Introduction
Atherosclerosis (AS) is a common pathological basis
of cardiovascular and cerebrovascular diseases, and inflammatory
reactions and lipid metabolism disorders serve an important role
(1). AS is prevalent in the elderly,
and its incidence increases with age. Therefore, aging is a key
causal factor of AS (2). In
particular, a study has previously reported that cell senescence
may be observed in atherosclerotic plaques in patients with AS
(3). Oxidative stress damage and
vascular inflammation caused by senescence can cause arterial
dysfunction (4). Cyclin dependent
kinase inhibitor 1A (P21) and P53 are considered to be marker
proteins for senescence (5).
Autophagy protects the body from stress damage and
delays the development of aging-associated diseases such as AS
(6). Autophagy is an effective
response against inflammation and oxidative stress in AS plaque
cells, and thus it is considered to serve an important role in the
initiation and development of AS (7). Autophagosomes observed under a
transmission electron microscope (TEM) are regarded as the gold
standard for autophagy detection (8). mTOR is a member of the PI3K-related
family of protein kinases. mTOR integrates multiple upstream
signaling pathways, interacts with regulator proteins and is a key
regulatory molecule in the induction of autophagy (9). Microtubule associated protein 1 light
chain 3α (LC3) is a cytosolic ubiquitin-like protein that binds to
phosphatidylethanolamine to form autophagosomes (10). 3-methyladenine (3-MA) is a widely
used autophagy inhibitor, which inhibits class III PI3K and blocks
the early stage of autophagy (8).
The Traditional Chinese Medicine (TCM) monomer
quercetin (QUE), a flavonoid compound with anti-inflammatory,
antioxidative and lipid metabolism-modulating properties, has been
demonstrated to have therapeutic effects in AS treatment (11). Previous studies have revealed that
QUE can effectively interfere with AS development by regulating
inflammatory mediator production and promoting cholesterol efflux
(12,13). A previous study our laboratory
revealed that QUE can effectively reduce oxidized-low density
lipoprotein (ox-LDL)-induced RAW264.7 cell damage, reduce lipid
accumulation and delay cell senescence (14). Additionally, it has been demonstrated
that QUE serves an important role in the prevention and treatment
of AS by upregulating autophagy in endothelial cells to protect
cells from stress damage (15).
However, it is unclear whether QUE exerts anti-atherosclerotic
effects by regulating autophagy in ApoE−/− mouse models
of AS induced by a high-fat diet (HFD).
In the present study, using an AS model in
ApoE−/− mice, aorta autophagosomes were analyzed using
TEM, protein levels of mTOR, LC3 II/I ratio, P53 and P21 in the
aorta were examined using western blotting analysis, levels of the
cytokines tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β)
and interleukin-18 (IL-18) were assessed using ELISA assays, and
aorta pathology, lipid accumulation and collagen deposition were
investigated using hematoxylin and eosin (H&E), Oil Red O and
Masson staining, respectively. Based on the results of these
analyses, deeper mechanistic insights into QUE-mediated inhibition
of AS development were gained.
Materials and methods
Experimental animals
A total of 96 specific pathogen-free (SPF) male
ApoE−/− mice [age, 12 weeks; weight, 20±5 g; animal
license no. SCXK (Jing) 2016-0006] and 24 age-matched male
wild-type C57BL/6 mice [animal license no. SCXK (Jing) 2016-0006]
were purchased from Beijing Vital River Laboratory Animal
Technology Co., Ltd. The mice were kept in an SPF grade animal
facility at the Animal Center of the Shanghai University of
Traditional Chinese Medicine (Shanghai, China). The room
temperature was kept constant at 24±1°C, the relative humidity was
50–70% with a 12 h light:dark cycle. All mice had free access to
food and water. After 12 weeks, all mice were treated with cervical
dislocation following anesthesia to collect samples. All animal
experiments strictly followed the Guide for the Care and Use of
Medical Laboratory Animals (16).
The present study was approved by the Laboratory Animal Welfare and
Ethics Committee of Shanghai University of Traditional Chinese
Medicine (approval no. PZSHUTCM18113002).
Drugs and major reagents
The following drugs and reagents were used in the
current study: QUE (cat. no. B20527; Shanghai Yuanye Bio-Technology
Co., Ltd.), anti-rabbit mTOR (cat. no. 2983S; Cell Signaling
Technology, Inc.), anti-rabbit LC3B (cat. no. 3868S; Cell Signaling
Technology, Inc.), anti-rabbit P53 (cat. no. ab31333; Abcam),
anti-rabbit P21 (cat. no. ab188224; Abcam), anti-rabbit GAPDH (cat.
no. 5174S; Cell Signaling Technology, Inc.), IRDye®
800CW-conjugated goat anti-rabbit secondary antibody (cat. no.
926-32211; LI-COR Biosciences), IRDye® 800CW-conjugated
goat anti-mouse secondary antibody (cat. no. 926-32210; LI-COR
Biosciences), RIPA buffer (Beyotime Institute of Biotechnology),
PMSF (Beyotime Institute of Biotechnology), bicinchoninic acid
(BCA) protein assay kit (Beyotime Institute of Biotechnology),
protein ladder (Thermo Fisher Scientific, Inc.), SDS-PAGE Gel
Preparation kit (Beyotime Institute of Biotechnology). All ELISA
kits were purchased from EK-Bioscience: Mouse total cholesterol
(TC) ELISA kit (cat. no. Ek-M20591; Ek-Bioscience), mouse
triglyceride (TG) ELISA kit (cat. no. Ek-M20590; Ek-Bioscience),
mouse high density lipoprotein (HDL-C) ELISA kit (cat. no.
Ek-M20589; Ek-Bioscience), mouse low density lipoprotein (LDL-C)
ELISA kit (cat. no. Ek-M20588; Ek-Bioscience), mouse TNF-a ELISA
kit (cat. no. EK-M21159; Ek-Bioscience), mouse IL-1β ELISA kit
(cat. no. Ek-M20166; Ek-Bioscience) and mouse IL-18 ELISA kit (cat.
no. Ek-M20162; Ek-Bioscience).
