Introduction
During the initial stages of vision development,
lens opacity may arise, which is mainly caused by congenital
cataracts and may lead to deprivation amblyopia (1). According to the literature,
approximately one-third of infantile blindness cases are caused by
congenital cataracts (2,3). The incidence of congenital cataracts is
6.31/100,000 individuals (4) in
industrialized countries, while that in developing countries is
assumed to be notably higher (5,6).
As the worldwide leading cause of impaired vision in
children, the hereditary modes of congenital cataracts include
autosomal dominant, autosomal recessive and X-linked hereditary
modes. Among these modes, autosomal-dominant congenital cataract
(ADCC) is most common (4); variable
phenotypes occur in different families (7,8). To
date, ≥23 genes have been associated with ADCC. These genes are
mainly involved in the formation of the lens, including the
crystallin (CRY) genes [α-CRY (CRYA), β-CRY (CRYB) and γ-CRY
(CRYG)], lens-specific connexin (Cx) genes (Cx43, Cx46 and Cx50),
major intrinsic protein gene or aquaporine, cytoskeletal structural
protein genes, paired-like homeodomain transcription factor 3,
avian musculoaponeurotic fibrosarcoma, heat shock transcription
factor 4, beaded filament structural protein 2 and non-muscle
myosin heavy chain IIA (MYH9) (7,9,10). In addition, ephrin receptor subfamily
(EPHA)1 and −2, RPGR-interacting protein 1 (RPGRIP1) and paired box
6 (PAX6) serve a vital role in the pathogenesis of cataracts.
Of note, mutations in the same gene may lead to
different phenotypes (7,8). For individuals in the same family, ADCC
may present with different clinical features. The present study
reported variable clinical features in patients from the same
family as well as from different families. Therefore, in the
present study, targeted gene capture was performed using a
hereditary-eye-disease-enriching panel and next-generation
sequencing to identify the mutations of six Chinese families with
ADCC, including two novel mutations in MYH9 (c.4150G>C) and
CRYBA4; c.169T>C). The results of the present study may provide
insight into the mutations associated with the development of
ADCC.
Subjects and methods
Recruitment of patients and clinical
evaluation
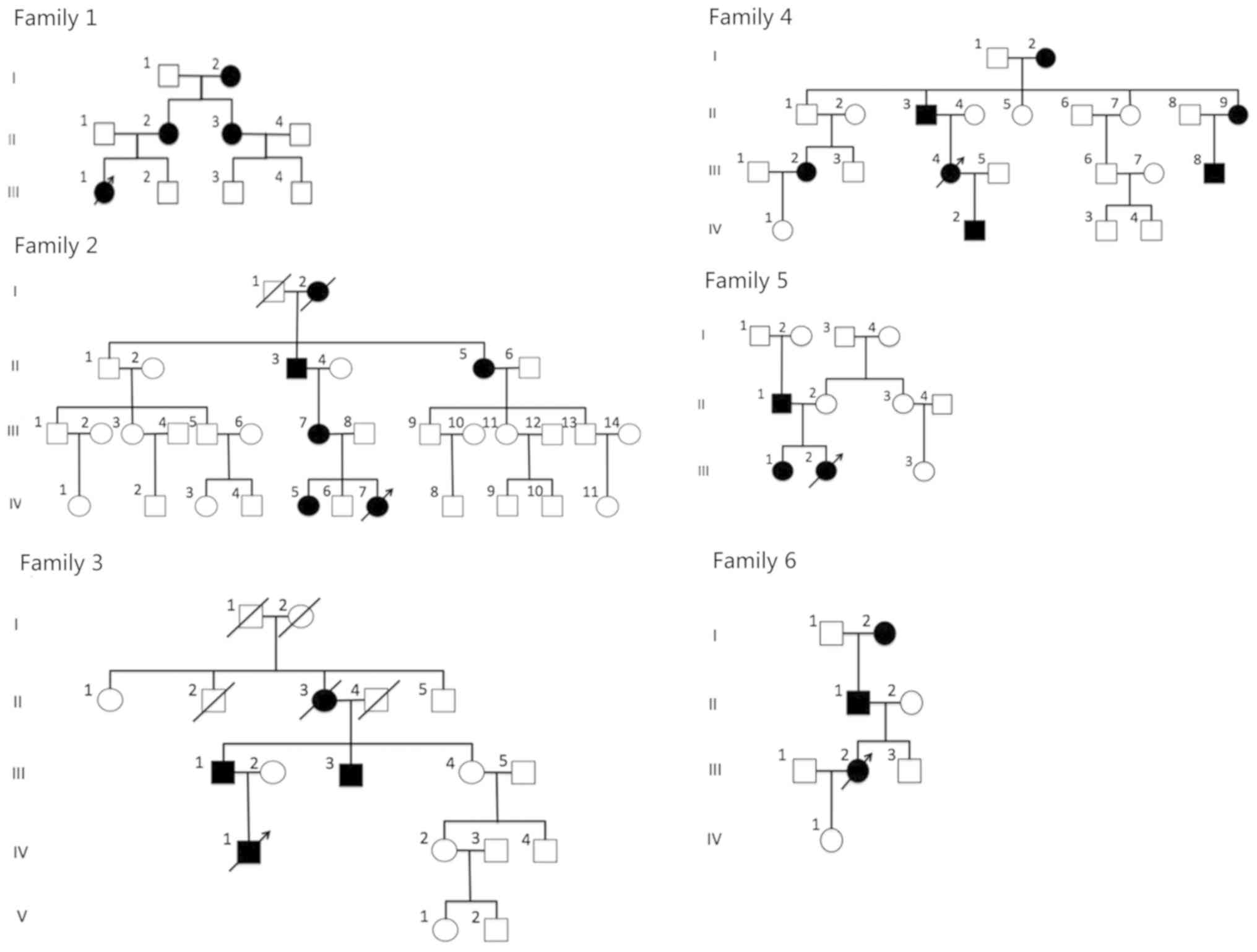
A total of 6 Chinese families with 103 members in
total (96 members alive) were recruited from the Peking University
Third Hospital (Beijing, China). The pedigree charts are provided
in Fig. 1. The probands were as
follows: II-1 in family 1; IV-7 in family 2; IV-1 in family 3;
III-4 in family 4; III-2 in family 5 and III-2 in family 6. A total
of 27 patients were affected by ADCC (4 patients from family 1; 6
patients from family 2; 4 patients from family 3; 7 patients from
family 4; 3 patients from family 5 and 3 patients from family 6).
