Introduction
The worldwide prevalence of chronic obstructive
pulmonary disease (COPD) and associated mortality have been
increasing (1). According to the
Global Disease Burden Research Project and the World Health
Organization, COPD will become the third largest cause of death in
the world and the fifth largest economic burden in the world by
2020 (2). COPD is a universally
preventable and treatable disease characterized by persistent
airflow limitation, and is usually associated with progressive
development and an increase in the chronic inflammatory response in
the respiratory tract, including the lung (3). Bronchoscopy reveals infiltration of
neutrophils, lymphocytes and macrophages in the airways of affected
patients (4). These inflammatory
cells produce multiple immune mediators (5). A large number of studies have indicated
that the immune response has an important role in the pathogenesis
of COPD (4,6,7).
Adaptive immunity, and particularly innate immunity, may be
involved in the abnormal inflammatory response and tissue damage
during COPD.
Interleukin (IL)-33 is a member of the IL-1 cytokine
family and may bind with the heterodimeric receptor composed of ST2
[also known as IL-33 receptor (IL-33R) or IL1R-like 1] and
IL-1R-combined protein (IL-1RAcP) to induce multiple immune
responses (8). The IL-33/ST2
signaling pathway is involved in various inflammatory diseases,
including asthma, influenza-associated airway hyper responsiveness
and allergic rhinitis (9–11). In addition, IL-33 has an important
role in the development of COPD (4).
Kearley et al (12) indicated
that cigarette smoke may cause changes in the lung microenvironment
and promote IL-33-dependent inflammatory responses, resulting in
exacerbations of COPD. IL-33 is highly expressed in the lung tissue
of patients with severe COPD and is associated with IL-13 and
airway mucin expression, which in turn causes COPD (12). In addition, bronchial epithelial
cell-derived IL-33 is involved in regulating the innate immune
response of COPD through IL-13 (13), causing airway mucus secretion and
airway hyper reactivity (14,15). The
receptor ST2 is highly expressed on dendritic cells, natural killer
T cells, innate lymphocytes and basophils, but not on naïve
CD4+ T cells (16). To
date, it has remained elusive how epithelial-derived IL-33 induces
innate immune cells to produce type 2 T-helper (Th2) cell
cytokines, and thus participate in the disease process of COPD.
Type 2 innate lymphoid cells (ILC2s) are one of the
major types of Th2 cell of the innate immune system (17). ILC2s were once known as natural
helper cells (18), nuocytes
(19), or Ih2 cells (20). The development of ILC2 relies on the
transcription factor RORA (21) and
the differentiation of ILC2 depends on inhibitor of DNA binding 2
and GATA binding protein 3 (GATA3). ILC2s are mainly distributed in
the intestinal mucosa, blood, lungs and airway mucosa of humans and
mice (22). ILC2s exhibit
morphological features of lymphocytes, but do not express lineage
(Lin) markers or antigen heterologous receptors (T-cell receptor or
B-cell receptor) (23). The
molecules on the surface of ILC2 mainly include CD45, IL-7Rα
(CD127), prostaglandin D2 receptor 2 (CRTH2), T1/ST2, IL-17RB,
CD25, CD38 and CD69 (24). Although
certain studies have described the important role of ILC2 in
helminth infections, respiratory inflammatory responses and atopic
dermatitis (18–20), the function of ILC2 in COPD has
remained to be fully investigated (25).
It has been indicated that IL-33 is the major
predisposing factor for human ILC2s (17). IL-33 may induce ILC2 to produce IL-4,
IL-5, IL-10 and IL-13 to participate in Th2-type immune responses
(26). However, the biological role
of peripheral blood ILC2s and IL-33 in patients with COPD has
remained elusive.
In the present study, the proportion and
characteristics of ILC2s in peripheral blood of patients with COPD
were examined. The results indicated that the expression of GATA3
and ST2 in ILC2s cells was significantly increased. Further in
vitro experiments suggested that IL-33 induced the release of a
large number of Th2 cytokines from ILC2. Therefore, IL-33 and ILC2s
have a role in the regulation of COPD inflammation and may be
potential therapeutic targets for COPD.
Materials and methods
Patients
In total, 107 patients with COPD and 110 matched
control subjects were recruited (Table
I) from January 2017 to September 2018 at the Traditional
Chinese Medicine Hospital Affiliated to Xinjiang Medical
University. COPD patients were diagnosed according to the criteria
proposed by the Global Initiative for Chronic Obstructive Lung
Disease (GOLD) guidelines (1) and
were free of exacerbation for at least 4 weeks prior to the study.
Healthy control subjects were age and sex matched who visited the
hospital for routine physical examination. Patients with COPD and
controls all had a history of smoking with a pack-year index of
>20. Inclusion criteria for COPD were as follows: i) Patients
with COPD diagnosed based on clinical manifestations (e.g., chronic
cough, cough and/or dyspnea), history of exposure to risk factors,
physical signs and pulmonary function tests; ii) patients with
incomplete reversible airflow limitation (forced expiratory volume
in one second/forced vital capacity <70% after administration of
bronchodilator). The exclusion criteria were as follows: i)
Comorbidities of severe lung diseases, including pneumothorax, lung
cancer and pulmonary tuberculosis; ii) intake of inhaled
corticosteroids or oral theophylline, anti-inflammatory therapy or
oral steroids for chronic inflammatory diseases during the previous
4 weeks; iii) patients with primary diseases, including severe
cardiovascular diseases, hepatorenal diseases and hematopoietic
system diseases; iv) pregnant and lactating females. Peripheral
blood was obtained from all the subjects.
 | Table I.Demographics and clinical
characteristics of the study cohort. |
Table I.