Modeling and grouping
After 1 week of acclimatization on a standard mouse
diet, 96 male, 12-week-old ApoE−/− mice were randomly
divided into four experimental groups (n=24 mice/group) using a
random number table method as described previously (17): i) ApoE−/− mice + HFD
(Model group); ii) ApoE−/− mice + HFD + QUE (QUE group);
iii) apoE−/− mice + HFD + 3-MA (3-MA group); and iv)
apoE−/− mice + HFD + 3-MA + QUE (QUE + 3-MA group). In
addition, 24 age-matched wild-type C57BL/6 mice were used as normal
controls (Control group). The HFD was established by adding 21% fat
and 0.5% cholesterol to the standard mouse basal diet. The number
of samples per group for each experiment was 6. The health and
behavioral status of mice were monitored every 2 weeks. The QUE
group was treated by daily oral gavage of a QUE solution (12.5
mg/kg), and the 3-MA group was treated by peritoneal injection of a
water solution containing 3-MA (15 mg/kg) every other day, as
previously described (13,18). Mice in the Model and Control groups
were given an equal volume of distilled water by oral gavage on a
daily basis. HFD feeding to establish the AS model and drug
regimens started at the same time and lasted for 12 weeks.
Detection of AS indicators
Aorta pathology
Mice were sacrificed by cervical dislocation
following anesthesia. Then, the heart and aorta from the thoracic
to abdominal sections were removed and fixed in 10% formalin (room
temperature; 1 day) solution for later paraffinization (six mice
per group; thickness of sections, 5 µm; for H&E and Masson
staining) or frozen sectioning (six mice per group; thickness of
sections, 10–15 µm; for Oil Red O staining). H&E, Oil Red O and
Masson staining (room temperature) were performed on the aorta
section samples to determine aorta lipid plaque areas, lipid
accumulation and collagen fiber content, respectively. Images were
acquired using a digital scanning system (Pannoramic MIDI;
3DHISTECH, Ltd.). Semi-quantitative analysis was conducted as
follows: Plaque area (PA; mm2)=aorta vessel area (VA;
mm2)-aorta lumen area (LA; mm2), with
normalized plaque area=PA/VA-LA; collagen fiber=collagen fiber area
(mm2)/aorta lumen area (mm2); lipid
content=Oil Red O positive area (mm2)/aorta lumen area
(mm2).
Analysis of autophagosomes by TEM
Following anesthesia with pentobarbital sodium (50
mg/kg; i.p.), mice were sacrificed by cervical dislocation and the
aortic arch was quickly isolated. Six mice per group were used for
these experiments. A small piece of tissue was cut out and fixed in
2% glutaraldehyde-OsO4 for 2 h. Subsequent to ethanol
series dehydration, epoxy resin embedding (37°C; 3 h), ultra-thin
sectioning (70 nm), uranyl acetate (30 min) and citric acid
staining (10 min), autophagosomes were observed using a TEM (JEOL
JEM-1230, JEOL, Ltd.). All aforementioned procedures were performed
at room temperature.
Serum lipid analysis by ELISA
Following anesthesia with pentobarbital sodium (50
mg/kg; i.p.), mice were sacrificed by cervical dislocation. Blood
was collected using an orbital sinus blood collection method for
ELISA analyses, and fresh tissue samples were collected for western
blot analysis described below. Six mice per group were used for
these experiments. Blood was allowed to set for 12 h prior to being
centrifuged at 13,523 × g at 4°C for 30 min to obtain serum. TC,
TG, HDL-C and LDL-C levels were measured using ELISA kits following
the manufacturer's protocols. Optical density (OD)450
values of samples were acquired using a microplate reader
(PowerWave™ XS; BioTek Instruments, Inc.) and used to calculate
concentrations based on standard curves generated using serially
diluted standards.
Serum TNF-α, IL-1β and IL-18 analysis
by ELISA
Serum samples were prepared as described above.
TNF-α, IL-1β and IL-18 levels were measured using ELISA kits
according to the manufacturer's protocols. OD450 values
of samples were acquired using a microplate reader (PowerWave™ XS;
BioTek Instruments, Inc.) and used for calculating concentrations
based on standard curves generated using serially diluted
standards.
Measurement of Aorta mTOR, LC3, P53
and P21 protein levels by western blotting
Proteins were extracted from aorta tissues with RIPA
lysis buffer containing PMSF. Following centrifugation at 13,523 ×
g at 4°C for 30 min, the supernatant was collected for protein
quantification using the BCA method. Samples were mixed with 5X
loading buffer (cat. no. P0015; Beyotime Institute of
Biotechnology) and heated in boiling water for 10 min to denature
proteins. These treated samples (amount of protein, 30 µg) were
separated on SDS-PAGE gels (percentage of the gel, 12%) and then
transferred to PVDF membranes (cat. no. IPVH00010; Millipore). The
membranes were blocked with 5% skimmed milk (cat. no. C500625;
Sangon Biotech) for 2 h at room temperature and subsequently
incubated in the aforementioned primary antibody solutions (1:1,000
dilutions; 4°C) overnight. After washing, membranes were incubated
with the aforementioned secondary antibodies (1:1,000) for 1 h at
room temperature. An Odyssey far-infrared luminescence scanner
(cat. no. 9120; Li-COR Biosciences) was used to capture the image.
Protein band images were acquired and analyzed as integrated
absorbance (IA; IA=mean OD × area) using Image J (version 1.8.0;
National Institutes of Health), and the relative levels of target
proteins were normalized to GAPDH (target protein IA/GAPDH IA).
Statistical analysis
Statistical analysis was conducted using SPSS
software (version 23.0; IBM Corp.), and figures were generated
using GraphPad Prism 5 software (GraphPad Software, Inc.). Results
are presented as the mean ± SD. Each experiment was replicated ≥3
times. Differences between two groups were determined by unpaired
t-tests and differences among three or more groups were assessed
using one-way ANOVA followed by Student-Newman-Keuls test was then
utilized. P<0.05 was considered to indicate a statistically
significant difference.
Results
Serum lipid profiles
The Model group exhibited higher serum levels of TC
and LDL-C, and lower levels of HDL-C compared with the Control
group (P<0.01; Table I). Compared
with the Model group, the QUE group had lower serum levels of TC
and LDL-C (P<0.01; Table I).