Detailed family and medical histories, and a series of results from
ophthalmic examinations, were obtained for the family members,
including visual acuity, slit lamp examination and fundus
examination with dilated pupils. A total of 100 normal controls
were also recruited. All participating individuals provided
informed consent in accordance with the Declaration of Helsinki.
The present study was approved by the Peking University Third
Hospital Medical Ethics Committee (Beijing, China).
Genomic DNA extraction
Venous blood (2 ml) was collected from the
participating family members and was stored in BD Vacutainers (BD
Biosciences) containing EDTA to prevent coagulation. Genomic DNA
was extracted from the white blood cells using a DNA Extraction kit
(Tiangen Biotech Co., Ltd.), and was quantified with a NanoDrop
2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
Mutation screening
Following the extraction of DNA from the white blood
cells of the probands from in each family, a specific eye disease
enrichment panel was used to capture the gene mutations in the
samples (cat. no. OT021-29; MyGenostics GenCap Enrichment
Technologies, Inc.). A minimum of 3 µg DNA was used for analysis in
the indexed Illumina libraries according to the manufacturer's
protocols (MyGenostics GenCap Enrichment Technologies, Inc.). The
target genes in the enriched libraries were captured in accordance
with the MyGenostics Targeted Genes Capture protocols and were then
sequenced on an Illumina NextSeq 500 sequencer (Illumina, Inc.) for
paired-end reads of 150 bp.
A total of 663 disease-associated genes in the panel
were linked to hereditary eye diseases. Among these genes, 135 were
associated with cataracts (57 genes were associated with congenital
cataracts; the others were associated with hereditary eye diseases
with opacified lens).
Following sequencing, raw image files were processed
using Bcl2Fastq software (Bcl2Fastq 2.18.0.12; Illumina, Inc.) for
base calling and raw data generation. Low-quality variations were
filtered out using a quality score ≥20. Short Oligonucleotide
Analysis Package (SOAP) aligner software (SOAP2.21; soap.genomics.org.cn/soapsnp.html) was
then used to align the clean reads to the reference human genome.
PCR duplicates were removed and single nucleotide polymorphisms
(SNPs) were identified by the GATK (version 4.1.2.0; http://www.broadinstitute.org/gsa/wiki/index.php/Home_Page)
and the SOAPsnp (http://soap.genomics.org.cn/soapsnp.html) programs.
Identified SNPs and insertion/deletions were annotated using the
Exome-assistant program (http://122.228.158.106/exomeassistant).
DNA samples from other individuals of the families
were used to validate all mutations identified by Sanger sequencing
on an ABI3730XL analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The coding regions of the candidate genes [MYH9,
CRYBA4 (c.169T>C), RPGRRIP1, wolframin (WFS1), CRYBA4
(c.26C>T), EPHA2 and PAX6] were amplified by PCR: An initial
denaturation of 98°C for 30 sec, 15 cycles of denaturation at 98°C
for 25 sec, annealing at 65°C for 30 sec, extension at 72°C for 30
sec, and a final extension of 72°C for 5 min; primers are listed in
Table I. The coding regions were
then screened by using bidirectional sequencing followed by
analysis with Chromas 2.33 (http://technelysium.com.au/wp/chromas/); comparisons
were made using reference sequences in the National Center for
Biotechnology Information (NCBI) database.
 | Table I.Primer sequences for candidate genes
for amplification and sequencing. |
Table I.
Primer sequences for candidate genes
for amplification and sequencing.
| Gene name | Location of the
mutation | Forward primer
name | Forward primer
sequence (5′-3′) | Reverse primer
name | Reverse primer
sequence (3′-5′) |
|---|
| MYH9 | chr22-36688226 | F086-A1_F |
AGCCCAGGCTTTCTCTGATG | R086-A1_R |
TTTCATAACTGGGCAGATCCC |
| CRYBA4 | chr22-27021455 | F434-E9_F |
AAAAATGTCTCCAGCCATCG | R434-E9_R |
GCCCCATTTCAAGATGAAGA |
| RPGRIP1 | chr14-21794291 | F163-C3_F |
CACCACAGATCCTAGGCTTCA | R163-C3_R |
TCTGCTCTGTTGCTCTTGACA |
| WFS1 | chr4-6302757 | F165-H2_F |
TCCCGCTGGTCATCTTCTAC | R165-H2_R |
CTTCAGGTAGGGCCAATTCA |
| CRYBA4 | chr22-27018586 | F429-D7_F |
AGAGTGGGGCTCAGAGTCAA | R429-D7_R |
GGTCAACTTTGGGAACCAGA |
| EPHA2 | chr1-16455928 | F386-F1_F |
CAAAGAGAGGAGCATTGAGGG | R386-F1_R |
AGGTTAGGGAGCAGCAGGTG |
| PAX6 | chr11-31824384 | F411-F3_F |
CAGTAAGAAATGAAGAGAGGGCG | R411-F3_R |
GATGAGGATGCATTGTGGTTG |
Bioinformatics analysis
Based on the results obtained from the mutation
analysis, several bioinformatics analyses were performed. The
potential effects of an amino acid substitution on the structure
and function of a protein were predicted using Protein Variation
Effect Analyzer (PROVEAN v1.1.3; http://provean.jcvi.org/index.php) (11) Sorting intolerant from tolerant (SIFT;
http://sift.bii.a-star.edu.sg/)
(12), Mutation Taster (http://www.mutationtaster.org) (13), Polymorphism and Phenotyping version 2
(PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/) (14) and Swiss model (https://swissmodel.expasy.org) (15).