Demographics and clinical
characteristics of the study cohort.
| Variable | Control
(n=110) | COPD (n=107) | P-value |
|---|
| Age (years) | 67±8.9 | 67±8.3 | 0.64 |
| Sex
(male/female) | 79/31 | 83/24 | 0.53 |
| Smoking index (pack
years) | 30.46±16.91 | 31.05±17.57 | 0.67 |
| FEV1 (%
predicted) | 78.5±14.2 |
58.3±21.3a | <0.01 |
| FEV1/FVC (%) | 80.2±11.2 | 66.4±10.0 | <0.01 |
| GOLD grade n
(%) |
|
| <0.01 |
| Grade
I | 94 (85.5) | 5 (4.6) |
|
| Grade
II | 16 (14.5) | 94 (87.8) |
|
| Grade
III | 0 | 8 (7.4) |
|
| Grade
IV | 0 | 0 |
|
Reagents
The anti-human antibodies included FITC-labeled CD3
(cat. no. 561807), CD19 (cat. no. 555412), CD123 (cat. no. 561694),
CD11b (cat. no. 562793), CD11c (cat. no. 561355), CD8 (cat. no.
557085), FceR1 (cat. no. 12-5899-41; Thermo Fisher Scientific,
Inc.), CD14 (cat. no. 555397), CD4 (cat. no. 561005), CD56 (cat.
no. 562794); APC-CY7 labeled CD45 (cat. no. 557748); PerCP-CY5.5
labeled CRTH2 (cat. no. 558042); and PE-CY7 labeled IL-7Rα (cat.
no. 557938) and IL-13 (cat. no. JES10-5A2; all from BD Bioscience,
except FceR1). IL-33 (cat. no. EK133), IL-4 (cat. no. EK1042), IL-5
(cat. no. EK1052), IL-6 (cat. no. EK1062), IL-13 (cat. no. EK1132)
and sST2 (cat. no. EK11634) ELISA kits were purchased from Hangzhou
Lianke Biotechnology Co., Ltd. The EasySep™ Human ILC2 Isolation
kit was purchased from STEMCELL™ Technologies (cat. no. 17972;
Vancouver, British Columbia, Canada). Iscove's modified Dulbecco's
medium (IMDM; cat. no. 12440061) was from Thermo Fisher Scientific,
Inc. Penicillin-streptomycin was from Gibco (Thermo Fisher
Scientific, Inc.). Human IL-2 was from Peprotech. Human recombinant
(r)IL-33 cytokine (cat. no. 3625-IL-010), human ST2/IL-33R
neutralizing antibody (cat. no. MAB523) and normal goat IgG control
(cat. no. AB-108-C) were purchased from R&D Systems. A Cell
Counting Kit-8 was purchased from Dongren Chemical Technology Co.,
Ltd. (Dojindo Laboratories). TRIzol LS reagent was from Invitrogen
(Thermo Fisher Scientific, Inc.). The Transcriptor first strand
complementary (c)DNA synthesis kit was from Roche Diagnostics (cat.
no. 4379012001; Roche Diagnostics). Ficoll-Hypaque Solution (cat.
no. P8610) was obtained from Beijing Solarbio Science &
Technology Co., Ltd.
Flow cytometry, cell sorting and cell
culture
Peripheral blood mononuclear cells (PBMCs) were
isolated from whole blood using the Ficoll density gradient method.
Samples were diluted with PBS at 1:1 ratio, mixed with equal
volumes of Ficoll-Hypaque Solution and centrifuged at 750 × g for
22 min at room temperature. The white supernatant consisted of
PBMCs. PBMCs were then rinsed twice with PBS and then centrifuged
at 500 × g for 10 min at room temperature to collect the cell
pellets. Then the cells were re-suspended in PBS to yield a cell
concentration of 1×106 cells/ml. The antibodies,
including Lin-(CD3, CD19, CD123, CD11b, CD11c, CD8, FceR1, CD14,
CD4, CD56)-FITC, CD45-APC-CY7, PerCP-CY5.5-CRTH2, CD127-PE-CY7 and
ST2-APC, were added, followed by incubation at room temperature for
30 min in the dark. Isotype control was added to reduce
non-specific binding. The cells were washed with PBS twice. Samples
were detected by flow cytometry (LSR II; BD Bioscience), and
analyzed using FlowJo software version 7.6 (FlowJo, LLC).
As the percentage of ILC2 in peripheral blood was
low, the ILC2 cells were separated from the mixed blood of 5–6
subjects (total volume, 200 ml) and a total of 10 mixed blood
samples were used. ILC2 cells were identified as
Lin−CD45+CD127+ (IL7Rα), and
CRTH2+ and ST2+ cells were identified as
Lin−CD45+CD127+ (IL7Rα)
CRTH2+ST2+. The isolation of ILC2 cells from
the mixed peripheral blood of COPD patients was performed using the
human ILC2 isolation kit according to the manufacturer's protocol.
ILC2 cells were then seeded into 96-well plates at 500 cells/well,
mixed with gamma-irradiated PBMCs (24) from three healthy volunteers
(2×106 cells/ml), and cultured in the presence of 500
ng/ml IL-2 in IMDM (Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (Sigma-Aldrich; Merck KGaA) and 10 ml/l
penicillin-streptomycin.
Cell stimulation
After 4–6 weeks, the growing ILC2 cells were seeded
into 24-well plates at 1×104 cells/well and divided into
a control group, an IL-33 stimulation group and an anti-ST-2
neutralization group. Cells in the control group were treated with
PBS. Cells in the IL-33 stimulation group were stimulated with
human rIL-33 (10, 50 or 100 ng/ml) for 48 h. In the anti-ST-2
neutralization group, cells were pre-treated with human ST2/IL-33R
neutralizing antibody (1 µg/ml) or normal goat IgG control (20
µg/ml) for 1 h and then treated with human rIL-33 (10, 50 or 100
ng/ml) for 48 h. The culture supernatants were stored at −20°C for
analysis by ELISA.