Compared with the 3-MA group, the QUE + 3-MA group had lower serum
levels of TC and LDL-C (P<0.01; Table
I). No statistically significant differences in TG content
between the groups were observed.
 | Table I.Comparison of serum lipid levels
among groups. |
Table I.
Comparison of serum lipid levels
among groups.
| Group | Number of mice
used | TC (mmol/l) | TG (mmol/l) | LDL-C (mmol/l) | HDL-C (mmol/l) |
|---|
| Control | 6 | 2.10±0.09 | 2.14±0.03 | 1.92±0.07 | 2.04±0.03 |
| Model | 6 |
6.86±0.12a | 2.21±0.06 |
5.28±0.04a |
1.30±0.01a |
| QUE | 6 |
5.81±0.05b | 2.15±0.04 |
4.48±0.06b | 1.32±0.02 |
| 3-MA | 6 |
7.23±0.07b | 2.26±0.05 |
5.74±0.07b | 1.29±0.05 |
| QUE + 3-MA | 6 |
6.74±0.07c | 2.20±0.03 |
4.69±0.04c | 1.31±0.02 |
Serum TNF-α, IL-1β and IL-18
levels
The Model group exhibited higher serum levels of
TNF-α, IL-1β and IL-18 compared with the Control group (P<0.01;
Table II). Compared with the Model
group, serum levels of TNF-α, IL-1β and IL-18 were lower in the QUE
group (P<0.01; Table II) but
higher in the 3-MA group (P<0.01; Table II). The serum levels of all examined
cytokines were lower in the QUE + 3-MA group compared with the 3-MA
group (P<0.01; Table II).
| Table II.Comparison of serum TNF-α, IL-1β and
IL-18 levels among groups. |
Table II.
Comparison of serum TNF-α, IL-1β and
IL-18 levels among groups.
| Group | Number of mice
used | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-18 (pg/ml) |
|---|
| Control | 6 | 333.51±7.57 | 58.97±1.77 | 92.17±1.17 |
| Model | 6 |
528.62±7.15a |
110.44±0.88a |
138.78±0.93a |
| QUE | 6 |
458.95±29.51b |
97.32±1.02b |
121.51±0.44b |
| 3-MA | 6 |
611.89±12.02b |
117.57±1.38b |
141.79±0.86b |
| QUE + 3-MA | 6 |
486.27±7.41c |
102.95±1.46c |
126.90±1.82c |
Aorta pathology
H&E staining
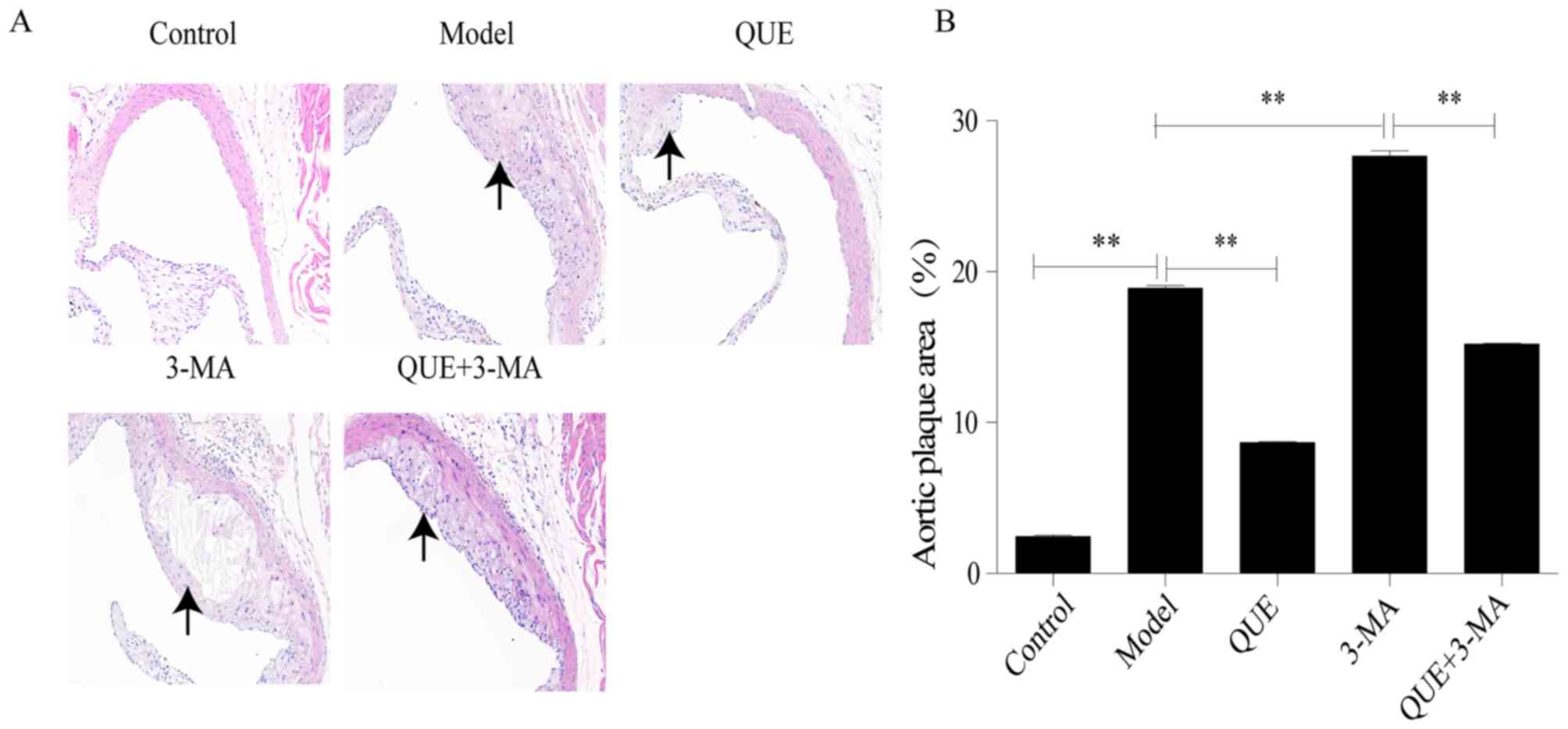
Following HFD feeding for 12 weeks, mouse aortic
roots exhibited significant formation of AS plaque compared with
the Control group (P<0.01; Fig. 1A
and B). Compared with the Model group, areas of AS plaque were
significantly smaller in the QUE group (P<0.01; Fig. 1A and B), and significantly larger in
the 3-MA group (P<0.01; Fig. 1A and
B). Compared with the 3-MA group, the QUE + 3MA group exhibited
significantly smaller areas of AS plaque (P<0.01; Fig. 1A and B).