Results
Clinical features
Analysis of the family history and medical history
indicated that none of the patients had any other systemic diseases
that may be associated with the development of cataracts or
ophthalmic diseases. In the present study, 27 patients with ADCC
(25 alive) in 6 Chinese families were identified, including 9 males
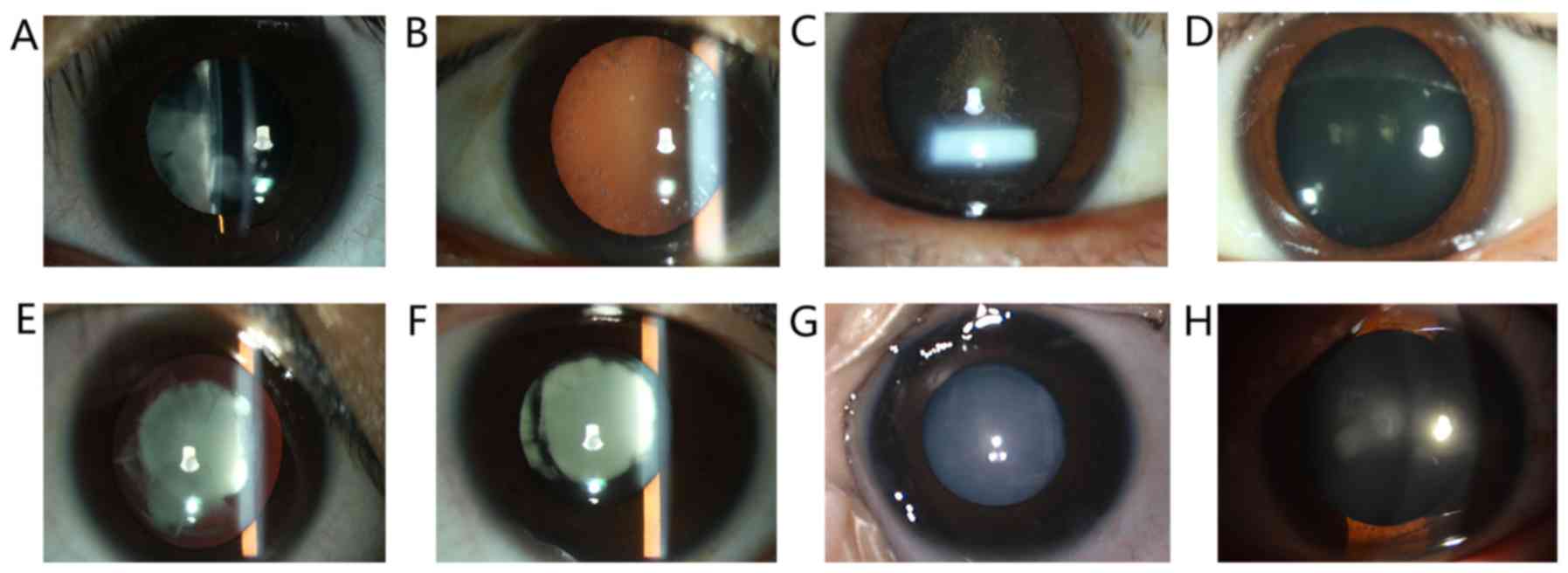
and 18 females (16 alive). Their slit lamp examination images are
presented in Fig. 2. Among these
patients, opacified lens was observed at a young age in certain
subjects, including patients in families 2, 4, 5 and 6. The
youngest patient of the 6 families was 3 months old. The parents
observed an opacified lens for the first time when the patient was
2 months old. The non-transparent area quickly progressed in the
past 2 months and eventually, the whole lens was opacified
(Fig. 2G); patients in families 1
and 3 gradually developed symptoms of ADCC after 11 years of
age.
Of note, the proband of family 1 and their mother
(suffering from ADCC) presented with a dissimilar clinical feature
that affected her visual acuity in a different manner (Table II). The proband of family 1 was a
13-year-old female with irregular nuclear cataracts in the
bilateral eyes (Fig. 2A) and her
visual acuity was 20/200 OU. However, her mother with the same
mutation in the MYH9 gene only presented with mild symptoms of
ADCC; spot-like opacity was observed in the peripheral area of the
lens (Fig. 2B), which resulted in a
slight reduction in visual acuity (20/25 OU).
| Table II.Clinical features of selected
patients. |
Table II.
Clinical features of selected
patients.
| Family number | Affected
individual | Gender | Age | Visual acuity | Phenotype |
|---|
| 1 | III-1 | Female | 13 y | 20/200 | Nuclear
cataract |
| 1 | II-2 | Female | 35 y | 20/25 | Spot-like cataract
in the peripheral area of the lens |
| 2 | IV-7 | Female | 3 m | Not determined | Irregular spot-like
cataract in the middle of the lens |
| 3 | IV-1 | Male | 14 y | 20/40 | Two round-shaped
opacifications in the middle of the lens |
| 4 | III-4 | Female | 22 y | 20/200 | Irregular nuclear
cataract |
| 4 | II-3 | Male | 53 y | 20/200 | Irregular nuclear
cataract |
| 5 | III-2 | Female | 4 m | Not determined | Extensive
opacification of the lens |
| 6 | III-2 | Female | 23 y | 20/100 | Round-shaped
opacification in the middle of the lens |
Mutation screening and bioinformatics
analysis
High-throughput screening of the blood samples of
all of the probands was performed in the present study. Compared
with the normal gene sequences, each proband had >3,000
nucleotide alterations in hereditary eye disease-associated genes.
Few of these nucleotide alterations did not result in changes in
the amino acid sequence or were not in accordance with the
hereditary model of ADCC; thus, these alterations were excluded.
Only the genes known to be associated with ADCC were selected.
Among them, variants in MYH9, RPGRRIP1, WFS1, EPHA2 and PAX6 of the
probands were indicated to be potentially pathogenic. Sanger
sequencing was performed with the blood samples of other
individuals in the families. The results revealed that heterozygous
MYH9 c.4150G>C, CRYBA4 c.169T>C, RPGRIP1 c.2669G>A, WFS1
c.1235T>C, CRYBA4 c.26C>T, EPHA2 c.2663+1G>A and PAX6
c.11-2A>G mutations were only detected in affected individuals.
These mutations were not detected in the 100 controls and were
considered to be associated with ADCC (Fig. 3). The evolutionary conservation
results of these genes were also shown in Fig. 4.