Measurement of cytokines by ELISA
The concentration of IL-33, IL-4, IL-5, IL-6 and
IL-13 in serum and the concentration of IL-4, IL-5, IL-6, IL-13 and
soluble (s)ST2 in cell culture supernatant were determined by ELISA
using human ELISA kits, according to the manufacturer's
instructions. The limits of detection for the IL-33, IL-4, IL-5,
IL-6, IL-13 and sST2 ELISA kits were 2.17, 0.11, 0.76, 0.90 and
1.10 pg/ml, respectively.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from PBMCs of the control
group, COPD groups and in vitro-stimulated ILC2 cells using
TRIzol LS. Furthermore, the cDNA was synthesized from the isolated
total RNA using a Transcriptor first strand cDNA synthesis kit. The
mRNA expression levels of CRTH2, GATA3, ST2 and RAR-related orphan
receptor (ROR)α were detected using qPCR. Real-time PCR was
performed in a BIO-RAD CFX96 detection system (Bio-Rad
Laboratories) with SYBR Premix Ex Taq (Takara Bio, Inc.). The PCR
parameters were 95°C for 5 min, followed by 40 cycles of 95°C for
30 sec and 60°C for 30 sec. Quantitative results were determined
using the 2−∆∆Cq method (27) and gene expression levels were
presented as the relative abundance level after normalizing to the
mRNA expression levels of GAPDH. The primers used for PCR are
listed in Table II.
| Table II.Primers for PCR. |
Table II.
Primers for PCR.
|
|
| Primer sequence
(5′→3′) |
|
|
|---|
|
|
|
|
|
|
|---|
| Gene | Genbank accession
no. | Forward | Reverse | Product size
(bp) | Tm (°C) |
|---|
| CRTH2 | NM_004778.2 |
CGCCACACTGAAGCCACTCTG |
GCGTGGTCGATGTAGCGGATG | 90 | 60 |
| RORA | NM_002943.3 |
CTGGTGTGCATAGCGGAGGTTG |
CCTGCGGACTGGCAATAATCGG | 101 | 60 |
| GATA3 | NM_001002295.1 |
GTGCATGACTCACTGGAGGACTTC |
CATGTGGCTGGAGTGGCTGAAG | 114 | 60 |
| ST2 | NM_001282408.1 |
CTTCACGGTCAAGGATGAGCAAGG |
CACAGGACGGCAGCCAAGAAC | 156 | 60 |
| GAPDH | NM_002046 |
CAGGAGGCATTGCTGATGAT |
GAAGGCTGGGGCTCATTT | 138 | 60 |
CCK-8 assay
Cell viability was detected by Cell Counting Kit-8.
In brief, 10 µl CCK8 reagent was added to 100 µl cell suspension at
1 h prior to the end of the culture. The absorbance was measured at
450 nm by using a spectrophotometer.
Statistical analysis
Values are expressed as the mean ± standard
deviation unless otherwise specified. Differences between groups
were assessed using an unpaired Student's t-test. One-way analysis
of variance were used for comparisons among groups followed by
Tukey's test. P<0.05 was considered to indicate statistical
significance. All data were analyzed with GraphPad Prism software
version 5.0 (GraphPad Software Inc.).
Results
IL-33 and ST2 are involved in the
immune response in COPD
A summary of patient demographics are presented in
Table I. No significant differences
were identified in the baseline demographic characteristics of the
two groups. Forced expiratory volume (FEV1) and FEV1/forced vital
capacity of COPD patients were significantly lower than those in
controls. In COPD patients, pulmonary function deteriorated
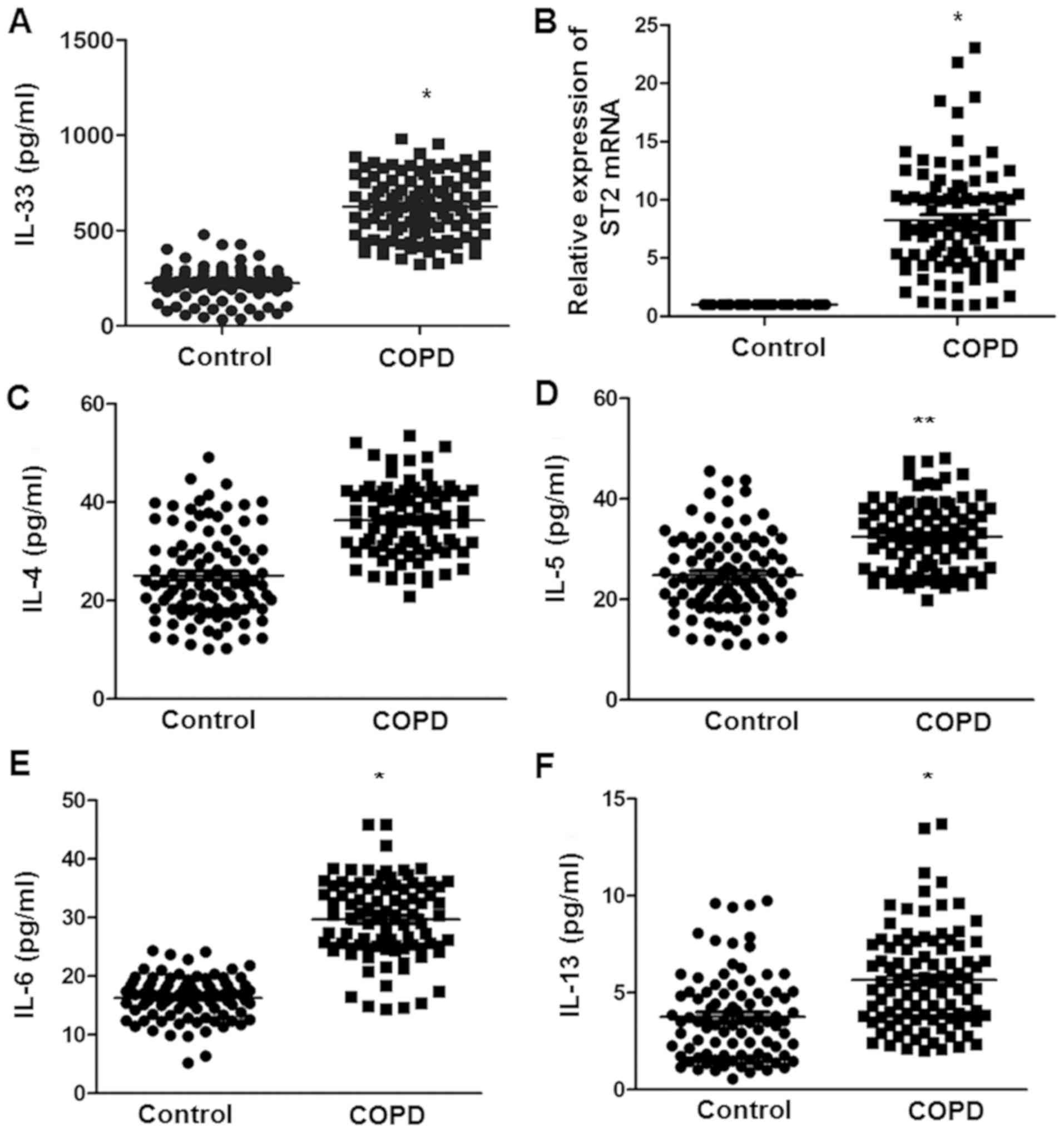
significantly compared with controls. The serum IL-33 levels were
assessed in control and COPD patients by using ELISA. The serum
levels of IL-33 in COPD patients were significantly higher than
those in the control group (Fig.
