Introduction
Retinal neovascularization is a common pathological
change in a number of eye diseases, resulting in severe eye injury
and possibly leaving the patient blind (1,2). The
upregulation of annexin A2 (A2) under a hypoxic and ischemic
microenvironment has been demonstrated to be a key factor in this
pathological process (3). It was
reported that mice deficient in the A2 gene were resistant
to hypoxia-related retinal neovascularization in a model of
diabetic retinopathy (3).
A2−/− newborn mice had a reduced neoangiogenic
response in the oxygen-induced retinopathy model (4). Additionally, A2−/−
mice also displayed reduced angiogenesis in three different models
of postnatal angiogenesis (5).
The human A2 gene is located on chromosome 15
(15q21) and is expressed in multiple human cells, including
endothelial cells, monocytes, macrophages, dendritic cells,
trophoblast cells, and tumor cells (6,7). As a
type of Ca2+-regulated, phospholipid-binding protein, A2
exists both as a free monomer in the cytoplasm or tethered to the
surface of the plasma membrane (6).
Accumulating evidence has demonstrated that A2 plays multifaceted
roles in membrane organization, signal transduction, cell
proliferation, migration, apoptosis, immunity and inflammation
(3,6,8). A2 has
also been reported to be closely associated with multiple
pathological processes, including thrombosis, vascular occlusion,
hyperfibrinolysis, hemorrhage and retinal neovascularization
(3,9). Knockdown of the A2 receptor (A2R)
significantly inhibited the proliferation, adhesion, migration and
tube formation of human umbilical vein endothelial cells (HUVECs),
which is usually used in the in vitro study of angiogenesis
(10). Disrupting A2R can inhibit
neovascularization in vivo via inactivating phosphorylation
of the protein kinase B (AKT) and extracellular signal regulated
kinase (ERK) signaling pathways (10). The authors' previous study
additionally demonstrated that A2 silencing inhibited the
proliferation and enhanced the apoptosis of HUVECs (11).
The underlying mechanism by which A2 is required for
retinal neovascularization remains largely unknown. In the present
study, oxygen-glucose deprivation (OGD) was used to mimic the
ischemic and hypoxic conditions and the role of A2 in retinal
neovascularization and autophagy was observed using human retinal
endothelial cells (HRECs). The present study is the first to
demonstrate that A2 may promote survival of HRECs under hypoxic and
ischemic conditions by inducing autophagy via the hypoxia-inducible
transcription factor-1α (HIF-1α) signaling pathway, to the best of
our knowledge. The present study also demonstrated a novel role of
A2 during retinal neovascularization under pathological conditions
and thus highlights a potential therapeutic target for treating
conditions where neovascularization of the eye is observed.
Materials and methods
Chemical reagents and plasmids
A HIF-1α inhibitor was purchased from Calbiochem
(cat. no. 400083; Merck KGaA). The autophagy inhibitor,
3-methyladenine (3-MA), was purchased from Sigma-Aldrich; Merck
KGaA. The concentrations of the inhibitors used were determined by
previous studies (12,13). The scramble control short hairpin RNA
(shRNA) plasmid, A2 shRNA plasmid, the wide-type A2 overexpression
plasmid (pcDNA3.1-A2) and control plasmid (pcDNA3.1) were
constructed by Shanghai GenePharma Co., Ltd.
Cell culture
Primary HRECs (ACBRI 181) were obtained from Cell
Systems and cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 20% fetal bovine serum (Thermo Fisher
Scientific, Inc.), 1% endothelial cell growth supplement
(Sigma-Aldrich; Merck KGaA) and 100 U/ml penicillin and 0.1 mg/ml
streptomycin (Beyotime Institute of Biotechnology) at 37°C in a
humidified incubator under 5% CO2. Cells which had been
passaged between 10 and 20 times were used for the following
experiments.
OGD treatment
At 80–90% confluence, the culture medium of HRECs
was replaced with serum-free and glucose-free DMEM (Gibco; Thermo
Fisher Scientific, Inc.), after which the cells were placed in a
hypoxic incubator chamber (1% O2, 5% CO2, 94%
N2; Forma Scientific; Thermo Fisher Scientific, Inc.)
for the indicated times (3, 6, 12 and 24 h). Control cells were
cultured with supplemented medium and incubated under normal oxygen
concentrations.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and 2 mg total RNA was
reverse transcribed using RevertAid™ First Strand cDNA Synthesis
Kit (Fermentas MBI; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. The temperature protocol for the
reverse transcription was 25°C for 10 min, 42°C, 60 min and finally
70°C for 10 min. qPCR was performed in triplicate using SYBR Green
PCR Master Mix (Toyobo Life Science) on a Mastercycler ep realplex
(Eppendorf). The primer sequences used were as follows: A2 forward,
5′-GTGAAGAGGAAAGGAACCGA-3′ and reverse 5′-CTTGATGCTCTCCAGCATGT-3′;
GAPDH forward, 5′-GCCTTCCGTGTTCCTACC-3′ and reverse
5′-AGAGTGGGAGTTGCTGTTG-3′. Thermocycling conditions consisted of an
initial denaturing step (95°C, 2 min), followed by 40 cycles of
denaturing (95°C, 15 sec), annealing (60°C, 15 sec) and extending
(72°C, 45 sec). The mRNA levels were normalized to GAPDH (internal
control) and their relative quantities were determined using the
2−ΔΔCq formula (14).