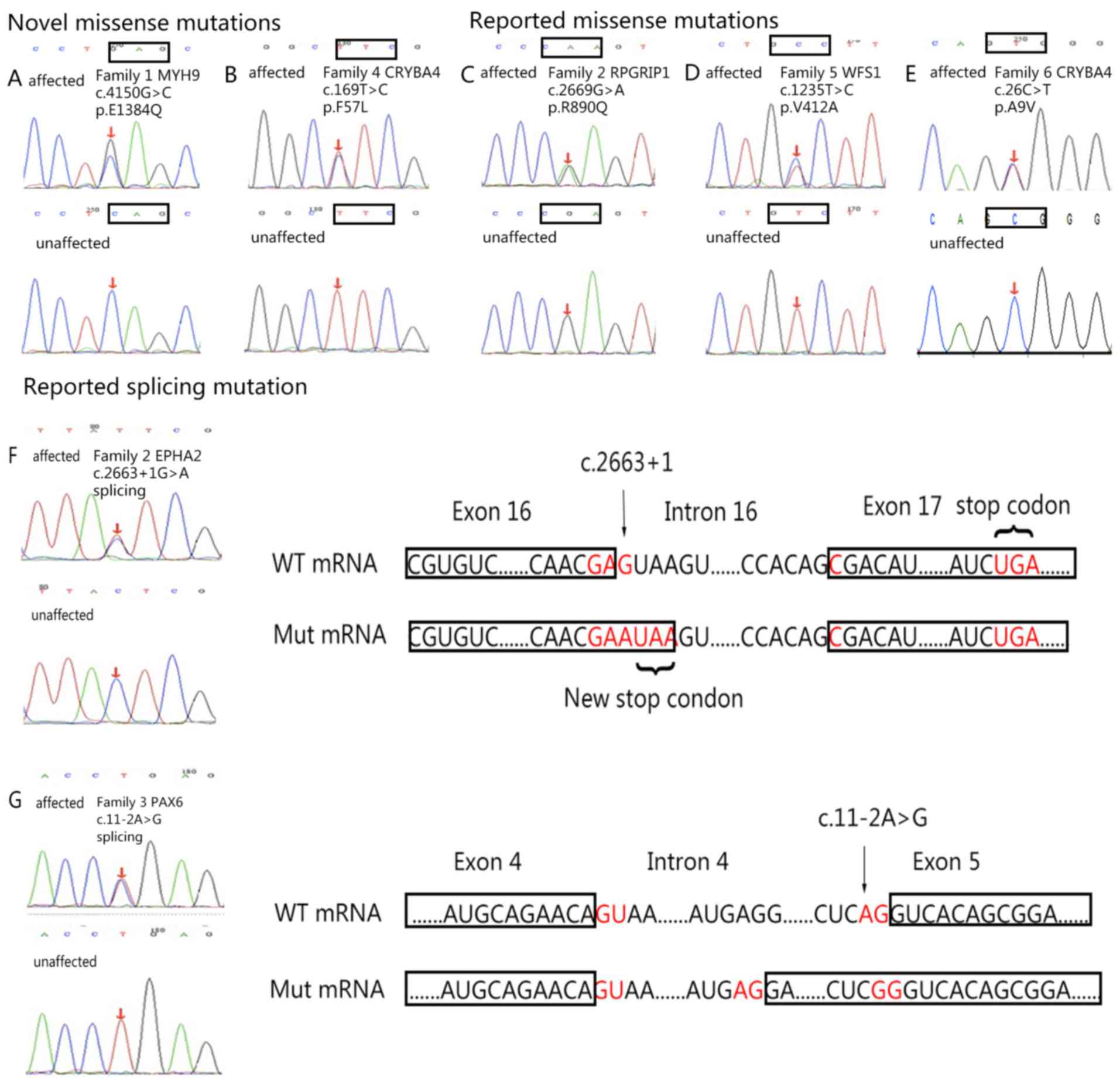 | Figure 3.DNA sequences of affected and
unaffected individuals. (A) MYH9 (c.4150G>C). (B) CRYBA4
(c.169T>C). (C) RPGRIP1 (c.2669G>A). (D) WFS1 (c.1235T>C).
(E) CRYBA4 (c.26C>T). (F) EPHA2 (c.2663+1G>A); the splicing
mutation was determined in the first loci of intron 16, which leads
to the substitution from aspartic acid (D, encoded by GAC) to
glutamate (E, encoded by GAA), as well as the generation of a new
stop codon. This new stop codon causes the loss of 34 amino acids
encoded by exon 17. (G) PAX6 (c.11-2A>G); the c.11-2A>G
mutation in the PAX6 gene leads to the substitution from serine (S,
encoded by AGU) to arginine (R, encoded by AGA) and the addition of
56 amino acids. Associated codons are indicated with rectangles.
CRYBA4, β-crystallin A4; EPHA2, EPH receptor 2; MYH9, myosin heavy
chain 9; PAX6, paired box 6; RPGR-interacting protein 1; WFS1,
wolframin; WT, wild-type; Mut, mutant. |
A novel damaging missense mutation:
MYH9 c.4150G>C
The mutation c.4150G>C in MYH9 was identified in
family 1 (Fig. 3A). This missense
mutation led to an amino acid substitution from glutamate to
glutamine at codon 1,384 (p.E1384Q) in MYH9.
According to the analysis with Mutation Taster,
glutamate was highly conserved at codon 1,384 of MYH9 among
different species (Fig. 4A).
Substituting glutamine at this codon was predicted to be pathogenic
by Mutation Taster (‘disease-causing’), PROVEAN (‘deleterious’ with
a score of −2.64), SIFT (‘damaging’ with a score of 0.023) and
PolyPhen-2 (‘probably damaging’ with a score of 1.000, sensitivity
of 0.00 and specificity of 1.00).
A novel damaging missense mutation:
CRYBA4 c.169T>C
The c.169T>C mutation in CRYBA4 was identified in
family 4 (Fig. 3B). This missense
mutation led to an amino acid substitution from phenylalanine to
leucine at codon 57 (p.F57L) in CRYBA4.
According to analysis with Mutation Taster,
phenylalanine is conserved at codon 57 of CRYBA4 in different
species (Fig. 4B). This mutation was
predicted to be pathogenic by Mutation Taster (‘disease-causing’),
PROVEAN (‘deleterious’ with a score of −5.08), SIFT (‘damaging’
with a score of 0.002) and PolyPhen-2 (‘probably damaging’ with a
score of 0.968, sensitivity of 0.74 and specificity of 0.96). The
three-dimensional (3D) models of wild-type and mutated
CRYBA4-encoding protein were then generated by Swiss model
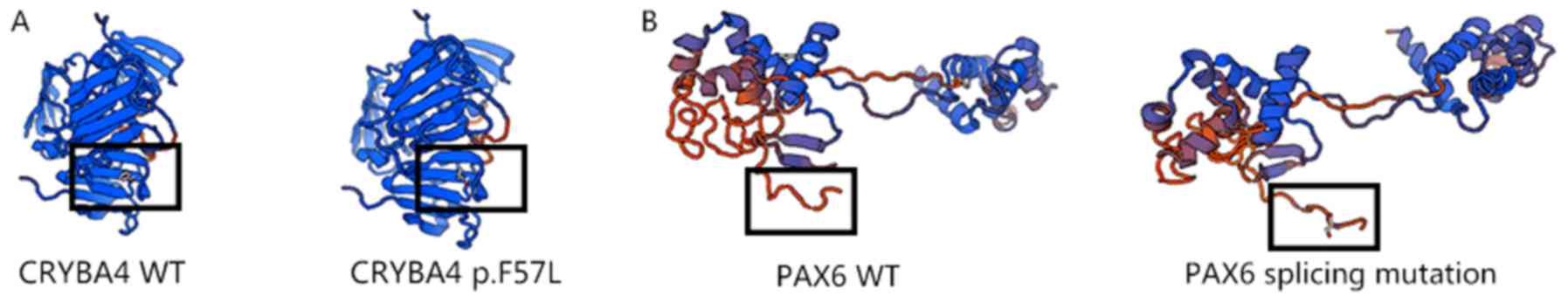
(Fig. 5).
