Introduction
The protease-activated receptor 2 (PAR) belongs to
the family of G protein-coupled receptors expressed in the cell
membrane, which is activated by proteolytic cleavage of the
N-terminal domain to unmask a tethered ligand (1). SLIGRL, a PAR2 agonist peptide
consisting of six amino acids, is able to selectively activate PAR2
in the absence of serine proteases. PAR2 activation has been shown
to play a role in inflammation (2),
effects that are attributed in part to a neurogenic mechanism
possibly involving activation of the transient receptor potential
vanilloid 1 (TRPV1) channels (3).
TRPV1 is a non-selective cation channel mainly expressed in primary
sensory neurons and sensory C- and Aδ-fibers (4) that play a role in inflammatory
processes through release of neuropeptides including calcitonin
gene-related peptide (CGRP) and substance P (SP) from the sensory
nerve endings (3). TRPV1 may be
activated by noxious heat and various inflammatory mediators such
as protons, lipoxygenase products, and other endogenous arachidonic
acid derivatives (5). PAR2 has been
shown to co-express with TRPV1 in neurons containing SP and CGRP in
dorsal root ganglia (DRG) (3).
Activation of PAR2 sensitizes TRPV1 expressed in DRG neurons in
vitro and in vivo (6) and
in particular, PAR2 agonists but not other PARs selectively
stimulate release of CGRP and SP from primary spinal afferent
neurons (6).
Activation of PAR2 with SLIGRL protects against
myocardial ischemia and reperfusion (I/R) injury (7). Our previous study demonstrated that
SLIGRL-induced protective effects in ex vivo myocardial I/R
injury model was impaired in TRPV1−/− hearts (8). The present study, using an in
vivo myocardial I/R injury model, investigated whether
PAR2-induced cardioprotection is mediated by activation of TRPV1
and release of CGRP and SP.
It has been shown that PAR2 protects against
myocardial I/R injury via lipoxygenase (LOX)-derived eicosanoids
released from endothelium to regulate the coronary circulation
(7,9). PAR-2 activation causes vasodilation,
which can be attenuated by inhibition of nitric oxide (NO) or
prostaglandins (PGs) synthesis (10). Vasodilation responses to
acetylcholine in the perfused heart were impaired after I/R injury
whereas vasodilatory response to PAR2 was preserved (7). Given that PGs and LOX products have
been indicated to be able to activate or sensitize TRPV1 channel
(11), PAR2-mediated activation of
the LOX pathways may contribute to cardioprotection after I/R
injury via activation of TRPV1. Despite a substantial body of
evidence demonstrates that arachidonic acid (AA) derivatives play
an important role in myocardial I/R injury (12), it is unknown whether LOX or
cyclooxygenase (COX) pathways contribute to PAR2-induced activation
or sensitization of TRPV1 channel.
In this study, we tested the hypothesis that the LOX
pathway mediates PAR2-induced activation of TRPV1 to protect
against myocardial I/R injury. We investigated the involvement of
TRPV1 channel and its induced CGRP and SP in PAR2-induced
cardioprotection in in vivo myocardial I/R injury model
using TRPV1 null (TRPV1−/−) mouse model and
pharmacological antagonists, respectively. The involvement of LOX
and COX in PAR2-induced activation of TRPV1 was dissected by using
various pharmacological inhibitors in ex vivo perfused heart
model.
Materials and methods
In vivo myocardial I/R model
All experimental procedures involving animals were
approved by the Michigan State University Animal Care and Use
Committee (East Lansing, USA) and conform to The National
Institutes of Health guidelines (Bethesda, MD, USA). The
12-week-old male TRPV1−/− strain
B6.129S4-TRPV1tm1Jul and matching control wild type (WT)
strain C57BL/6J mice (Jackson Laboratory) were used. Acute
myocardial I/R models were established as described (13). Briefly, mice were anesthetized by
intraperitoneal injection of pentobarbital sodium (50 mg/kg body
weight). The left anterior descending (LAD) artery was ligated with
an 8-0 silk suture for 45 min, and the myocardial ischemia was
confirmed by pale color in the occluded distal. Then, the ligature
was released for 3 h reperfusion, and the reperfusion was confirmed
by return of a red color in the pale region.