Western blotting
HRECs were rinsed three times with PBS and lysed in
RIPA buffer (cat. no. P0013C; Beyotime Institute of Biotechnology)
supplemented with 1% PMSF to obtain total cell lysates. The protein
concentration was qualified using the bicinchoninic acid assay
method and 50 µg proteins for each sample were separated on a 12%
SDS-PAGE, transferred to PVDF membranes and blocked with 5% non-fat
milk for 60 min at room temperature. The membranes were probed
overnight at 4°C with primary antibodies against A2 (cat. no. 8235;
1:1,000; Cell Signaling Technology, Inc.), LC3 (cat. no. 3868;
1:1,000; Cell Signaling Technology, Inc.), Beclin-1 (cat. no. 3495;
1:1,000; Cell Signaling Technology, Inc.), cleaved-caspase 3 (cat.
no. 9961; 1:1,000; Cell Signaling Technology, Inc.), HIF-1α (1:500;
Santa Cruz Biotechnology, Inc.) and β-actin (cat. no. A1978;
1:10,000; Sigma-Aldrich; Merck KGaA). Subsequently, the membranes
were incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. no. 610-1302; 1:5,000; Rockland Immunochemicals,
Inc.) for 2 h at room temperature. Finally, the blots were
visualized using an enhanced chemiluminescence reagent (BeyoECL
Plus Kit; cat. no. P0018; Beyotime Institute of Biotechnology).
Protein bands were quantified using the ImageJ software (Version
1.52p; National Institute of Health).
Cell transfection
Lentiviral vectors encoding shRNAs against human A2
(sh-A2, sequence 5′-TGAGGGTGACGTTAGCATTAC-3′ for A2 shRNA),
lentiviral control vectors (vector1, sequence
5′-GGATCATCATGCTATGCAGTT-3′ for control scramble shRNA), lentiviral
A2 overexpression vectors (OE-A2, primers used as follows, sense:
5′-CGGGGTACCATGTCTACTGTTCACGAAATCCTGT-3′, anti-sense:
5′-ATTGGGCCCGTCATCTCCACCACACAGGTAC-3′ and lentiviral control
vectors (vector2, pcDNA3.1 for the control plasmid) were packed and
obtained from OBiO Technology (Shanghai) Corp., Ltd. HRECs were
plated in 24-well plates at a density of 2×105
cells/well and allowed to grow at ~40% confluence. Cells were
infected with the aforementioned lentiviral vector (multiplicity of
infection, 50) for 48 h and the transfection efficiency was
evaluated by western blotting.
Luciferase assay
The A2 promotor-luciferase reporter and pRL-SV40-luc
plasmids were designed and purchased from Promega Corporation.
HRECs were plated in triplicate into a 24-well plate at a density
of 5×105 cells per well for overnight culture, after
which the cells in each well were infected with lentivirus
particles expressing the reporter plasmid [manufactured by OBiO
Technology (Shanghai) Corp., Ltd.]. Additionally, the lentivirus
particles expressing pRL-SV40-luc [manufactured by OBiO Technology
(Shanghai) Corp., Ltd.] was co-transfected to normalize the
transfection efficiency as an internal control (multiplicity of
infection, 50). Following transfection for 48 h, cells underwent
OGD for another 24 h, in the presence or absence of the HIF-1α
inhibitor (100 µmol/l). Luciferase activity was measured using the
dual luciferase assay system (Promega Corporation) and normalized
to a constitutively expressed Renilla reporter.
Cell viability assay
HRECs were infected with the lentivirus particles
expressing A2-shRNA, A2 overexpressing plasmid or control vector
for 48 h and plated in 96-well plates at the density of
4×104 per well in triplicate for overnight culture
(multiplicity of infection, 50). Transfected cells underwent OGD
for a further 24 h in the presence or absence of 3-MA (5 mM), after
which the cell viability was evaluated using Cell Counting Kit-8
(Dojindo Molecular Technologies, Inc.) according to the
manufacturer's protocol. The optical density was measured at a
wavelength of 450 nm using a synergy microplate reader (BioTek
China).
Cell apoptosis assay
Cell apoptosis was detected by measuring the protein
level of cleaved-caspase 3 as described previously (15,16).
HRECs were transfected with A2-shRNA, A2 overexpressing plasmid or
control vector for 48 h, after which the transfected cells
underwent OGD for another 24 h in the presence or absence of 3-MA
(5 mM). Cell lysates were collected and the protein expression
level of cleaved-caspase 3 was determined by western blotting.
Statistical analysis
Statistical significance between multiple
experimental groups was analyzed by an analysis of variance with a
post-hoc Tukey's test using SPSS statistics version 19.0 software
(IBM Corp.). Quantitative data were expressed as the mean ±
standard deviation of at least three experimental repeats.
P<0.05 was considered to indicate a statistically significant
difference.