A novel neutral missense mutation:
RPGRIP1 c.2669G>A
The c.2669G>A mutation in RPGRIP1 was identified
in family 2 (Fig. 3C). This missense
mutation may cause a ‘Neutral’ (PROVEAN prediction with a score of
1.41) and ‘Tolerated’ (SIFT prediction with a score of 0.867) amino
acid substitution from arginine to glutamine at codon 890 (p.R890Q)
in RPGRIP1. The arginine residue was not highly conserved in
different species (Fig. 4C). The
prediction made with Mutation Taster was ‘polymorphism’ and
PolyPhen-2 analysis scored this mutation as 0.000 (sensitivity:
1.00; specificity: 0.00).
A previously reported missense
mutation: WFS1 c.1235T>C
The mutation c.1235T>C in WFS1 was identified in
family 5 (Fig. 3D). This missense
mutation leads to an amino acid substitution from valine to alanine
at codon 412 (p.V412A).
Analysis with Mutation Taster revealed that valine
is conserved at codon 412 of WFS1 in different species (Fig. 4D); the mutation was then predicted to
be pathogenic by Mutation Taster (‘disease-causing’), SIFT
(‘damaging’ with a score of 0.021) and PolyPhen-2 (‘probably
damaging’ with a score of 0.981, sensitivity of 0.75 and
specificity of 0.96). However, analysis with PROVEAN indicated a
prediction of ‘neutral’ with a score of −2.29.
This mutation was first reported by Choi et
al (16) as a candidate gene for
familial nonsyndromic hearing loss; however, according to the
results for family 5, it may be possible that this mutation also
causes ADCC.
A reported missense mutation: CRYBA4
c.26C>T
The c.26C>T mutation was identified in CRYBA4 in
family 6 (Fig. 3E), which causes an
amino acid substitution from alanine to valine at codon 9 (p.A9V).
This mutation has been reported by Sun et al (17) and Zhai et al (18).
The present study reported the clinical features of
ADCC in the families analyzed; however, bioinformatics analysis
indicated that this mutation may not be pathogenic. According to
the results of bioinformatics analysis, it is possible that the
c.26C>T mutation is not responsible for the disease. According
to Mutation Taster, alanine is conserved in different species
(Fig. 4E). On the contrary, the
mutation was then predicted to cause a ‘polymorphism’ using
Mutation Taster. In addition, mutation of the amino acid (p.V412A)
was predicted by PROVEAN, SIFT and PolyPhen-2. Prediction with
PROVEAN suggested that this mutation is ‘neutral’ with a score of
−0.27. SIFT indicated that the mutation is a ‘tolerated’ mutation
with a score of 0.502. The prediction with PolyPhen-2 was ‘benign’
and this mutation was scored as 0.009 (sensitivity: 0.96;
specificity: 0.77).
Zhai et al (18) proposed co-segregation of this
mutation in ADCC; however, further investigation is required.
A reported splicing mutation: EPHA2
c.2663+1G>A
Following mutation screening, a splice donor site
mutation was determined in EPHA2 (c.2663+1G>A, chr-16455928;
Fig. 3F) in family 2. The screening
results revealed that the mutation is located in the first region
of Intron 16 and it was predicted to induce a substitution from
aspartic acid to glutamate. In addition, it was predicted to lead
to the formation of a stop codon, causing the loss of 34 amino
acids encoded by exon 17 (19).
It was not possible to predict the severe
consequences of this mutation using SIFT, PROVEAN and PolyPhen2;
however, the degree of damage associated with this mutation was
investigated using Mutation Taster and GERP++ prediction tools. The
mutation in EPHA2 received a GERP score of 5.77 and was considered
to be a conserved site. Mutation Taster analysis indicated
disease-causing effects, as sequence motif loss and loss of protein
were reported.
This mutation has been previously reported by Bu
et al (19); the mutation in
EPHA2 is located in chromosome 1-16455928. It is possible that
EPHA2 is able to exhibit a variety of mutation types. The present
study reported the mutation in EPHA2 as c.2663+1G>A using the
NCBI database.
A reported splicing mutation: PAX6
c.11-2A>G
The mutation in PAX6 was also identified in family 3
as a splicing mutation in intron 4, which is located in two regions
prior to the beginning of exon 5. This mutation was predicted to
lead to a substitution of serine (encoded by TCA) to arginine
(encoded by TCG). It was predicted to result in the addition of 56
amino acids (Fig. 3G) (20).
SIFT, PROVEAN and PolyPhen2 analyses were not able
to make any prediction regarding the mutation in PAX6; however,
according to the GERP++ score of 4.9 and Mutation Taster score of
1, the site was determined to be highly conserved among different
species. Mutation Taster predicted the mutation to be
‘disease-causing’. From the 3D models of wild-type and mutated
PAX6, the additional amino acids were observed to form an
abnormally long ‘tail’, which may affect the function of PAX6
(Fig. 5). This mutation was also
reported by Churchill et al (20).
Discussion
Congenital cataracts are the leading cause of
impaired vision in pediatric patients worldwide (1). According to recent studies, ~45% of
families with a history of cataracts have been reported to carry
mutations in the CRY genes; ~12% have mutations in the genes
encoding various growth or transcription factors. In addition, 16%
have mutations in Cx-encoding genes; ~5% possess mutations in
intermediate filament proteins, membrane proteins or protein
degradation-associated genes. Furthermore, ~8% have mutations in a
variety of other functionally diverse genes. Inheritance of the
same mutation in different families or in different individuals
within the same family may result in distinct cataract phenotypes
(phenotypic heterogeneity). This suggests that additional genetic
or environmental factors may affect the expression of the mutant
protein, which may be the primary cause of cataracts (7,8). On the
contrary, mutations in different genes involved in biological
processes that are potentially possibly not linked with each other
may lead to cataracts with similar or identical morphologies
(genotypic heterogeneity). This suggests that cataract may be the
ultimate common pathway for different spectra of initial insults
(21). For example, CRYBA4 belongs
to crystallin family. It is part of a gene cluster with beta-B1
(CRYBB1), beta-B2 (CRYBB2) and beta-B3 (CRYBB3). Any mutation of
these genes may affect the synthesis of crystallins, resulting in
opacification of lens (22).