1A). The expression of ST2 mRNA detected by RT-qPCR in PBMCs of
patients with COPD was also significantly higher than that in the
control group (Fig. 1B). In
addition, the levels of the cytokines IL-4, IL-5, IL-6 and IL-13 in
the serum of the control and COPD subjects were determined. No
significant difference was identified in the serum IL-4 levels
between the two groups (Fig. 1C).
The levels of IL-5, IL-6 and IL-13 in COPD patients were
significantly higher than those in the control group (Fig. 1D-F). These results indicate that
IL-33, ST2 and associated cytokines may be involved in the immune
response in COPD.
Peripheral blood ILC2 are
significantly increased in COPD patients
In the present study,
Lin−CD45+CD127+CRTH2+
cells were detected in peripheral blood by using flow cytometry,
and the changes in the percentage of ILC2 cells in peripheral blood
of control and COPD subjects were analyzed to determine whether
they are involved in the pathogenesis of COPD. The results
indicated that the proportion of ILC2 cells in the peripheral blood
of COPD patients was significantly higher than that in the control
group (Fig. 2A). Furthermore,
RT-qPCR was used to detect the mRNA expression levels of
transcription factors in PBMCs of control and COPD subjects. The
results indicated that the relative mRNA expression levels of CRTH2
and RORA in PBMCs of COPD patients were significantly higher than
those in the controls (P<0.05; Fig.
2B). The expression of ST2 on the surface of ILC2 cells was
then determined using flow cytometry. As presented in Fig. 2C, a high expression of ST2 on the
surface of ILC2 cells was identified in COPD patients, which was
significantly higher than that in the control group. The above
results indicate that ILC2s accounted for a large proportion of
PBMCs in COPD patients, suggesting a role of ILC2s in COPD.
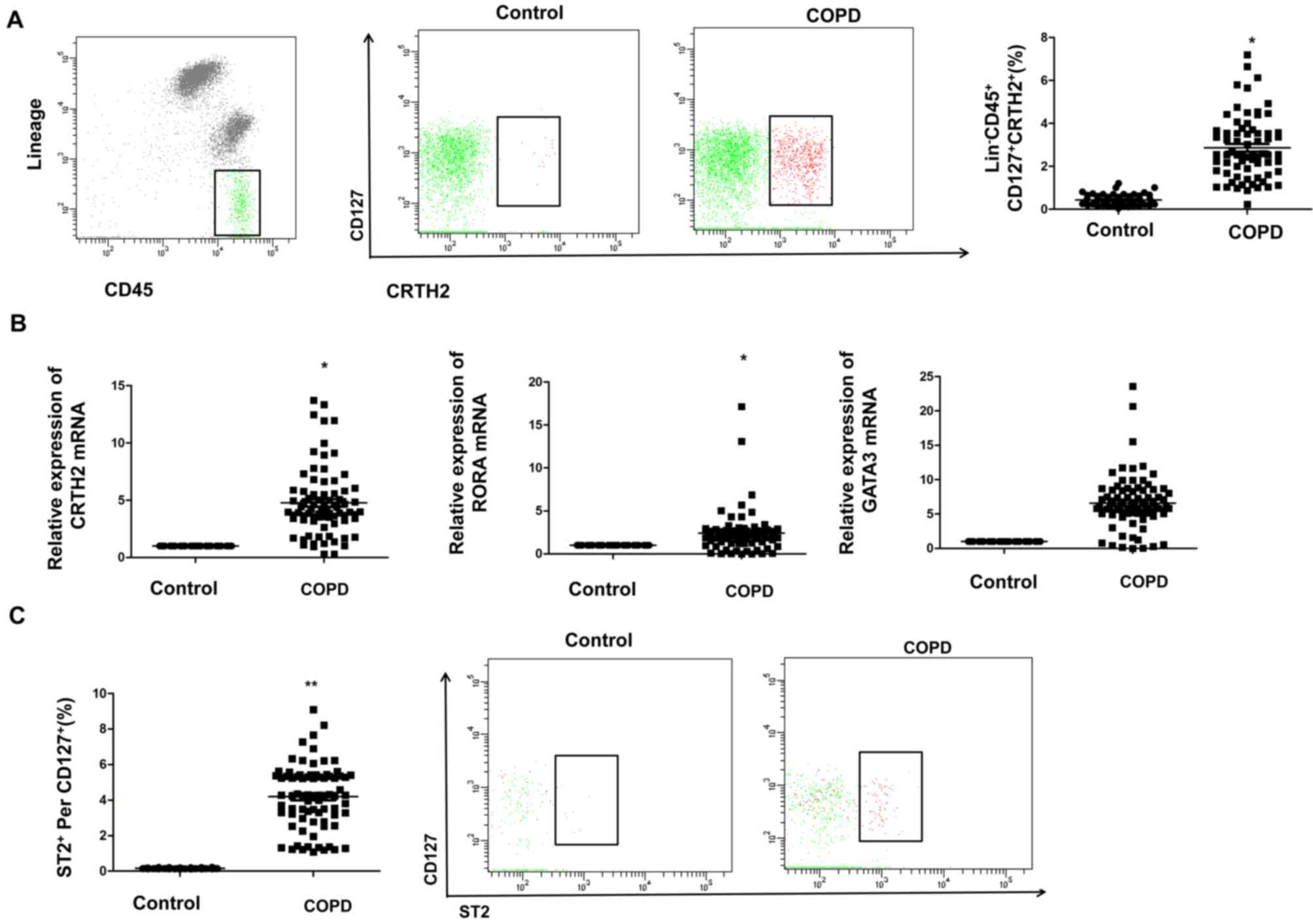 | Figure 2.Increased proportion of ILC2 cells,
nuclear transcription factor and ST2 expression in PBMCs of COPD
patients. (A) Percentage of ILC2s in the peripheral blood of
control subjects and COPD patients. (B) Relative mRNA expression of
CRTH2, RORA and GATA3 in PBMCs of control subjects and COPD
patients. Data were normalized to the control group and the mean
value was set as 1. (C) ST2 expression on the surface of peripheral
blood ILC2s from control subjects and COPD patients. Values are
expressed as the mean ± standard deviation. *P<0.05, **P<0.01
vs. control group. PBMCs, peripheral blood mononuclear cells; COPD,
chronic obstructive pulmonary disease; ILC2s, type 2 innate
lymphoid cells; Lin, lineage; GATA3, GATA binding protein 3; RORA,
RAR-related orphan receptor α; CRTH2, ST2 and prostaglandin D2
receptor 2. |
IL-33 stimulates PBMC derived ILC2s in
patients with COPD
In response to rIL-33 stimulation, the number of
ILC2s increased compared with the PBS control group, suggesting
that rIL-33 stimulates the differentiation of peripheral blood
ILC2s (Fig. 3A). There were
significant differences between each group, but the 50 ng/ml IL-33
exhibited the strongest differentiation effect on ILC2 cells. To
investigate the effects of rIL-33 and anti-ST-2 on the cell
viability of ILC2s in vitro, a CCK-8 assay was performed. As
presented in Fig. 3B, the cell
viability of ILC2s was significantly increased with 50 and 100
ng/ml of rIL-33 for 48 h. However, this increase was abolished by