Results
OGD treatment upregulates the
expression of A2 in HRECs
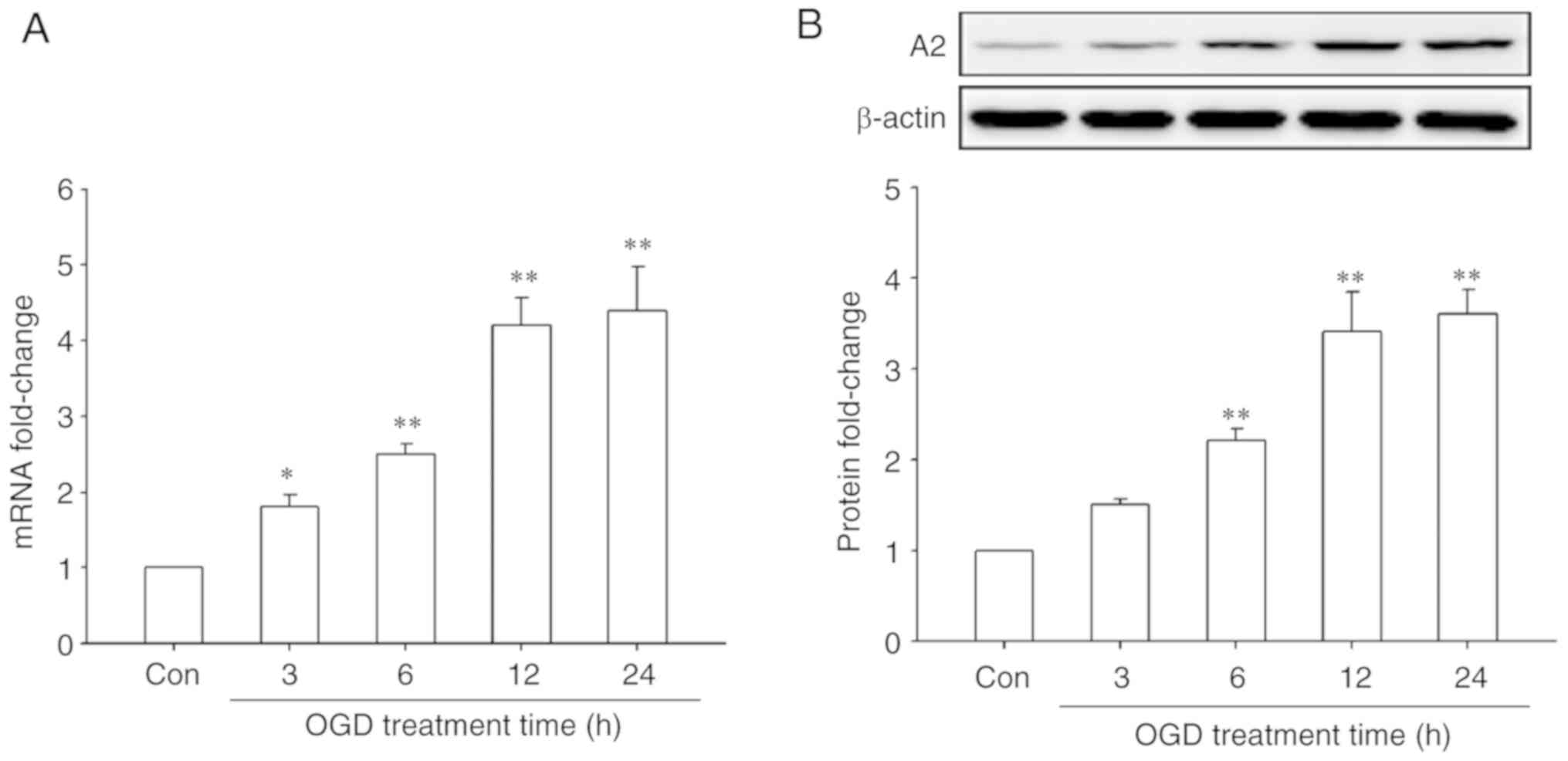
First, the effect of OGD treatment on A2 expression
in HRECs was investigated by RT-qPCR and western blotting. The
results showed that OGD treatment significantly upregulated the
mRNA and protein levels of A2 in a time-dependent manner
(P<0.05; Fig. 1). The mRNA levels
of A2 began to increase after OGD treatment for 3 h and reached a
maximal significant 4.4-fold increase compared with the control
cells after OGD treatment for 24 h (P<0.01; Fig. 1A). A similar result was observed at
the protein level, reaching a maximal significant 3.6-fold increase
compared with the control cells after OGD treatment for 24 h
(P<0.01; Fig. 1B). These data
indicated that OGD significantly upregulated the expression of A2
in HRECs.