In the present study, a specific panel that included
135 cataract-associated target genes was used to perform screening
tests on 6 Chinese families with congenital cataracts. A total of 7
suspected mutations were detected in six genes, including two novel
deleterious missense mutations (MYH9 c.4150G>C and CRYBA4
c.169T>C). Amongst them, MYH9 c.4150G>C, CRYBA4 c.169T>C,
RPGRRIP1 c.2669G>A, WFS1 c.1235T>C and CRYBA4 c.26C>T were
determined to lead to amino acid changes. EPHA2 c.2663+1G>A and
PAX6 c.11-2A>G were denoted as splicing mutations.
The c.4150G>C variant is a missense mutation in
exon 31 of MYH9 and leads to the substitution of glutamate to
glutamine at codon 1,384 (p.E1384Q) in MYH9. In addition, this
mutation was not detected in 100 normal controls, suggesting that
it is a pathogenic mutation rather than a polymorphism.
Bioinformatics analyses indicated that this mutation is a
disease-causing mutation. Thus, these results suggest that
congenital cataracts in family 1 may have been caused by the
mutation in exon 31 of MYH9.
Mutations in MYH9 may cause MYH9-associated
disorders, which are characterized by congenital
macrothrombocytopenia, accompanied with young-adult-onset
sensorineural hearing loss, nephropathy and congenital cataracts;
however, the etiologies of these diseases remain elusive (23). To date, several studies have
investigated mutations in MYH9 (23–33; Table III). According to Pecci et
al (34), cataracts did not
commonly occur in a family with a MYH9-associated disorder and were
only observed in 43 of 235 patients (18%); among them, there were
four congenital forms of disease. In the present study, family 1
had the ocular manifestation of congenital cataracts without renal
complications and hearing loss; the symptoms of these patients were
notably mild.
| Table III.Mutations in myosin heavy chain 9
associated with congenital cataracts. |
Table III.
Mutations in myosin heavy chain 9
associated with congenital cataracts.
| Amino acid
changes | Mutation type | Family origin | Pattern of
inheritance | (Refs.) |
|---|
|
p.E1066_A1072del | Deletion | Japanese | AD | Aoki T et al
(23) |
|
p.E1066_A1072del | Deletion | Italian | AD | Seri et al
(24) |
|
p.E1066_A1072del | Deletion | Japanese | AD | Miyazaki et
al (25) |
| p.Q1068_L1074
del | Deletion | French | AD | Saposnik et
al (26) |
|
p.E1066_A1072dup | Duplication | Italian | AD | De Rocco D et
al (27) |
| p.D1424N | Substitution | Japanese | AD | Wasano K et
al (28) |
| p.E1841K | Substitution | Sweden | AD | Zetterberg E et
al (29) |
| p.V1560G | Substitution | Sweden | AD | Zetterberg E et
al (29) |
| p.R1165C | Substitution | Japanese | AD | Okano S et
al (30) |
| p.V34G | Substitution | Italian | AD | De Rocco D et
al (31) |
| p.R702S | Substitution | Italian | AD | De Rocco D et
al (31) |
| p.M847_E853dup | Duplication | Italian | AD | De Rocco D et
al (31) |
|
p.K1048_E1054del | Deletion | Italian | AD | De Rocco D et
al (31) |
| p.D1447Y | Substitution | Italian | AD | De Rocco D et
al (31) |
| p.R1162T | Substitution | Italian | AD | Vettore S et
al (32) |
| p.V1516M | Substitution | Italian | AD | Pecci A et
al (33) |
| p.R1557L | Substitution | Italian | AD | Pecci A et
al (33) |
MYH9, mapped to chromosome 22q12.3, includes
47 exons, which encode MYH9 protein. The transcription of this gene
begins with the first ATG codon of the open reading frame in exon
2, which terminates at the stop codon in exon 41. The mutation
identified in the present study was located in exon 31, which may
affect the tail domain of MYH9. According to Pecci et al
(34), there was a strong
correlation between the genotype and clinical features of patients
with MYH9-associated disorders. In addition, mutations in the
rod-tail domain have been associated with a mild phenotype
(35). Thus, it may be proposed that
mutations in exon 31 may lead to hereditary disease, but with a
notably mild phenotype, as observed by the manifestations in family
1. Furthermore, this mutation was not detected in 100 normal
controls, suggesting that it is a pathogenic mutation rather than a
polymorphism. Bioinformatics analysis suggested that this mutation
was a disease-causing mutation. Thus, these results support that
congenital cataracts in family 1 may be caused by a mutation in
exon 31 of MYH9.
The missense mutation (c.169 T>C) in CRYBA4 was
reported to lead to an amino acid substitution from phenylalanine
to leucine at codon 57 (p.F57L) in CRYBA4. This mutation was not
detected in 100 normal controls and was predicted to be pathogenic
by various bioinformatics analyses.
CRYBA4 belongs to the CRYB family of proteins. The
CRYB family (35%), CRYA (40%) and CRYG are major members of the CRY
protein family detected in the mammalian lens (36). CRY proteins are markedly stable in
the lens for the maintenance of transparency and refractive
ability. CRYB comprises 7 protein forms, each of which is encoded
by six genes (CRYBA1, CRYBA2 and CRYBA4, as well as CRYBB1, CRYBB2
and CRYBB3). Among them, CRYBA4 constitutes 196 amino acids and
pathogenic mutations have been reported in exons 2 and 4 (18,37–39)
(Table IV). According to
Billingsley et al (36) and
Zhou et al (40), the
mutation in exon 4 serves an important role in the onset of
congenital cataracts. The novel mutation detected in the present
study is also in this region.
| Table IV.Mutations in β-crystallin A4
associated with congenital cataracts. |
Table IV.