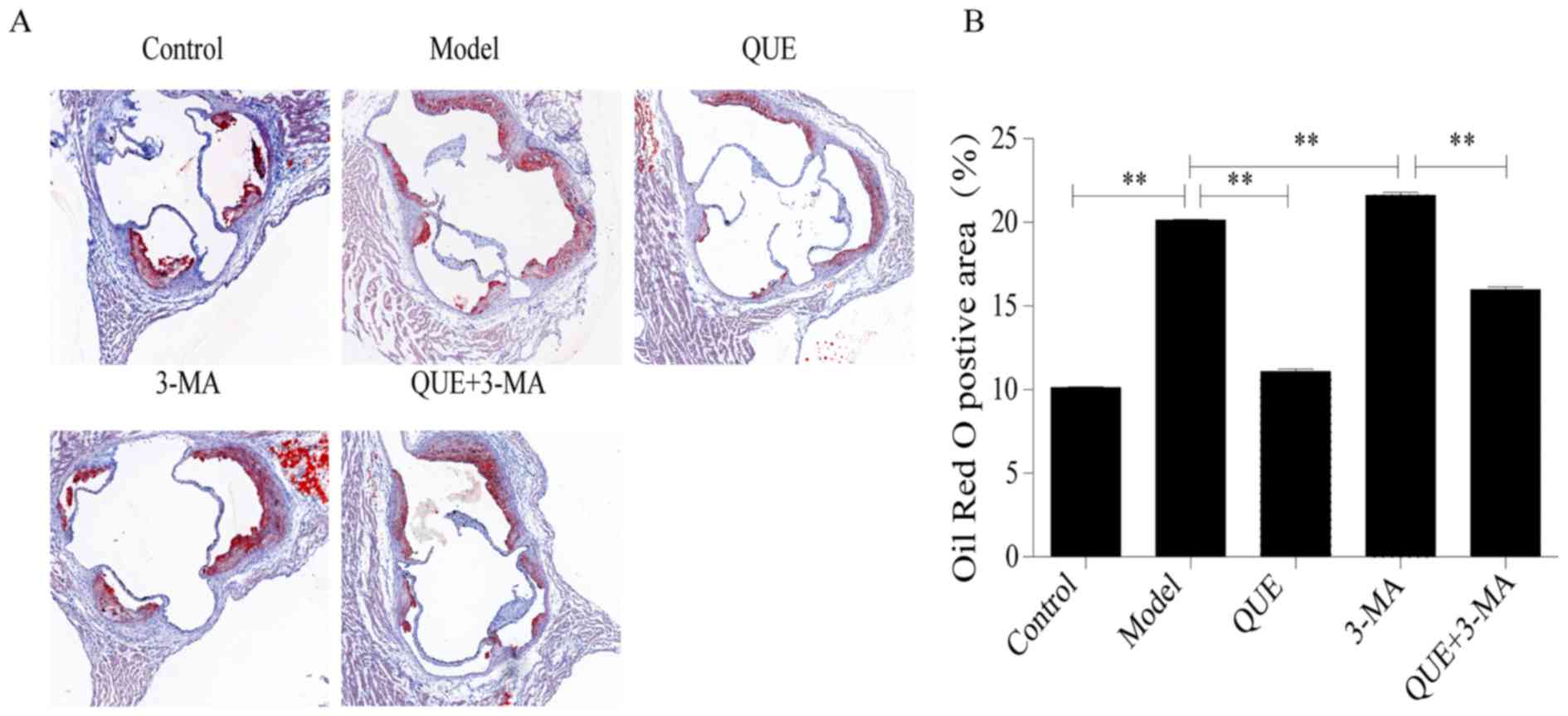
Oil red O staining
Aortic roots of mice in the Model group had larger
amounts of red-stained lipid accumulation compared with the Control
group. Compared with the Model group, lipid content was
significantly less abundant in the QUE group (P<0.01; Fig. 2A and B), and more abundant in the
3-MA group (P<0.01; Fig. 2A and
B). Lipid content in the QUE + 3-MA group were significantly
less abundant than those in the 3-MA group (P<0.01; Fig. 2A and B).