simultaneous anti-ST-2 treatment. No obvious cell proliferation was
observed in the PBS-treated group. Similarly, the levels of RORA,
GATA3, ST2 and CRTH2 mRNA were significantly increased after
stimulation with rIL-33 (Fig. 3C).
This increase was blocked byanti-ST2 treatment. These results
indicate that IL-33 increases the viability of ILC2s and it is
likely that this effect is exerted, at least partially, via ST2.
The current study also assessed the ability of ILC2 cells to
secrete cytokines after stimulation with rIL-33. The results
revealed that with the increase in the rIL-33 concentration, the
secretion of IL-4, IL-5, IL-6 and IL-13 (Fig. 3D) increased significantly. However,
the expression levels of these cytokines decreased significantly
after the addition of ST2-neutralizing antibody. The results also
indicated that, with the increase of the rIL-33 concentration, the
level of sST-2 increased, which may attenuate the effect of IL-33
and cause the decrease of cytokines observed. Thus, the in
vitro stimulation test further confirmed that IL-33 stimulates
the differentiation of peripheral blood ILC2s in patients with
COPD, and further induces ILC2s to produce the Th2 cytokines
partially through the IL-33/ST2 signaling pathway, thus
participating in the pathogenesis of COPD.
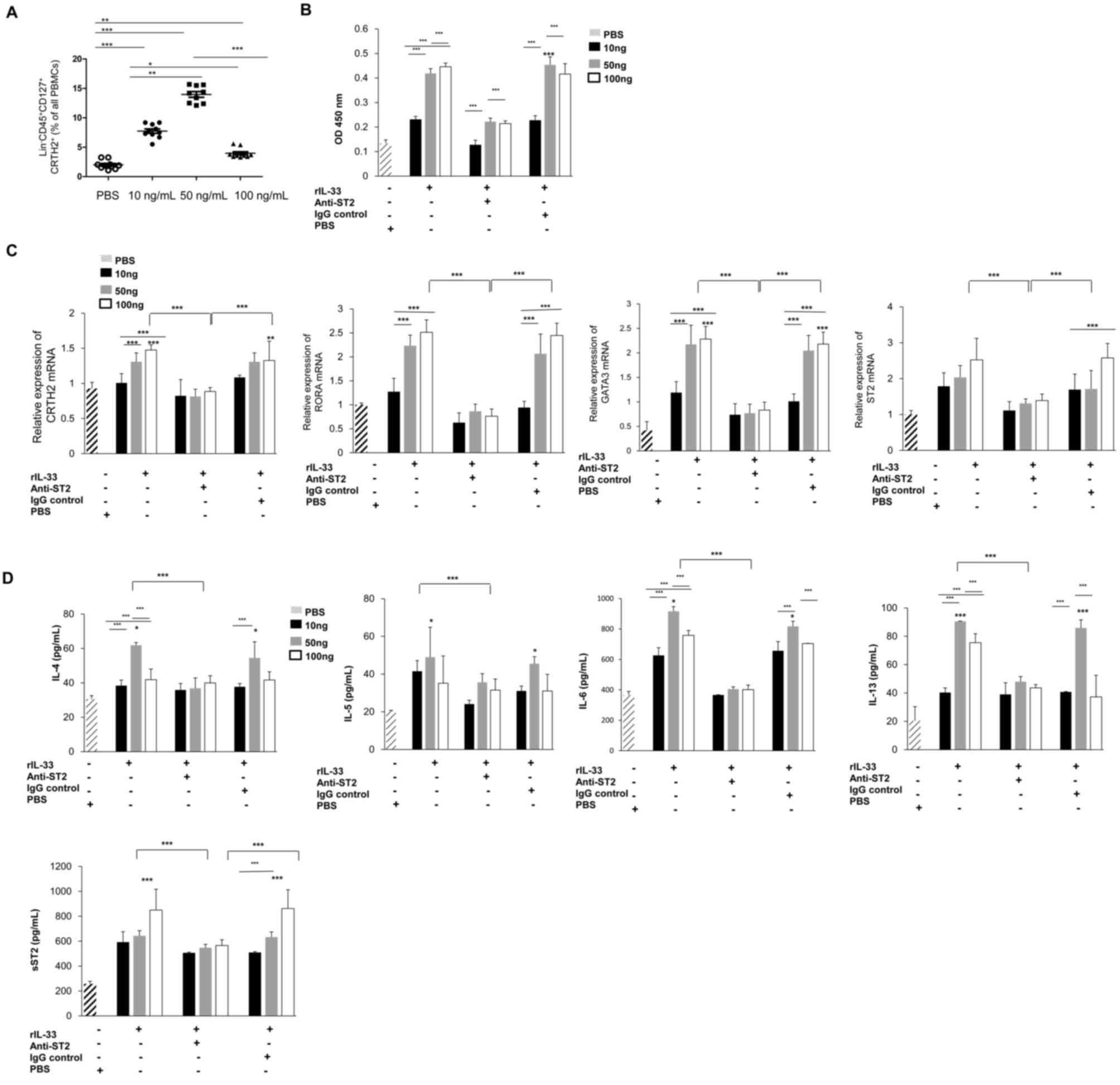 | Figure 3.Viability of ILC2 cells and
expression of cytokines after IL-33 stimulation. (A) The proportion
of ILC2 cells in peripheral blood mononuclear cells from patients
with COPD after stimulation with different concentrations of IL-33
for 48 h. (B) Cell viability was measured with a Cell Counting
Kit-8 assay. The OD value at 450 nm is provided. (C and D) ILC2s
from patients with COPD were stimulated with rIL-33, Anti-ST2, IgG
control or PBS alone for 48 h. (C) The relative expression of
CRTH2, RORA, GATA3 and ST2 mRNA was detected by reverse
transcription-quantitative PCR (n=10). (D) The supernatants were
collected to detect changes in the levels of IL-4, IL-5, IL-6,
IL-13 and sST2 (n=10). *P<0.05, **P<0.01, ***P<0.001 vs.
PBS group. OD, optical density; rIL, recombinant interleukin; sST2,
soluble ST2; COPD, chronic obstructive pulmonary disease; IgG,
immunoglobulin G; GATA3, GATA binding protein 3; RORA, RAR-related
orphan receptor α; CRTH2, ST2 and prostaglandin D2 receptor 2. |
Discussion
Abnormal immune responses to environmental factors
are considered to be the major cause of the pathogenesis of COPD
(28). There is increasing evidence
that the response of the innate immune system is crucial for the
development of COPD (16). IL-33 is
a pleiotropic cytokine that may coordinate a series of complex
immune responses in disease and the innate immune system (29,30).
IL-33 is thought to act as a pro-inflammatory factor and current
research mostly focuses on its role in the cardiovascular system,
autoimmune diseases, infectious diseases (8) and asthma (31,32).
However, there is currently a lack of research on the role of IL-33
and ILC2 cells in COPD. The results of the present study indicate
that the serum levels of IL-33 in COPD patients were significantly
higher than those in healthy individuals. In addition, the
expression of ST2 mRNA in PBMCs of COPD patients was significantly
increased. IL-33 activates mitogen-activated protein kinase and