Mutations in β-crystallin A4
associated with congenital cataracts.
| Amino acid
changes | Mutation type | Family origin | Pattern of
inheritance | (Refs.) |
|---|
| p.L69P | Substitution | India | AD | Vanita V et
al (37) |
| p.F94S | Substitution | India | AD | Vanita V et
al (37) |
| p.G2D | Substitution | China | AD | Kumar M et
al (38) |
| p.D3Y | Substitution | Honduras | AD | Kumar M et
al (38) |
| p.L11S | Substitution | Denmark | AD | Kumar M et
al (38) |
| p.T19M | Substitution | India | AD | Kumar M et
al (38) |
| p.V28M | Substitution | India | AD | Kumar M et
al (38) |
| p.F32L | Substitution | China | AD | Kumar M et
al (38) |
| p.R33L | Substitution | India | AD | Kumar M et
al (38) |
| p.V44M | Substitution | China | AD | Kumar M et
al (38) |
| p.V44M | Substitution | USA | AD | Kumar M et
al (38) |
| p.W45S | Substitution | China | AD | Kumar M et
al (38) |
| p.D47N | Substitution | China | AD | Kumar M et
al (38) |
| p.P59L | Substitution | USA | AD | Kumar M et
al (38) |
| p.A9V | Substitution | China | AD | Zhai Y et al
(18) |
| p.G147V | Substitution | Pakistan | AD | Chen J et al
(39) |
The 3D models of wild-type and mutated CRYBA4
revealed the substitution of phenylalanine at codon 57 with
leucine. The mutation was determined to be located in a β-sheet,
which is stabilized by hydrogen bonds. The substitution from
phenylalanine to leucine may break these hydrogen bonds, reducing
the stability of the secondary structure of the CRYBA4 protein.
In the present study, a c.1235T>C mutation was
identified in WFS1 in family 5. This missense mutation may cause an
amino acid substitution from valine to alanine at codon 412
(p.V412A). The mutation was predicted to be pathogenic by Mutation
Taster, SIFT and PolyPhen-2; however, PROVEAN suggested that the
mutation was ‘neutral’ with a score of −2.29.
The WFS1 gene encodes a transmembrane protein
expressed by brain, pancreatic, heart and insulinoma β cell lines.
Mutations in this gene may cause Wolfram syndrome, which leads to
numerous conditions, including diabetes insipidus, diabetes
mellitus, optic atrophy and deafness. This mutation was first
reported by Choi et al (16)
as a candidate gene for familial nonsyndromic hearing loss;
however, the present study reported the mutation to be associated
with the development of ADCC.
Considering the differences between splicing
mutations, and those that lead to amino acid substitutions and
alterations in protein structure, various bioinformatics tools were
employed to determine the consequences of these mutations. The
present study reported that, compared with the amino acid
substitution mutations, splicing mutations may be more dangerous,
as gene expression may be affected by newly formed stop codons or
the addition of amino acids. These alterations may cause the loss
of motifs, or changes in the structure and function of proteins.
The results of the present study revealed that the mutation EPHA2
c.2663+1G>A created a new stop codon, which resulted in the loss
of 34 amino acids encoded by exon 17. The mutation PAX6
c.11-2A>G was predicted to cause the addition of 56 amino acids.
Further investigation is required to determine the mechanisms
underlying the pathological effects of these mutations.
The methods applied to detect the mutations in the
present study are of high significance. Considering the number of
individuals in the families and the number of cataract-associated
genes analyzed, targeted exome sequencing to identify the mutations
of the probands and Sanger sequencing for the remaining patients
are highly efficient and economical (41). Of note, future studies will be
performed in co-operation with the Reproductive Medicine Center of
Peking University Third Hospital (Beijing, China) to develop
potential prenatal diagnostic strategies to inhibit the effects of
pathogenic mutations.
In addition, more information should be obtained to
verify whether the cataracts caused by these mutations are
progressive; however, it may not be possible to investigate certain
features of the phenotypes of the affected individuals in the
families in the future, as patients may undergo surgery to
eliminate cataracts.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Capital's Funds
for Health Improvement and Research (grant no. 2018-2-4093).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZW and CH analyzed the patient data and were major
contributors in writing the manuscript. YS, HL, MZ and XL performed
the ophthalmic examination of the patients. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All participating individuals provided informed
consent according to the tenets of the Declaration of Helsinki. The
Medical Ethics Committee of Peking University Third Hospital
(Beijing, China) approved all procedures of the present study.
Patient consent for publication
All participating individuals provided informed
consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hejtmancik JF: Congenital cataracts and
their molecular genetics. Semin Cell Dev Biol. 19:134–149. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Robinson GC, Jan JE and Kinnis C:
Congenital ocular blindness in children, 1945 to 1984. Am J Dis
Child. 141:1321–1324. 1987.PubMed/NCBI
|
|
3
|
Shiels A and Hejtmancik JF: Genetic
origins of cataract. Arch Ophthalmol. 125:165–173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang B and He W: Molecular
characteristics of inherited congenital cataracts. Eur J Med Genet.
53:347–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Francis PJ, Berry V, Bhattacharya SS and
Moore AT: The genetics of childhood cataract. J Med Genet.
37:481–488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Apple DJ, Ram J, Foster A and Peng Q:
Elimination of cataract blindness: A global perspective entering
the new millenium. Surv Ophthalmol. 45 (Suppl 1):S1–S196.
2000.PubMed/NCBI
|
|
7
|
Santana A and Waiswo M: The genetic and
molecular basis of congenital cataract. Arq Bras Oftalmol.
74:136–142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gill D, Klose R, Munier FL, McFadden M,
Priston M, Billingsley G, Ducrey N, Schorderet DF and Héon E:
Genetic heterogeneity of the Coppock-like cataract: A mutation in
CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci.
41:159–165. 2000.PubMed/NCBI
|
|
9
|
Liu Q, Wang KJ and Zhu SQ: A novel p.G112E
mutation in BFSP2 associated with autosomal dominant pulverulent
cataract with sutural opacities. Curr Eye Res. 39:1013–1019. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kunishima S, Kojima T, Matsushita T,
Tanaka T, Tsurusawa M, Furukawa Y, Nakamura Y, Okamura T, Amemiya
N, Nakayama T, et al: Mutations in the NMMHC-A gene cause autosomal
dominant macrothrombocytopenia with leukocyte inclusions
(May-Hegglin anomaly/Sebastian syndrome). Blood. 97:1147–1149.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40:W452–W457.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Waterhouse A, Bertoni M, Bienert S, Studer
G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C,
Bordoli L, et al: SWISS-MODEL: Homology modelling of protein
structures and complexes. Nucleic Acids Res. 46:W296–W303. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi BY, Park G, Gim J, Kim AR, Kim BJ,
Kim HS, Park JH, Park T, Oh SH, Han KH and Park WY: Diagnostic
application of targeted resequencing for familial nonsyndromic
hearing loss. PLoS One. 8:e686922013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun W, Xiao X, Li S, Guo X and Zhang Q:
Exome sequencing of 18 Chinese families with congenital cataracts:
A new sight of the NHS gene. PLoS One. 9:e1004552014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhai Y, Li J, Yu W, Zhu S, Yu Y, Wu M, Sun
G, Gong X and Yao K: Targeted exome sequencing of congenital
cataracts related genes: Broadening the mutation spectrum and
genotype-phenotype correlations in 27 Chinese Han families. Sci
Rep. 7:12192017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bu J, He S, Wang L, Li J, Liu J and Zhang
X: A novel splice donor site mutation in EPHA2 caused congenital
cataract in a Chinese family. Indian J Ophthalmol. 64:364–368.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Churchill AJ, Hanson IM and Markham AF:
Prenatal diagnosis of aniridia. Ophthalmology. 107:1153–1156. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shiels A and Hejtmancik JF: Mutations and
mechanisms in congenital and age-related cataracts. Exp Eye Res.