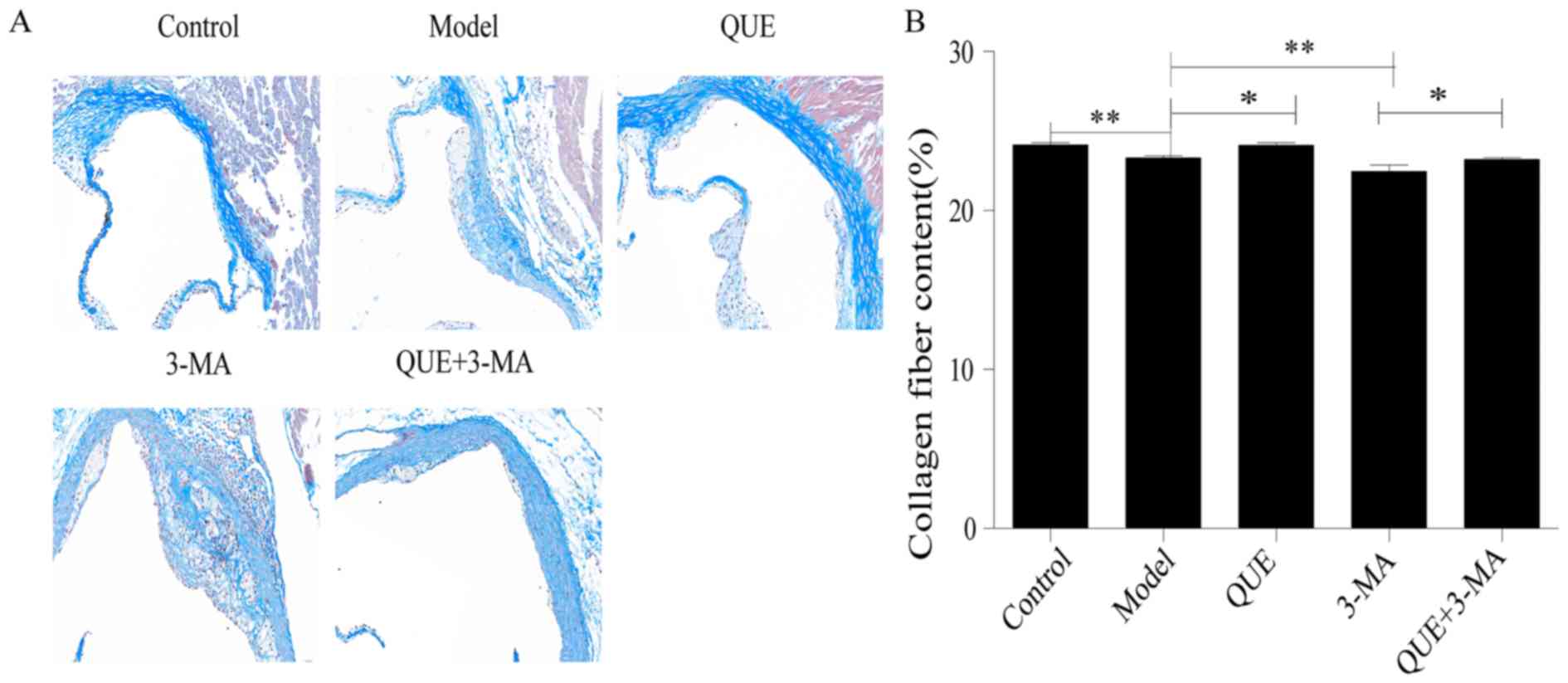
Masson staining
Aortic walls in the Control group exhibited large
amounts of collagen and smooth muscle fibers, while the AS plaques
in the Model group contained thin layers of fibrous caps and few
matrix fibers (P<0.01; Fig. 3A and
B). Compared with the Model group, the QUE group exhibited more
collagen fibers (P<0.05; Fig. 3A and
B), whilst the fibrous caps of plaques in the 3-MA group were
thinner and contained less collagen (P<0.01; Fig. 3A and B). The collagen fibers in
aortas from the QUE + 3-MA group were more abundant compared with
the 3-MA group (P<0.05; Fig. 3A and
B).
TEM observations
Macrophages in aortas of the Control group exhibited
intact structures and contained small numbers of autophagosomes,
which were less abundant in the Model group. Aortic macrophages in
the QUE group exhibited more autophagosomes Compared with the Model
group, whilst the 3-MA group exhibited no autophagosome formation.
However, unlike the 3-MA group, autophagosomes were found in the
aortic macrophages of the QUE + 3-MA group (Fig. 4A).
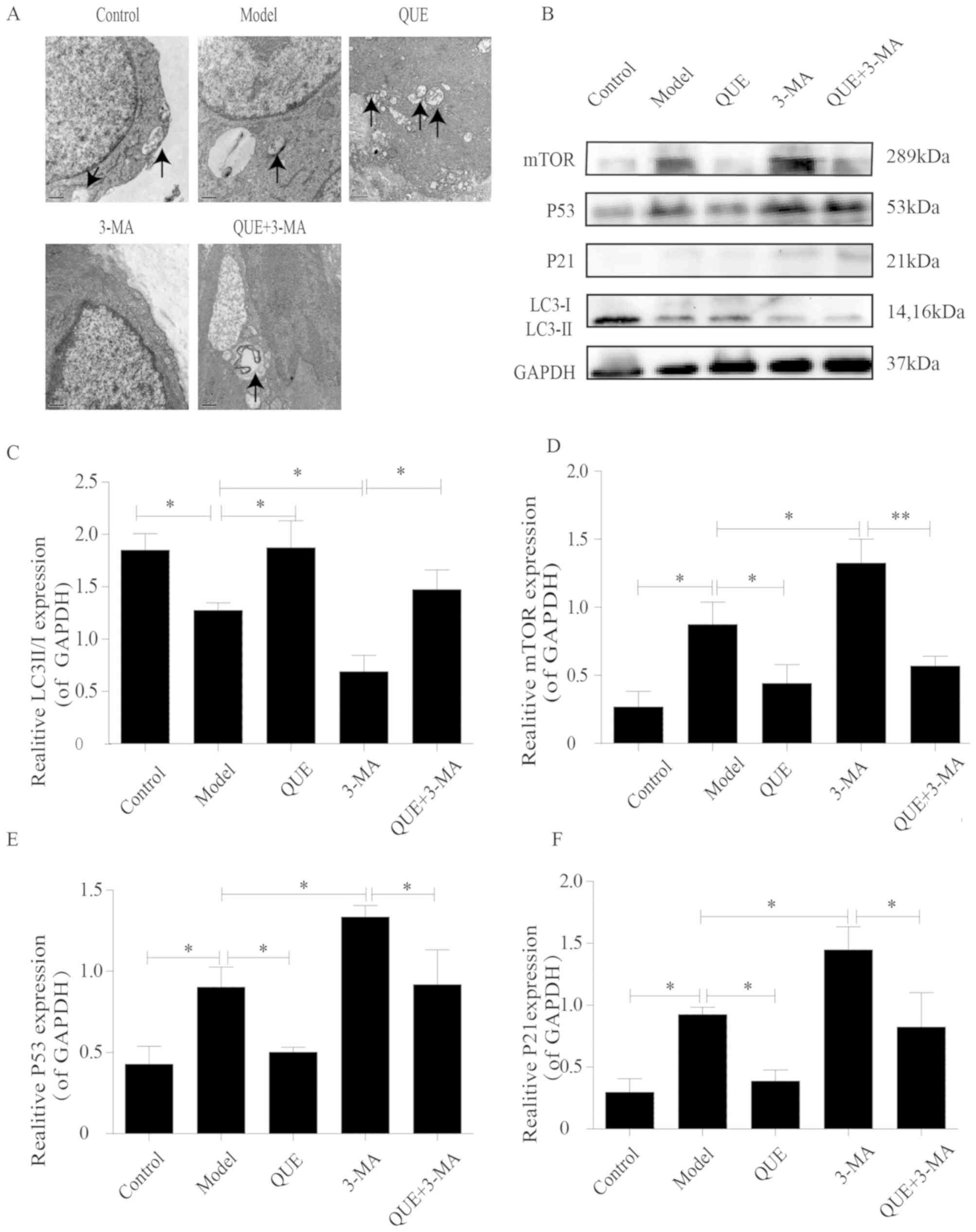 | Figure 4.Effects of QUE on autophagosomes and
protein expression levels of P53, P21 mTOR and LC3 II/I. (A) Effect
of QUE on autophagosomes of mouse aortic macrophages.