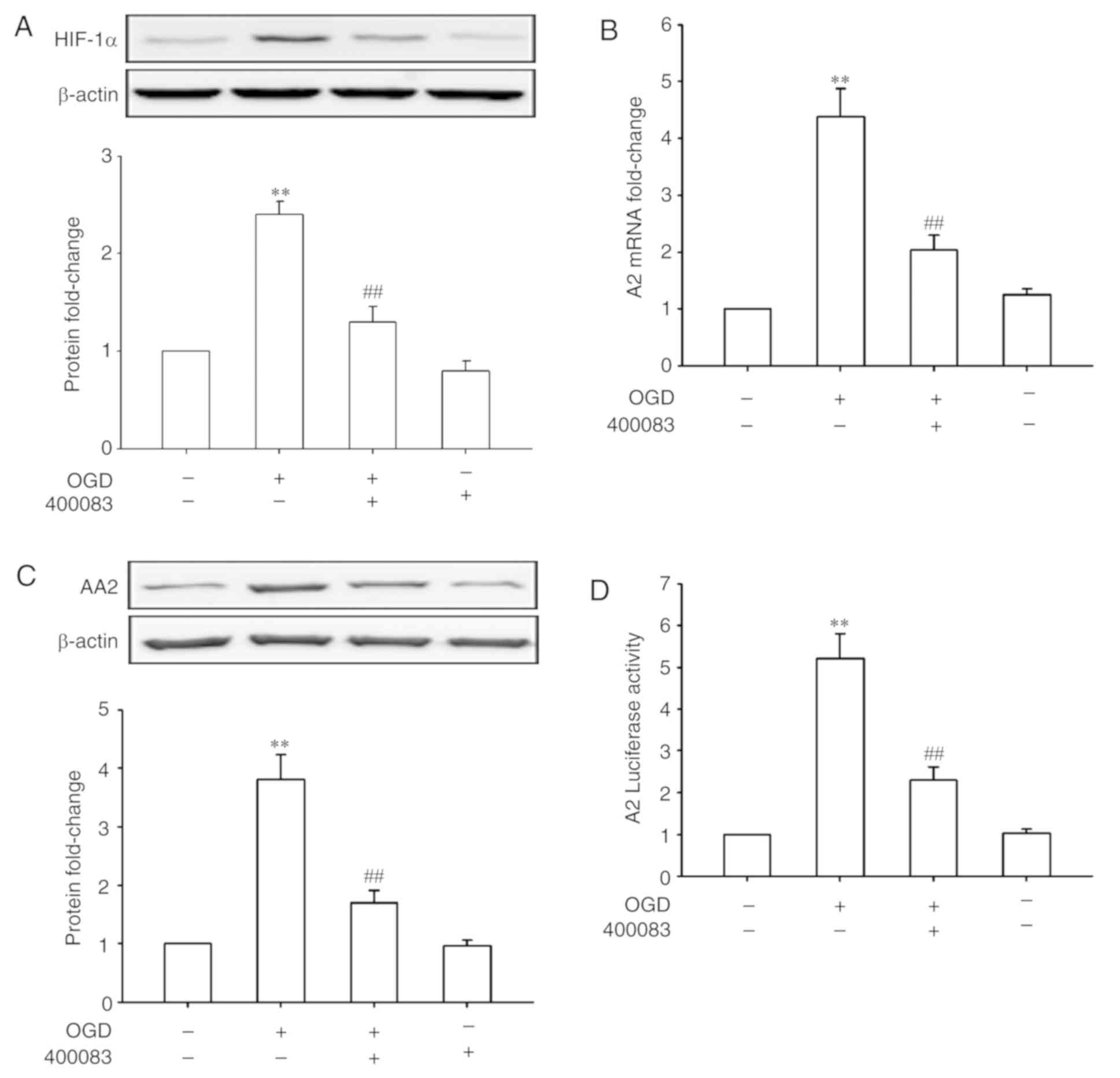
HIF-1α mediates the upregulation of A2
under OGD conditions
HIF-1 plays a central role in a number of hypoxic
events (17,18). Whether HIF-1 also has a role in
OGD-induced A2 expression in HRECs was further investigated. As
shown in Fig. 2A, OGD treatment for
24 h significantly increased the expression of HIF-1α (P<0.01).
Inhibiting HIF-1α using 400083 significantly inhibited OGD-induced
A2 upregulation at both the mRNA and protein levels (P<0.01;
Fig. 2B and C), suggesting that
HIF-1α regulated the expression of A2 at the transcriptional level.
To confirm this speculation, HRECs were transfected with an A2
promoter-luciferase reporter gene and treated by OGD in the
presence or absence of a HIF-1α inhibitor. As shown in Fig. 2D, OGD treatment for 24 h
significantly increased the A2 promoter activity (5.2-fold of
control, P<0.01), whereas the suppression of HIF-1α signaling
significantly reversed the OGD-induced promoter activity
(P<0.01). These data confirmed that upregulation of A2 by OGD
was mediated by transcriptional regulation of HIF-1α in HRECs.
A2 is required for OGD-induced
autophagy in HRECs
Next, the effects of OGD treatment on the activation
of autophagy were observed in HRECs by detecting the expression of
Beclin-1 and the conversion of LC3I to LC3II, two usually used
markers of autophagy (19), and the
role of A2 in this process. The results showed that OGD treatment
increased the levels of autophagy in a time-dependent manner in
HRECs (Fig. 3A). And, as shown in
Fig. 3C and E, OGD-induced autophagy
was significantly abrogated after A2 silencing (P<0.01) but
significantly enhanced by A2 overexpression (P<0.01), indicating
that A2 upregulation contributed to OGD-induced autophagy
activation in OGD-induced autophagy in HRECs. The knockdown and
overexpression efficiency of A2 were confirmed by western blotting
(Fig. 3B and D).