156:95–102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Graw J: Genetics of crystallins: Cataract
and beyond. Exp Eye Res. 88:173–189. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aoki T, Kunishima S, Yamashita Y,
Minamitani K and Ota S: Macrothrombocytopenia with congenital
bilateral cataracts: A phenotype of MYH9 disorder with Exon 24
indel mutations. J Pediatr Hematol Oncol. 40:76–78. 2018.PubMed/NCBI
|
|
24
|
Seri M, Pecci A, Di Bari F, Cusano R,
Savino M, Panza E, Nigro A, Noris P, Gangarossa S, Rocca B, et al:
MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome,
Fechtner syndrome, and Epstein syndrome are not distinct entities
but represent a variable expression of a single illness. Medicine
(Baltimore). 82:203–215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miyazaki K, Kunishima S, Fujii W and
Higashihara M: Identification of three in-frame deletion mutations
in MYH9 disorders suggesting an important hot spot for small
rearrangements in MYH9 exon 24. Eur J Haematol. 83:230–234. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saposnik B, Binard S, Fenneteau O, Nurden
A, Nurden P, Hurtaud-Roux MF and Schlegel N; FrenchMYH9 networka, :
Mutation spectrum and genotype-phenotype correlations in a large
French cohort of MYH9-Related Disorders. Mol Genet Genomic Med.
2:297–312. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
De Rocco D, Pujol-Moix N, Pecci A, Faletra
F, Bozzi V, Balduini CL and Savoia A: Identification of the first
duplication in MYH9-related disease: A hot spot for unequal
crossing-over within exon 24 of the MYH9 gene. Eur J Med Genet.
52:191–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wasano K, Matsunaga T, Ogawa K and
Kunishima S: Late onset and high-frequency dominant hearing loss in
a family with MYH9 disorder. Eur Arch Otorhinolaryngol.
273:3547–3552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zetterberg E, Carlsson Alle MS, Najm J and
Greinacher A: Thrombin generation in two families with MYH9-related
platelet disorder. Platelets. 27:264–267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Okano S, Takase M, Iseki K, Toriumi N,
Kaneda M and Kunishima S: Genotype-phenotype correlation of the
p.R1165C mutation in the MYH9 disorder: Report of a Japanese
pedigree. J Pediatr Hematol Oncol. 37:e352–e355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
De Rocco D, Zieger B, Platokouki H, Heller
PG, Pastore A, Bottega R, Noris P, Barozzi S, Glembotsky AC,
Pergantou H, et al: MYH9-related disease: Five novel mutations
expanding the spectrum of causative mutations and confirming
genotype/phenotype correlations. Eur J Med Genet. 56:7–12. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vettore S, De Rocco D, Gerber B,
Scandellari R, Bianco AM, Balduini CL, Pecci A, Fabris F and Savoia
A: A G to C transversion at the last nucleotide of exon 25 of the
MYH9 gene results in a missense mutation rather than in a splicing
defect. Eur J Med Genet. 53:256–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pecci A, Panza E, De Rocco D, Pujol-Moix
N, Girotto G, Podda L, Paparo C, Bozzi V, Pastore A, Balduini CL,
et al: MYH9 related disease: Four novel mutations of the tail
domain of myosin-9 correlating with a mild clinical phenotype. Eur
J Haematol. 84:291–297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pecci A, Klersy C, Gresele P, Lee KJ, De
Rocco D, Bozzi V, Russo G, Heller PG, Loffredo G, Ballmaier M, et
al: MYH9-related disease: A novel prognostic model to predict the
clinical evolution of the disease based on genotype-phenotype
correlations. Hum Mutat. 35:236–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pecci A, Panza E, Pujol-Moix N, Klersy C,
Di Bari F, Bozzi V, Gresele P, Lethagen S, Fabris F, Dufour C, et
al: Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA)
mutations predicts the natural history of MYH9-related disease. Hum
Mutat. 29:409–417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Billingsley G, Santhiya ST, Paterson AD,
Ogata K, Wodak S, Hosseini SM, Manisastry SM, Vijayalakshmi P,
Gopinath PM, Graw J and Héon E: CRYBA4, a novel human cataract
gene, is also involved in microphthalmia. Am J Hum Genet.
79:702–709. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vanita V, Guo G, Singh D, Ott CE and
Robinson PN: Differential effect of cataract-associated mutations
in MAF on transactivation of MAF target genes. Mol Cell Biochem.
396:137–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumar M, Agarwal T, Kaur P, Kumar M,
Khokhar S and Dada R: Molecular and structural analysis of genetic
variations in congenital cataract. Mol Vis. 19:2436–2450.
2013.PubMed/NCBI
|
|
39
|
Chen J, Wang Q, Cabrera PE, Zhong Z, Sun
W, Jiao X, Chen Y, Govindarajan G, Naeem MA, Khan SN, et al:
Molecular genetic analysis of Pakistani families with autosomal
recessive congenital cataracts by homozygosity screening. Invest
Ophthalmol Vis Sci. 58:2207–2217. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou G, Zhou N, Hu S, Zhao L, Zhang C and
Qi Y: A missense mutation in CRYBA4 associated with congenital
cataract and microcornea. Mol Vis. 16:1019–1024. 2010.PubMed/NCBI
|
|
41
|
Chen K, Zhou YX, Li K, Qi LX, Zhang QF,
Wang MC and Xiao JH: A novel three-round multiplex PCR for SNP
genotyping with next generation sequencing. Anal Bioanal Chem.
408:4371–4377. 2016. View Article : Google Scholar : PubMed/NCBI
|