Magnification, ×20,000. n=3. (B) Western blotting results for LC3
II/I, mTOR, P53 and P21 expression in the mouse aorta. (C-F)
Expression levels of (C) LC3 II/I, (D) mTOR, (E) P53 and (F) P21.
Data are presented as the means ± SD, n=3. *P<0.05 and
**P<0.01. 3-MA, 3-methyladenine; LC3, microtubule associated
protein 1 light chain 3α; P21, cyclin dependent kinase inhibitor
1A; QUE, quercetin. |
Aortic expression of the
autophagy-associated proteins mTOR, LC3, P53 and P21
The aortas of mice in the Control group exhibited
higher of LC3 II/I ratios and lower expression levels of mTOR, P53
and P21 compared with the Model group, which had lower LC3 II/I
ratio (P<0.05; Fig. 4B and C) and
higher expression levels of mTOR (P<0.05; Fig. 4B and D), P53 (P<0.05; Fig. 4B and E) and P21 (P<0.05; Fig. 4B and F). Compared with the Model
group, the QUE group exhibited higher ratio of LC3 II/I and lower
expression of mTOR, P53, and P21 (P<0.05; Fig. 4B-F); and the 3-MA group exhibited
lower ratios of LC3 II/I and higher expression levels of mTOR, P53
and P21 (P<0.05; Fig. 4B-F).
Compared with the 3-MA group, the QUE + 3-MA group exhibited higher
ratios of LC3 II/I (P<0.05; Fig. 4B
and C) and lower expression levels of mTOR (P<0.01; Fig. 4B and D), P53 and P21 (P<0.05;
Fig. 4B, E and F).
Discussion
AS is a potential pathological basis of
cardiovascular and cerebrovascular events, and inflammation and
lipid metabolism serve an important role in its occurrence and
development (19). Autophagy is a
lysosomal degradation process that inhibits cellular senescence by
removing damaged organelles and long-lived proteins (20). Autophagy is a metabolic process that
degrades damaged cells and proteins, and defects in autophagy are
closely associated with senescence. Studies have indicated that
enhancing autophagy can prevent senescence and reduce
age-associated pathological alterations in the heart and kidney,
leading to improved health status in mice (21,22).
Autophagy, a process known to be protective in age-associated
cardiovascular disease, may serve an important role in the
initiation and development of AS (23). The pathology of AS is characterized
by vascular smooth muscle cell (VSMC) apoptosis, vascular
endothelial cell (VEC) remodeling and macrophage-mediated
inflammation (2). Weakened autophagy
takes place in all major cell types in AS plaques, resulting in
impaired macrophage apoptosis and increased senescence of blood
vessel endothelial and smooth muscle cells (2). Since VMSCs are the only cells in the
plaque fibrous cap that generate collagen fibers, promoting
autophagy of VSMCs helps stabilize the plaque and prevents its
rupture (24). It has been
demonstrated that the presence of senescent VSMCs in plaques is
associated with AS development, and impaired autophagy may increase
the senescence process of VSMCs to drive the formation of plaque
and the development AS (25).
Similarly, inhibiting autophagy promotes the senescence of VECs to
increase AS development (23).
TEM can provide an accurate and detailed view of
autophagosome structures, and it is thus taken as the gold standard
for determining autophagy levels (8). mTOR is a key molecule in the process of
autophagy induction. When energy is sufficient, mTORC1 is
activated, and autophagy is inhibited by highly phosphorylated
unc-51 like autophagy activating kinase 1 (ULK1) complex and
autophagy related 13 (Atg13) (26).
When energy is low, mTORC1 activity is inhibited, the
dephosphorylated Atg13 and ULK1 are inhibited, and subsequently
form a complex with RB1 inducible coiled-coil 1 to induce
nucleation and elongation of autophagosomes (26). LC3 is a molecular marker of
autophagy, and LC3-I is modified and transformed by ubiquitin to
become lipid-soluble LC3-II, which is involved in the formation of
autophagosomes (27). As a
transcription factor, P53 accumulates in senescent cells and
directly leads to cell senescence (28). P21 is a negative regulator of the
cell cycle, and serves a key role in cellular senescence (29). In the present study, an AS animal
model was induced by feeding ApoE−/− mice a HFD for 12
weeks. Compared with the Control group, the aortas of model mice
exhibited typical AS plaques. Oil Red O staining and serum lipid
analysis suggested a disorder in lipid metabolism; TEM results
revealed fewer autophagosomes and altered autophagy marker
proteins. Western blotting revealed increased expression in
senescence factors P53 and P21. Following treatment with 3-MA, the
area of aortic plaques increased compared with Model group. In
addition, the lipid metabolism disorder was further aggravated and
protein expression levels of mTOR, P53 or P21 were increased
further whereas the LC3 II/I ratio was decreased, no autophagosomes
were observed in the cells from the 3-MA group.
AS is a chronic inflammatory disease mediated by a
network of pro-inflammatory cytokines, including the interleukin
and TNF families (30). TNF-α is a
subclinical indicator for predicting AS. As a major
pro-inflammatory mediator found in AS plaques, TNF-α activates the
secretion of other pro-inflammatory mediators, prevents the
synthesis of the extracellular matrix, and thus negatively impacts
plaque stability (31). The
proinflammatory cytokine IL-1β serves a key role in the initiation
and development of AS. IL-1β promotes the release of
metalloproteinases which destroy the connective tissue matrix in
plaques to render them frail (32).