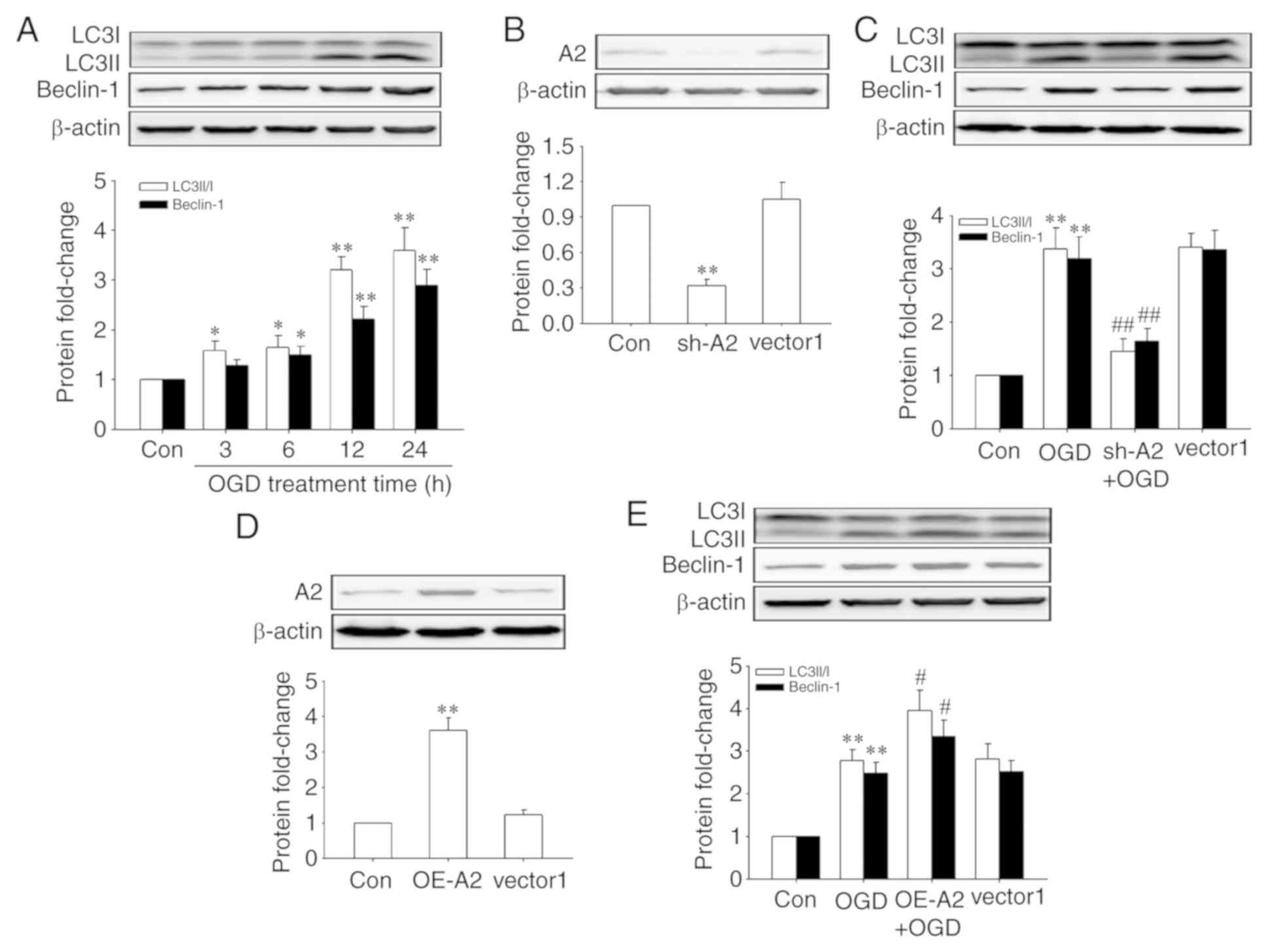 | Figure 3.Role of A2 in OGD-induced activation
of autophagy in human retinal endothelial cells. Cells were
infected with lentiviral vectors encoding shRNAs against human A2,
lentiviral scramble control vectors (vector1), lentiviral A2
expression vectors and lentiviral control vectors (vector2) for 48
h before OGD treatment. Protein levels were detected by western
blotting. (A) Expression of Beclin-1, LC3I and LC3II when cells
underwent OGD for different periods of time. (B) Transfection
efficiency of sh-A2. (C) Expression of Beclin-1, LC3I and LC3II
following knockdown of A2. (D) Transfection efficiency of OE-A2.
(E) Expression of Beclin-1, LC3I and LC3II after A2 overexpression.
*P<0.05 and **P<0.01 vs. Con group; #P<0.05 and
##P<0.01 vs. OGD group. A2, Annexin A2; sh-A2,
lentiviral vectors encoding short hairpin RNAs against human A2;
OE-A2, lentiviral A2 overexpression vectors; OGD, oxygen-glucose
deprivation; Con, control. |
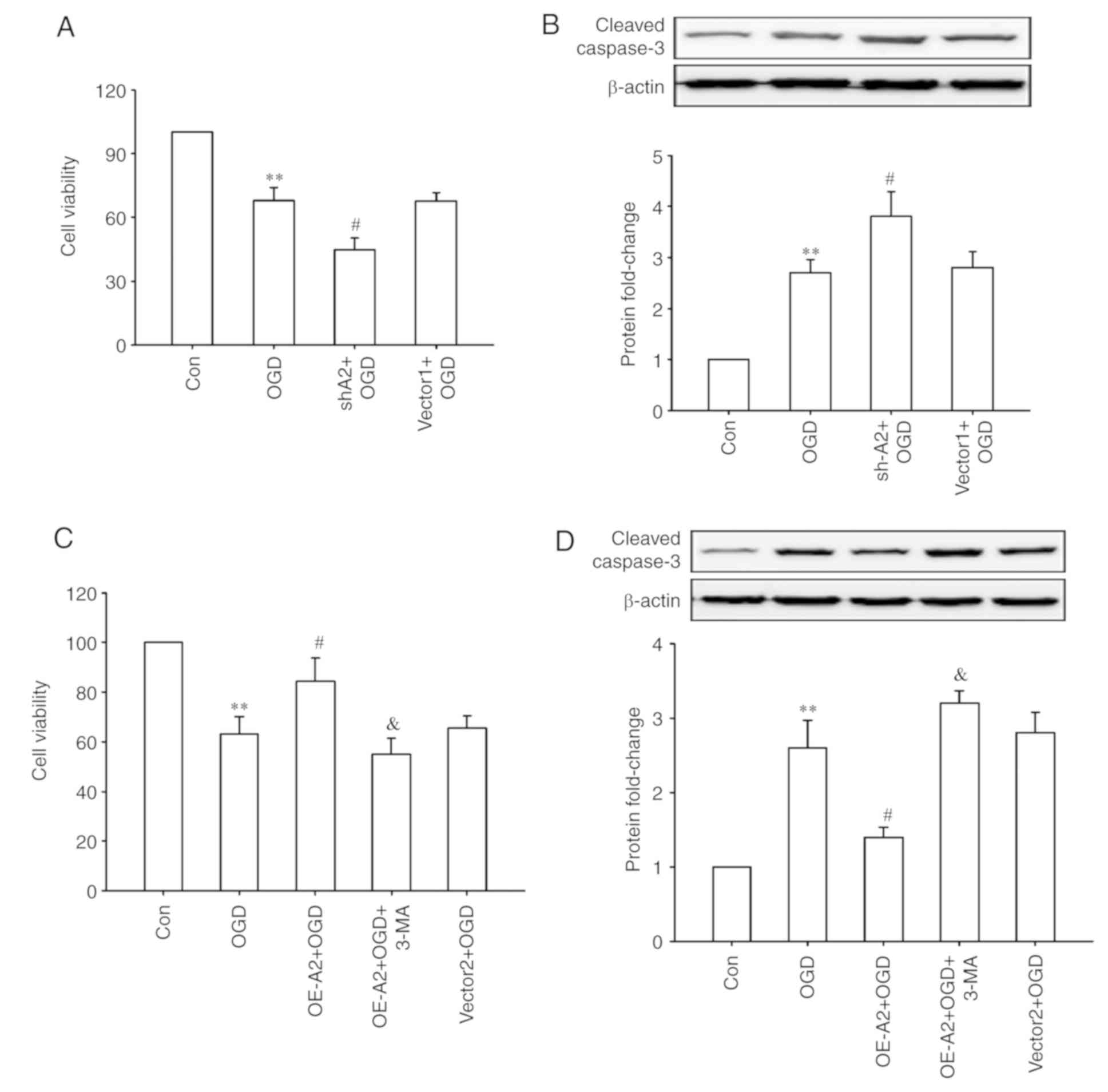
A2 upregulation protects HRECs against
OGD injury through autophagy activation
Finally, whether A2-mediated cell autophagy has a
protective effect on OGD injury was observed in HRECs. As shown in
Fig. 4, OGD treatment significantly
decreased the cell viability and increased the expression of
cleaved-caspase 3 (all P<0.01; Fig.
4A and B), which was further enhanced after A2 knockdown (all
P<0.05; Fig. 4A and B).
Overexpression of A2 alleviated these OGD-induced changes, whereas
inhibiting cell autophagy with a chemical inhibitor (3-MA)
significantly abrogated the pro-survival effects of overexpression
of A2 in HRECs (P<0.05; Fig. 4C and
D). These results indicated that upregulation of A2 protected
HRECs against OGD-induced injury potentially through autophagy
activation.
Discussion
Accumulating evidence highlighted that the role of
A2 in retinal neovascularization, which results in a number of