NF-κB signaling pathways by binding to the receptors ST2 and
IL-1RAcP, and promotes the expression of Th2 cell-associated
cytokines, including IL-4, IL-5 and IL-13 (33). Similarly, the present study
determined that the serum levels of IL-4, IL-6 and IL-13 in COPD
patients were significantly higher than those in healthy
individuals, suggesting that IL-33 may have a role in the
pathogenesis of COPD through the ST2 signaling pathway.
ILC2s are a newly discovered type of innate immune
cells characterized by the absence of lymphocyte markers and
production of Th2-type cytokines, which may link innate immune
responses and adaptive immune responses in diseases involving
various Th2-type immune responses (34). The cells involved in the inflammatory
response to COPD mainly include neutrophils, CD4+ T
cells, CD8+ T cells, macrophages and dendritic cells
(35). It has been indicated that
ILC2 may also be involved in the pathogenesis of COPD (25). ILC2s that are already there, but in
an inactive state, are activated by inflammatory mediators released
by lung epithelial cells and other structural cells, as well as by
immune cells, and promote the pathological inflammatory response in
the lungs (36,37). It has been reported that ILC2s
express CRTH2 and promote the production of IL-13 after IL-33
stimulation (38). Furthermore,
IL-33 promotes the egress of ILC2 cells from the bone marrow
(39). These results suggest that
human airway ILC2s may also respond to IL-33. Studies using mouse
models have also indicated that IL-33 induces type 2 pneumonia by
activating ILC2s (40,41). Activation of ILC2s results in rapid
and robust release of IL-4, IL-5 and IL-13, which are known to
participate in airway eosinophilia, mucus production, airway hyper
responsiveness and tissue remodeling (17). The results of the present study
indicated that the peripheral blood ILC2s in patients with COPD
were significantly elevated, and the mRNA expression levels of
CRTH2 and RORA in PBMCs were significantly higher than those in
healthy individuals. In addition, the flow cytometry results
indicated that the proportion of ILC2 ST2+ cells in
peripheral blood of patients with COPD was significantly higher
than that of healthy individuals, suggesting that ILC2 cells may
have a role in promoting the Th2-type immune response in COPD.
IL-33 stimulation of non-T, B cells in mice led to
rapid production of Th2 cytokines (42). Therefore, IL-33 may cause Th2
cytokine production through Th2 cell-independent pathways involved
in pathological processes. IL-33 is one of the major triggers of
human ILC2 activation (43). In the
present study, it was determined that IL-33 stimulated the
proliferation and differentiation of ILC2s from patients with COPD
in vitro, along with elevated mRNA expression of RORA,
GATA3, ST2 and CRTH2 and upregulation of Th2 cytokines, including
IL-4, IL-5, IL-6 and IL-13, leading to sustained inflammation and
contributing to disease progression. sST2 is a decoy receptor
regulating the activity of IL-33 (44). In the present study, 50 ng/ml IL-33
had the strongest differentiation effect on ILC2 cells. However,
the dose effect of IL-33 on ILC2 cells still requires further
study. Furthermore, the levels of sST2 increased as the IL-33
concentration increased. The increased sST2 may attenuate the
effect of IL-33, thus causing the observed decrease in cytokine
levels.