IL-18 is a novel member of the IL-1 family that has been
demonstrated to stimulate the expression of the inflammatory
cytokines TNF-α and interferon-γ and promote the pathogenesis of AS
(33). Higher expression levels of
IL-18 in AS plaques have been reported and are considered to
contribute to weakened stability of plaques (33). Autophagy, an important protective
response to pathological stress, serves a role in regulating
inflammation. Defects in autophagy cause the production and release
of pro-inflammatory mediators, and conversely, the induction of
autophagy helps eliminate inflammatory aggregation (34). A previous study revealed that
autophagy has an anti-AS effect by inhibiting AS-associated
inflammation, the lipid-lowering drug atorvastatin stabilizes AS
plaques and inhibits AS development by reducing the production of
the pro-inflammatory cytokines TNF-α, IL-1β and IL-18, a mechanism
which involves the induction of autophagy (35). In agreement with these findings, the
present study revealed that the mice in the Model group exhibited
higher serum levels of TNF-α, IL-1β and IL-18 compared with the
Control group, and serum levels of these cytokines were further
increased following treatment with the autophagy inhibitor
3-MA.
QUE is a flavonoid compound with multiple biological
activities, including anti-inflammatory, anti-senescence and
cardiovascular protective properties (36). TCM herbs South Dodder Seed Chinese
Dodder Seed and Taxillus sutchuenensis (Lecomte) Danser
contain QUE (37). A previous study
in our laboratory reported that QUE reduced ox-LDL-induced damage
in RAW264.7 cells, reduced lipid precipitation and delayed cellular
senescence (14). In a
peroxide-induced oxidative stress model in human umbilical vein
endothelial cells, QUE has been demonstrated to inhibit vascular
cell adhesion molecule 1 and CD80 expression, resulting in an
anti-AS effect (38). In a study
using HFD-induced AS in ApoE−/− mice, QUE was found to
exert its anti-AS effects by suppressing scavenger receptor
expression and reducing ox-LDL absorption (39). It has previously been reported that
QUE protects endothelial cells and effectively prevents AS by
upregulating autophagy via the ERK signaling pathway (15). In an in vitro study using
RSC96 cells incubated in high-glucose medium, QUE reduced cell
damage, which was associated with upregulated expression levels of
LC3 and increased numbers of autophagosomes (40).
The present study demonstrated that QUE could
alleviate AS lesions induced by a HFD in ApoE−/− mice,
reduce lipid accumulation in aortic roots and reduce serum levels
of TC and LDL-C, as well as the expression levels of TNF-α, IL-1β
and IL-18. TEM revealed an increased quantity of autophagosomes in
the aortas of mice in the QUE group compared with the Model group.
In addition, the ratio of LC3 II/I in mouse aortas was
significantly increased, while mTOR, P53 and P21 protein expression
levels were downregulated compared with the Model group. The
aforementioned results suggested that the mechanism of QUE in the
inhibition of AS may be associated with the enhancement of
autophagy and the delay of senescence.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81873348),
the Shanghai Nature Science Fund (grant no. 16ZR1433900) and the
Shanghai Health and Family Planning Commission Fund (grant no.
201640217).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
DS, HC, CC and SX conceived the experiments and
experimental plan. HC, QJ and LY performed the experiments and
collected and analyzed the data. HC and QJ wrote the manuscript,
DS, HC and QJ reviewed and edited the manuscript. All authors
approved the final version of these manuscript and figures.
Ethics approval and consent to
participate
The present study was approved by the Laboratory
Animal Welfare and Ethics Committee of Shanghai University of
Traditional Chinese Medicine (approval no. PZSHUTCM18113002;
Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grootaert MOJ, Lynn R, Schrijvers DM, De
Meyer GRY and Martinet W: Defective autophagy in atherosclerosis:
To die or to senesce? Oxid Med Cell Longe 2018. 76870832018.
|
|
3
|
Wang JC and Bennett M: Aging and
atherosclerosis: Mechanisms, functional consequences, and potential
therapeutics for cellular senescence. Cir Res. 111:245–259. 2012.
View Article : Google Scholar
|
|
4
|
Sun L, Dou F, Chen J, Chi H, Xing S, Liu
T, Sun S and Chen C: Salidroside slows the progression of EA.hy926
cell senescence by regulating the cell cycle in an atherosclerosis
model. Mol Med Rep. 17:257–263. 2018.PubMed/NCBI
|
|
5
|
Kim YY, Jee HJ, Um JH, Kim YM, Bae SS and
Yun J: Cooperation between p21 and Akt is required for
p53-dependent cellular senescence. Aging Cell. 16:1094–1103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kitada M, Ogura Y and Koya D: The
protective role of Sirt1 in vascular tissue: Its relationship to
vascular aging and atherosclerosis. Aging (Albany NY). 8:2290–2307.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Netea-Maier RT, Plantinga TS, van de
Veerdonk FL, Smit JW and Netea MG: Modulation of inflammation by
autophagy: Consequences for human disease. Autophagy. 12:245–260.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gurumurthy S, Xie SZ, Alagesan B, Kim J,
Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, et al:
The Lkb1 metabolic sensor maintains haematopoietic stem cell
survival. Nature. 468:659–663. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jacquin E, Leclerc-Mercier S, Judon C,
Blanchard E, Fraitag S and Florey O: Pharmacological modulators of
autophagy activate a parallel noncanonical pathway driving
unconventional LC3 lipidation. Autophagy. 13:854–867. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li C, Zhang WJ and Frei B: Quercetin
inhibits LPS-induced adhesion molecule expression and oxidant
production in human aortic endothelial cells by p38-mediated Nrf2
activation and antioxidant enzyme induction. Redox Biol. 9:104–113.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhaskar S, Sudhakaran PR and Helen A:
Quercetin attenuates atherosclerotic inflammation and adhesion
molecule expression by modulating TLR-NF-κB signaling pathway. Cell
Immunol. 310:131–140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cui Y, Hou P, Li F, Liu Q, Qin S, Zhou G,
Xu X, Si Y and Guo S: Quercetin improves macrophage reverse
cholesterol transport in apolipoprotein E-deficient mice fed a
high-fat diet. Lipids Health Dis. 16:92017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li S, Cao H, Shen D, Jia Q, Chen C and
Xing SL: Quercetin protects against ox-LDL-induced injury via
regulation of ABCAl, LXR-α and PCSK9 in RAW264.7 macrophages. Mol
Med Rep. 18:799–806. 2018.PubMed/NCBI
|
|
15
|
Zhi K, Li M, Bai J, Wu Y, Zhou S, Zhang X
and Qu L: Quercitrin treatment protects endothelial progenitor
cells from oxidative damage via inducing autophagy through
extracellular signal-regulated kinase. Angiogenesis. 19:311–324.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J: Guide for the care and use of
medical laboratory animals. Shanghai Sci Tech Publishers; 2012
|
|
17
|
Xiye F: Medical laboratory zoology.