severe eye diseases and even potentially blindness (3,11,20).
Ischemic and hypoxic stimulation are common pathological states in
many eye diseases associated with retinal neovascularization. In
the present study, OGD was used to mimic the ischemic and hypoxic
microenvironment and demonstrated for the first time the expression
of A2 was upregulated under OGD conditions both at mRNA and protein
levels in HRECs, to the best of our knowledge.
HIF-1 is a heterodimeric protein that consists of
HIF-1α and HIF-1β subunits serves a central role in many hypoxic
events (17,18). Hypoxia increases the protein
expression level of HIF-1α by stabilizing it, whereas HIF-1β is
constitutively expressed (21). To
further determine whether HIF-1 serves a role in OGD-induced A2
expression in HRECs, the effect of OGD treatment on the expression
of HIF-1α and the effect of HIF-1α inhibition on A2 expression
under OGD conditions in HRECs was determined. The results showed
that OGD treatment increased the expression of HIF-1α in HRECs.
Inhibiting the HIF-1α using 400083 decreased the mRNA and protein
levels of A2 induced by OGD. To confirm that HIF-1α regulated A2
expression at the transcriptional level, HRECs were transfected
with an A2 promoter-luciferase reporter gene and treated with OGD
in the presence or absence of the HIF-1α inhibitor. The results
showed that OGD treatment increased the activity of the A2
promoter, whereas inhibition of HIF-1α signaling reversed the
OGD-induced promoter activity. Together, these data suggest that
upregulation of A2 by OGD was mediated by transcriptional
regulation of HIF-1α in HRECs. Previously, Huang et al
(22) reported that transcriptional
induction of the A2 gene was required in the oxygen-induced
retinopathy mice. However, HIF-1α-mediated transcriptional
regulation of the A2 gene requires additional investigation in
multiple cell lines.