In the present study, according to GOLD staging, 5
patients with grade I pulmonary function, 94 with grade II
pulmonary function and 8 patients with grade III pulmonary function
were included. However, no patient with grade IV pulmonary function
was included. As subgroup analysis may be incomplete without the
group of patients with grade IV pulmonary function, no subgroup
analysis was performed. A further study with a subgroup analysis is
required.
In conclusion, IL-33 promotes the differentiation of
ILC2s and the secretion of IL-4, IL-5 and IL-6 in PBMCs from
patients with COPD. IL-33 and ILC2s may have important roles in the
development of COPD. Further in-depth study of the mechanism of
action of IL-33 and ILC2s cells in COPD is required. The present
study may provide experimental evidence for the development of cell
immunotherapy for COPD.
Acknowledgements
Not applicable.
Funding
The current study was supported by the National
Natural Science Foundation Regional Fund Project (grant no.
81760793).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
Conceived and designed the experiments: JD and FL.
Performed the experiments: MJ, ST, SZ and FZ. Analyzed the data:
JW. Wrote the manuscript: MJ, JD. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of the Affiliated Hospital of Traditional Chinese
Medicine, Xinjiang Medical University (Urumqi, China). In addition,
written informed consent was obtained from each subject.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Blanchette CM, Gross NJ and Altman P:
Rising costs of COPD and the potential for maintenance therapy to
slow the trend. Am Health Drug Benefits. 7:98–106. 2014.PubMed/NCBI
|
|
2
|
Gupta D, Agarwal R, Aggarwal AN, Maturu
VN, Dhooria S, Prasad KT, Sehgal IS, Yenge LB, Jindal A, Singh N,
et al: Guidelines for diagnosis and management of chronic
obstructive pulmonary disease: Joint ICS/NCCP (I) recommendations.
Lung India. 30:228–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Z, Cheng X, Yue L, Cui W, Zhou W,
Gao J and Yao H: Molecular pathogenesis in chronic obstructive
pulmonary disease and therapeutic potential by targeting
AMP-activated protein kinase. J Cell Physiol. 233:1999–2006. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim SW, Rhee CK, Kim KU, Lee SH, Hwang HG,
Kim YI, Kim DK, Lee SD, Oh YM and Yoon HK: Factors associated with
plasma IL-33 levels in patients with chronic obstructive pulmonary
disease. Int J Chron Obstruct Pulmon Dis. 12:395–402. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hogg JC, Chu F, Utokaparch S, Woods R,
Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson
HO and Paré PD: The nature of small-airway obstruction in chronic
obstructive pulmonary disease. New Engl J Med. 350:2645–2653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brusselle GG, Joos GF and Bracke KR: New
insights into the immunology of chronic obstructive pulmonary
disease. Lancet. 378:1015–1026. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee N, Shin MS and Kang I: T-cell biology
in aging, with a focus on lung disease. J Gerontol A Biol Sci Med
Sci. 67:254–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Molofsky AB, Savage AK and Locksley RM:
Interleukin-33 in tissue homeostasis, injury, and inflammation.
Immunity. 42:1005–1019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gordon ED, Simpson LJ, Rios CL, Ringel L,
Lachowicz-Scroggins ME, Peters MC, Wesolowska-Andersen A, Gonzalez
JR, MacLeod HJ, Christian LS, et al: Alternative splicing of
interleukin-33 and type 2 inflammation in asthma. Proc Natl Acad
Sci USA. 113:8765–8770. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nasr WF, Sorour SS, El Bahrawy AT,
Boghdadi GS and El Shahaway AA: The role of the level of
interleukin-33 in the therapeutic outcomes of immunotherapy in
patients with allergic rhinitis. Int Arch Otorhinolaryngol.
22:152–156. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shan S, Li Y, Wang J, Lv Z, Yi D, Huang Q,
Corrigan CJ, Wang W, Quangeng Z and Ying S: Nasal administration of
interleukin-33 induces airways angiogenesis and expression of
multiple angiogenic factors in a murine asthma surrogate.
Immunology. 148:83–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kearley J, Silver JS, Sanden C, Liu Z,
Berlin AA, White N, Mori M, Pham TH, Ward CK, Criner GJ, et al:
Cigarette smoke silences innate lymphoid cell function and
facilitates an exacerbated type I interleukin-33-dependent response
to infection. Immunity. 42:566–579. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia J, Zhao J, Shang J, Li M, Zeng Z, Zhao
J, Wang J, Xu Y and Xie J: Increased IL-33 expression in chronic
obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol.
308:L619–L627. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agapov E, Battaile JT, Tidwell R, Hachem
R, Patterson GA, Pierce RA, Atkinson JJ and Holtzman MJ: Macrophage
chitinase 1 stratifies chronic obstructive lung disease. Am J
Respir Cell Mol Biol. 41:379–384. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim EY, Battaile JT, Patel AC, You Y,
Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, et
al: Persistent activation of an innate immune response translates
respiratory viral infection into chronic lung disease. Nat Med.
14:633–640. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shaykhiev R and Crystal RG: Innate
immunity and chronic obstructive pulmonary disease: A mini-review.
