Izikson L, Klein RS, Charo IF, Weiner HL
and Luster AD: Resistance to experimental autoimmune
encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2.
J Exp Med. 192:1075–1080. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Semple BD, Bye N, Rancan M, Ziebell JM and
Morganti-Kossmann MC: Role of CCL2 (MCP-1) in traumatic brain
injury (TBI): Evidence from severe TBI patients and CCL2-/- mice. J
Cerebr Blood Flow Metab. 30:769–782. 2010. View Article : Google Scholar
|
|
53
|
Shechter R, London A, Varol C, Raposo C,
Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, et al:
Infiltrating blood-derived macrophages are vital cells playing an
anti-inflammatory role in recovery from spinal cord injury in mice.
PLoS Med. 6:e10001132009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Weber MS, Prod'homme T, Youssef S, Dunn
SE, Rundle CD, Lee L, Patarroyo JC, Stüve O, Sobel RA, Steinman L
and Zamvil SS: Type II monocytes modulate T cell-mediated central
nervous system autoimmune disease. Nat Med. 13:935–943. 2007.
View Article : Google Scholar : PubMed/NCBI
|