Regarding the biological significance of A2
upregulation, several studies have reported the role of A2 in
regulating autophagy. Li et al (23) demonstrated that A2 positively
regulated autophagy in the MH-S alveolar macrophages, thereby
contributing to host immunity against bacteria through the
AKT1-mammalian target of rapamicin-ULK1/2 signaling pathway. Moreau
et al (24) showed that A2
modulated the starvation-induced autophagy in HeLa cells and the
process was dependent on the A2 effectors, ARP2 and Spire1.
Notably, Zhang et al (25)
found that overexpression of A2R could also induce autophagy and
increase autophagy flux in Mum2C uveal melanoma cells. Consistent
with previous studies (23–25), upregulation of A2 contributed to the
OGD-induced autophagy in HRECs in the present study, suggesting
that A2 can function as an autophagy modulator. However, the
underlying mechanism involving A2 in OGD-induced autophagy is still
unknown.
Autophagy is a key mechanism for the maintenance of
cellular homeostasis by which damaged organelles and unused
proteins are digested and recycled to provide nutrients to promote
cell survival (26). A2-regulated
autophagy under OGD conditions was associated with cell survival in
HRECs. The data in the present study demonstrated that A2
positively regulated autophagy and negatively regulated apoptosis,
thereby affecting cell survival. In addition, autophagy inhibition
promoted cell apoptosis and decreased cell survival, indicating
that A2-regulated autophagy served a cytoprotective role in HRECs
under OGD conditions. Previous studies have demonstrated that A2
could negatively modulate apoptosis in HUVECs and human lung cancer
A549, AGZY83-a, Anip973 and BE1 cells (11,27,28).
Additionally, studies showed that A2 negatively modulated cell
apoptosis through affecting the expression and activity of p53, as
well as the downstream proteins in the signaling pathway, such as
Bcl2, Bax and the caspase family of proteins (28,29). The
results of the present study only demonstrated the anti-apoptotic
role of A2 in vitro and the exact role of A2 in regulating
endothelial cell apoptosis in vivo remains unknown. The
underlying molecular mechanism in which A2 modulated endothelial
cell apoptosis also remains unknown. Therefore, further
investigations concerning the role of A2 in apoptosis in gene
knock-out mice and its mechanism requires attention.
The roles of vascular endothelial growth factor
(VEGF), another HIF-1α target gene, in angiogenesis had been well
studied previously. VEGF promotes cell proliferation, migration and
tube formation, and regulates cell apoptosis, permeability,
metabolism and autophagy in retinal endothelial cells (30–32). A
number of studies have reported the role of the interaction of VEGF
and A2 signaling and demonstrated that A2 was upregulated by VEGF
treatment in monkey retinal vascular endothelial RF/6A cells and
osteoblastic MC3T3-E1 cells (33,34).
Together, these previous studies and the data presented in the
present study suggest that HIF-1α/A2 signaling and HIF-1α-regulated
autocrine VEGF/A2 signaling may synergistically reinforce
neoangiogenesis under ischemic and hypoxic conditions.
In conclusion, the expression of A2 was increased
under OGD conditions in HRECs, which is mediated through the
activation of the HIF-1 signaling pathway. Upregulation of A2
regulated the activation of cell autophagy and protected HRECs from
OGD-induced injury. Therefore, the hypoxic and ischemic environment
may trigger the HIF-1/A2 signaling pathway, which can further
activate cell autophagy and promote survival of retinal endothelial
cells. The data in the present study revealed a novel role and
mechanism of A2, which is involved in retinal angiogenesis by
regulating autophagy under pathological conditions and provides a
potential therapeutic target for treating patients with eye
diseases associated with angiogenesis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SJ and YX designed the present study and analyzed
the data. SJ performed the experiments.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang SX, Ma JH, Bhatta M, Fliesler SJ and
Wang JJ: The unfolded protein response in retinal vascular
diseases: Implications and therapeutic potential beyond protein
folding. Prog Retin Eye Res. 45:111–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mao XB, You ZP, Wu C and Huang J:
Potential suppression of the high glucose and insulin-induced
retinal neovascularization by Sirtuin 3 in the human retinal
endothelial cells. Biochem Biophys Res Commun. 482:341–345. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hajjar KA: The biology of Annexin A2: From
vascular fibrinolysis to innate immunity. Trans Am Clin Climatol
Assoc. 126:144–155. 2015.PubMed/NCBI
|
|
4
|
Smith LE, Wesolowski E, McLellan A, Kostyk
SK, D'Amato R, Sullivan R and D'Amore PA: Oxygen-induced
retinopathy in the mouse. Invest Ophthalmol Vis Sci. 35:101–111.
1994.PubMed/NCBI
|
|
5
|
Ling Q, Jacovina AT, Deora A, Febbraio M,
Simantov R, Silverstein RL, Hempstead B, Mark WH and Hajjar KA:
Annexin II regulates fibrin homeostasis and neoangiogenesis in
vivo. J Clin Invest. 113:38–48. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gerke V, Creutz CE and Moss SE: Annexins:
Linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell
Biol. 6:449–461. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grindheim AK, Saraste J and Vedeler A:
Protein phosphorylation and its role in the regulation of Annexin
A2 function. Biochim Biophys Acta Gen Subj. 1861:2515–2529. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moss SE and Morgan RO: The annexins.
Genome Biol. 5:2192004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo M and Hajjar KA: Annexin A2 system in
human biology: Cell surface and beyond. Semin Thromb Hemost.
39:338–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song H, Pan D, Sun W, Gu C, Zhang Y, Zhao
P, Qi Z and Zhao S: SiRNA directed against annexin II receptor
inhibits angiogenesis via suppressing MMP2 and MMP9 expression.