Gerontology. 59:481–489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karta MR, Broide DH and Doherty TA:
Insights into group 2 innate lymphoid cells in human airway
disease. Curr Allergy Asthma Rep. 16:82016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moro K, Yamada T, Tanabe M, Takeuchi T,
Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H and Koyasu S:
Innate production of T(H)2 cytokines by adipose tissue-associated
c-Kit(+)Sca-1(+) lymphoid cells. Nature. 463:540–544. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Neill DR, Wong SH, Bellosi A, Flynn RJ,
Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, et al:
Nuocytes represent a new innate effector leukocyte that mediates
type-2 immunity. Nature. 464:1367–1370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Price AE, Liang HE, Sullivan BM, Reinhardt
RL, Eisley CJ, Erle DJ and Locksley RM: Systemically dispersed
innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad
Sci USA. 107:11489–11494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wong SH, Walker JA, Jolin HE, Drynan LF,
Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, et al:
Transcription factor RORα is critical for nuocyte development. Nat
Immunol. 13:229–236. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Doherty TA: At the bench: Understanding
group 2 innate lymphoid cells in disease. J Leukoc Biol.
97:455–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang Y and Paul WE: Inflammatory group 2
innate lymphoid cells. Int Immunol. 28:23–28. 2016.PubMed/NCBI
|
|
24
|
Salimi M, Barlow JL, Saunders SP, Xue L,
Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST,
McKenzie AN, et al: A role for IL-25 and IL-33-driven type-2 innate
lymphoid cells in atopic dermatitis. J Exp Med. 210:2939–2950.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Grove KC, Provoost S, Verhamme FM,
Bracke KR, Joos GF, Maes T and Brusselle GG: Characterization and
quantification of innate lymphoid cell subsets in human lung. PLoS
One. 11:e01459612016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oczypok EA, Milutinovic PS, Alcorn JF,
Khare A, Crum LT, Manni ML, Epperly MW, Pawluk AM, Ray A and Oury
TD: Pulmonary receptor for advanced glycation end-products promotes
asthma pathogenesis through IL-33 and accumulation of group 2
innate lymphoid cells. J Allergy Clin Immunol. 136:747–756.e4.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhat TA, Panzica L, Kalathil SG and
Thanavala Y: Immune dysfunction in patients with chronic
obstructive pulmonary disease. Ann Am Thorac Soc. 12 (Suppl
2):S169–S175. 2015.PubMed/NCBI
|
|
29
|
Lefrançais E, Duval A, Mirey E, Roga S,
Espinosa E, Cayrol C and Girard JP: Central domain of IL-33 is
cleaved by mast cell proteases for potent activation of group-2
innate lymphoid cells. Proc Natl Acad Sci USA. 111:15502–15507.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Monticelli LA, Osborne LC, Noti M, Tran
SV, Zaiss DM and Artis D: IL-33 promotes an innate immune pathway
of intestinal tissue protection dependent on amphiregulin-EGFR
interactions. Proc Natl Acad Sci USA. 112:10762–10767. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mizutani N, Nabe T and Yoshino S:
Interleukin-33 and alveolar macrophages contribute to the
mechanisms underlying the exacerbation of IgE-mediated airway
inflammation and remodelling in mice. Immunology. 139:205–218.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith D, Helgason H, Sulem P, Bjornsdottir
US, Lim AC, Sveinbjornsson G, Hasegawa H, Brown M, Ketchem RR,
Gavala M, et al: A rare IL33 loss-of-function mutation reduces
blood eosinophil counts and protects from asthma. PLoS Genet.
13:e10066592017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu H, Turnquist HR, Hoffman R and Billiar
TR: Role of the IL-33-ST2 axis in sepsis. Mil Med Res. 4:32017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McKenzie AN: Type-2 innate lymphoid cells
in asthma and allergy. Ann Am Thorac Soc. 11 (Suppl 5):S263–S270.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mortaz E, Folkerts G and Redegeld F: Mast
cells and COPD. Pulm Pharmacol Ther. 24:367–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mohapatra A, Van Dyken SJ, Schneider C,
Nussbaum JC, Liang HE and Locksley RM: Group 2 innate lymphoid
cells utilize the IRF4-IL-9 module to coordinate epithelial cell
maintenance of lung homeostasis. Mucosal Immunol. 9:275–286. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Silver JS, Kearley J, Copenhaver AM,
Sanden C, Mori M, Yu L, Pritchard GH, Berlin AA, Hunter CA, Bowler
R, et al: Inflammatory triggers associated with exacerbations of
COPD orchestrate plasticity of group 2 innate lymphoid cells in the
lungs. Nat Immunol. 17:626–635. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mjösberg JM, Trifari S, Crellin NK, Peters
CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T and Spits H: Human
IL-25- and IL-33-responsive type 2 innate lymphoid cells are
defined by expression of CRTH2 and CD161. Nat Immunol.
12:1055–1062. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stier MT, Zhang J, Goleniewska K, Cephus
JY, Rusznak M, Wu L, Van Kaer L, Zhou B, Newcomb DC and Peebles RS
Jr: IL-33 promotes the egress of group 2 innate lymphoid cells from
the bone marrow. J Exp Med. 215:263–281. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Doherty TA, Khorram N, Chang JE, Kim HK,
Rosenthal P, Croft M and Broide DH: STAT6 regulates natural helper
cell proliferation during lung inflammation initiated by
Alternaria. Am J Physiol Lung Cell Mol Physiol. 303:L577–L588.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Riedel JH, Becker M, Kopp K, Düster M,
Brix SR, Meyer-Schwesinger C, Kluth LA, Gnirck AC, Attar M, Krohn
S, et al: IL-33-mediated expansion of type 2 innate lymphoid cells
protects from progressive glomerulosclerosis. J Am Soc Nephrol.
28:2068–2080. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Furusawa J, Moro K, Motomura Y, Okamoto K,
Zhu J, Takayanagi H, Kubo M and Koyasu S: Critical role of p38 and
GATA3 in natural helper cell function. J Immunol. 191:1818–1826.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang J, Xia YF, Zhang MZ and Zhang LM:
IL-33 signaling in lung injury. Transl Perioper Pain Med. 1:24–32.
2016.PubMed/NCBI
|
|
44
|
Hayakawa H, Hayakawa M and Tominaga SI:
Soluble ST2 suppresses the effect of interleukin-33 on lung type 2
innate lymphoid cells. Biochem Biophys Rep. 5:401–407.
2016.PubMed/NCBI
|