People's Health Press. 156:1995.
|
|
18
|
Wu X, He L, Chen F, He X, Cai Y, Zhang G,
Yi Q, He M and Luo J: Impaired autophagy contributes to adverse
cardiac remodeling in acute myocardial infarction. PLoS One.
9:e1128912014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zimmer S, Grebe A, Bakke SS, Bode N,
Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek
A, et al: Cyclodextrin promotes atherosclerosis regression via
macrophage reprogramming. Sci Transl Medi. 8:333ra502016.
View Article : Google Scholar
|
|
20
|
Kwon Y, Kim JW, Jeoung JA, Kim MS and Kang
C: Autophagy is pro-senescence when seen in close-up, but
anti-senescence in long-shot. Mol Cells. 40:607–612.
2017.PubMed/NCBI
|
|
21
|
Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi
M, McMillan KL, He C, Ting T, Liu Y, Chiang WC, et al: Disruption
of the beclin 1-BCL2 autophagy regulatory complex promotes
longevity in mice. Nature. 558:136–140. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Revuelta M and Matheu A: Autophagy in stem
cell aging. Aging Cell. 16:912–915. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiong Y, Yepuri G, Forbiteh M, Yu Y,
Montani JP, Yang Z and Ming XF: ARG2 impairs endothelial autophagy
through regulation of MTOR and PRKAA/AMPK signaling in advanced
atherosclerosis. Autophagy. 10:2223–2238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tai S, Hu XQ, Peng DQ, Zhou SH and Zheng
XL: The roles of autophagy in vascular smooth muscle cells. Int J
Cardiol. 211:1–6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grootaert MO, da Costa Martins PA, Bitsch
N, Pintelon I, De Meyer GR, Martinet W and Schrijvers DM: Defective
autophagy in vascular smooth muscle cells accelerates senescence
and promotes neointima formation and atherogenesis. Autophagy.
11:2014–2032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan HX, Russell RC and Guan KL:
Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient
stress-induced autophagy. Autophagy. 9:1983–1995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu J, Shen Y, Qian HY, Liu LJ, Zhou BC,
Xiao Y, Mao JN, An GY, Rui MZ, Wang T and Zhu CL: Effects of mild
hypothermia on the ROS and expression of caspase-3 mRNA and LC3 of
hippocampus nerve cells in rats after cardiopulmonary
resuscitation. World J Emerg Med. 5:298–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tran D, Bergholz J, Zhang H, He H, Wang Y,
Zhang Y, Li Q, Kirkland JL and Xiao ZX: Insulin-like growth
factor-1 regulates the SIRT1-p53 pathway in cellular senescence.
Aging Cell. 13:669–678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Si X, Shao C, Li J, Jia S, Tang W, Zhang
J, Yang J, Wu X and Luo Y: Loss of p21 promoted tumorigenesis in
the background of telomere dysfunctions induced by TRF2 and Wrn
deficiency. Int J Biol Sci. 14:165–177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
ten Kate GL, Sijbrands EJ, Staub D, Coll
B, ten Cate FJ, Feinstein SB and Schinkel AF: Noninvasive imaging
of the vulnerable atherosclerotic plaque. Curr Probl Cardiol.
35:556–591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee AS, Kim JS, Lee YJ, Kang DG and Lee
HS: Anti-TNF-α activity of Portulaca oleracea in vascular
endothelial cells. Int J Mol Sci. 13:5628–5644. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu Z, Lerman LO, Tang H, Barber C, Wan L,
Hui MM, Furenlid LR and Woolfenden JM: Inflammation imaging of
atherosclerosis in Apo-E-deficient mice using a (99m)Tc-labeled
dual-domain cytokine ligand. Nucl Med Biol. 41:785–792. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Şahin M, Ugan Y, Tunç ŞE, Akın Ş, Köroğlu
B, Kutlucan A, Sütçü R, Yeşildağ A and Kılbaş A: Potential role of
interleukin-18 in patients with rheumatoid arthritis-associated
carotid intima-media thickness but not insulin resistance. Eur J
Rheumatol. 1:135–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Wang G, Feng D, Zu G, Li Y, Shi X,
Zhao Y, Jing H, Ning S, Le W, et al: Targeting the
miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis
in intestinal ischemia/reperfusion. Cell Death Dis. 9:4832018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng S, Xu LW, Che XY, Xiao QQ, Pu J, Shao
Q and He B: Atorvastatin inhibits inflammatory response, attenuates
lipid deposition, and improves the stability of vulnerable
atherosclerotic plaques by modulating autophagy. Front Pharmacol.
9:4382018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang M, Xie Z, Gao W, Pu L, Wei J and Guo
C: Quercetin regulates hepatic cholesterol metabolism by promoting
cholesterol-to-bile acid conversion and cholesterol efflux in rats.
Nutr Res. 36:271–279. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun X, Yamasaki M, Katsube T and Shiwaku
K: Effects of quercetin derivatives from mulberry leaves: Improved
gene expression related hepatic lipid and glucose metabolism in
short-term high-fat fed mice. Nutr Res Pract. 9:137–143. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang D, Liu X, Liu M, Chi H, Liu J and Han
H: Protective effects of quercetin and taraxasterol against
H2O2-induced human umbilical vein endothelial
cell injury in vitro. Exp Ther Med. 10:1253–1260. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu L, Gao C, Yao P and Gong Z: Quercetin
alleviates high-fat diet-induced oxidized low-density lipoprotein
accumulation in the liver: Implication for autophagy regulation.
Biomed Res Int 2015. 6075312015.
|
|
40
|
Qu L, Xiao-Chun L, Gu B, Zhang H, Dai W
and SHI Y: Quercetin up-regulates autophagy in RSC96 cells
culutured in high glucose via the pathway of Akt-Mtor. Basic Clin
Med. 5:596–602. 2015.
|