Cell Physiol Biochem. 35:875–884. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang SL, Pan DY, Gu C, Qin HF and Zhao
SH: Annexin A2 silencing enhances apoptosis of human umbilical vein
endothelial cells in vitro. Asian Pac J Trop Med. 8:952–957. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang G, Su J, Zhang M, Jin Y, Wang Y,
Zhou P and Lu J: RhoB regulates the function of macrophages in the
hypoxia-induced inflammatory response. Cell Mol Immunol.
14:265–275. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li R, Wang LZ, Du JH, Zhao L and Yao Y:
Autophagy activation and the mechanism of retinal microvascular
endothelial cells in hypoxia. Int J Ophthalmol. 11:567–574.
2018.PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu D, Luo N, Wang L, Zhao Z, Bu H, Xu G,
Yan Y, Che X, Jiao Z, Zhao T, et al: Hydrogen sulfide ameliorates
chronic renal failure in rats by inhibiting apoptosis and
inflammation through ROS/MAPK and NF-κB signaling pathways. Sci
Rep. 7:4552017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
You P, Wu H, Deng M, Peng J, Li F and Yang
Y: Brevilin A induces apoptosis and autophagy of colon
adenocarcinoma cell CT26 via mitochondrial pathway and
PI3K/AKT/mTOR inactivation. Biomed Pharmacother. 98:619–625. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Riboldi E, Porta C, Morlacchi S, Viola A,
Mantovani A and Sica A: Hypoxia-mediated regulation of macrophage
functions in pathophysiology. Int Immunol. 25:67–75. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo T, Song H, Zhao Z, Qi Z and Zhao S:
Overexpression of Annexin A2 receptor inhibits neovascularization
via the promotion of krüppel-like transcription factor 2. Cell
Physiol Biochem. 46:1617–1627. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zagórska A and Dulak J: HIF-1: The knowns
and unknowns of hypoxia sensing. Acta Biochim Pol. 51:563–585.
2004.PubMed/NCBI
|
|
22
|
Huang B, Deora AB, He KL, Chen K, Sui G,
Jacovina AT, Almeida D, Hong P, Burgman P and Hajjar KA:
Hypoxia-inducible factor-1 drives annexin A2 system-mediated
perivascular fibrin clearance in oxygen-induced retinopathy in
mice. Blood. 118:2918–2929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li R, Tan S, Yu M, Jundt MC, Zhang S and
Wu M: Annexin A2 regulates autophagy in pseudomonas aeruginosa
infection through the Akt1-mTOR-ULK1/2 signaling pathway. J
Immunol. 195:3901–3911. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moreau K, Ghislat G, Hochfeld W, Renna M,
Zavodszky E, Runwal G, Puri C, Lee S, Siddiqi F, Menzies FM, et al:
Transcriptional regulation of Annexin A2 promotes
starvation-induced autophagy. Nat Commun. 6:80452015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Song H, Guo T, Zhu Y, Tang H, Qi
Z, Zhao P and Zhao S: Overexpression of Annexin II receptor-induced
autophagy protects against apoptosis in uveal melanoma cells.
Cancer Biother Radiopharm. 31:145–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi Y, Bowman JW and Jung JU: Autophagy
during viral infection-a double-edged sword. Nat Rev Microbiol.
16:341–354. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang Y, Jin Y, Yan CH, Yu Y, Bai J, Chen
F, Zhao YZ and Fu SB: Involvement of Annexin A2 in p53 induced
apoptosis in lung cancer. Mol Cell Biochem. 309:117–123. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang YX, Lv H, Li ZX, Li C and Wu XY:
Effect of shRNA mediated down-regulation of Annexin A2 on
biological behavior of human lung adencarcinoma cells A549. Pathol
Oncol Res. 18:183–190. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sharathchandra A, Lal R, Khan D and Das S:
Annexin A2 and PSF proteins interact with p53 IRES and regulate
translation of p53 mRNA. RNA Biol. 9:1429–1439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y, Yang C, Gu Q, Sims M, Gu W,
Pfeffer LM and Yue J: KLF4 promotes angiogenesis by activating VEGF
signaling in human retinal microvascular endothelial cells. PLoS
One. 10:e01303412015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jo DH, Bae J, Chae S and Kim JH, Han JH,
Hwang D, Lee SW and Kim JH: Quantitative proteomics reveals β2
integrin-mediated cytoskeletal rearrangement in vascular
endothelial growth factor (VEGF)-induced retinal vascular
hyperpermeability. Mol Cell Proteomics. 15:1681–1691. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Domigan CK, Warren CM, Antanesian V,
Happel K, Ziyad S, Lee S, Krall A, Duan L, Torres-Collado AX,
Castellani LW, et al: Autocrine VEGF maintains endothelial survival
through regulation of metabolism and autophagy. J Cell Sci.
128:2236–2248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao S, Huang L, Wu J, Zhang Y, Pan D and
Liu X: Vascular endothelial growth factor upregulates expression of
annexin A2 in vitro and in a mouse model of ischemic retinopathy.
Mol Vis. 15:1231–1242. 2009.PubMed/NCBI
|
|
34
|
Genetos DC, Wong A, Watari S and Yellowley
CE: Hypoxia increases Annexin A2 expression in osteoblastic cells
via VEGF and ERK. Bone. 47:1013–1019. 2010. View Article : Google Scholar : PubMed/NCBI
|