Experimental protocols in vivo
Mice were randomly assigned to one of following
treatment, all peptides injection combined with amastatin (1.25
mg/kg), an aminopeptidase inhibitor given simultaneously to avoid
peptide degradation; all treatments were given through
intraperitoneal injection (i.p.) 10 min before reperfusion. Control
groups: LSIGRL (2.5 mg/kg, i.p.) a SLIGRL negative control peptide
(Peptides international Inc.) was given; PAR2 activation group:
SLIGRL (2.5 mg/kg, i.p., EC50=5 µmol/l) was given 10 min
before reperfusion; RP67580 pretreatment group: RP67580 (2 mg/kg,
i.p.), a NK1 receptor antagonist was given 5 min before SLIGRL;
CGRP8-37 pretreatment group: CGRP8-37, a CGRP receptor antagonist
(1 mg/kg, i.p., IC50=4.9 nmol/l) was given 5 min before
SLIGRL and then CGRP8-37 was continually intravenous injection at 2
nmol/kg/min. The dosage of was determined by our pilot study and
based on previous report (14).
Evaluation of myocardial infarct
size
Infarct size was measured according to the method as
described (13). Briefly, after 3 h
of reperfusion, the LAD was occluded with a suture at the same site
of the initial ligation. To demarcate the ischemic area at risk,
Evans blue dye (1%) was perfused into the aorta. Then hearts were
excised and sliced into five cross sections below the ligature. The
heart sections were then incubated with a 1% triphenyltetrazolium
chloride (TTC) for 15 min at 37°C. Once the color was established,
the slices were fixed in 10% formalin for 24 h and weighed. Both
sides of each slice were quantified by Image J. The infarcted area
to risk area ratio (% infarct size) was calculated and multiplied
by the weight of the slice.
Measurement of SP and CGRP (15)
The blood samples were collected at 10 min after
SLIGRL treatment. Commercially available CGRP and SP
radioimmunoassay kits (Peninsula Laboratories Inc.) were used to
determine the concentrations of CGRP and SP. The plasma samples
were purified by the supplier recommended methods.
Langendorff heart preparation and
measurements of cardiac function (13,15)
Mice (12-week-old) were heparinized (500 U/kg i.p.)
and anesthetized with pentobarbital sodium (50 mg/kg i.p.).
Isolated hearts from TRPV1−/− and WT mice were perfused
at 37°C and 80 mmHg with Krebs-Henseleit buffer (118 mmol/l NaCl,
4.7 mmol/l KCl, 1.2 mmol/l MgSO4, 1.2 mmol/l
KH2PO4, 2.5 mmol/l CaCl2, 25
mmol/l NaHCO3, 0.5 mmol/l Na-EDTA, and 11 mmol/l
glucose, saturated with 95% O2−5% CO2, pH
7.4) with Langendorff apparatus. A water-filled balloon was
inserted into the left ventricle and adjusted to a left ventricular
end-diastolic pressure (LVEDP) of 5–8 mmHg. The distal end of the
catheter was connected to a Digi-Med Heart Performance Analyzer via
a pressure transducer. Coronary flow (CF) was continuously measured
using an ultrasonic flow probe. Hearts were paced at 400 bpm except
during sustained global ischemia, and pacing was reinitiated 3 min
after reperfusion. Left ventricular developed pressure (LVDP), left
ventricular peak positive dP/dt (+dP/dt) during isovolumic
contraction were used as indices of left ventricular (LV) systolic
function; LVEDP were used as indices of LV diastolic function.
Experimental protocols ex vivo
All hearts were allowed to stabilize for 25 min,
then perfused at 1% of the coronary flow rate with LSIGRL control
(10−7 M), vehicle control (DMSO), or SLIGRL
(10−7 M). For the groups of SLIGRL plus inhibitors, LOX
inhibitor nordihydroguaiaretic acid (NDGA, 5×10−6 M),
12-LOX inhibitor baicalein (1×10−5 M), COX inhibitor
indomethacin (1×10−5 M) (16), CYP450 inhibitor miconazole
(1×10−6 M), and selective CYP450 epoxygenase inhibitor
N-methylsulphonyl-6-(2-proparglyloxy-phenyl) hexanamide (PPOH,
2×10−5 M) (17) were
added into the perfusate 5 min before adding SLIGRL and continued
for additional 5 min after SLIGRL perfusion. Hearts were
subsequently subjected to 35 min of no-flow normothermic global
ischemia followed by 40 min of reperfusion.
Statistical analysis
All values are expressed as the mean ± standard
error of the mean. Comparisons among groups measured at the end of
the I/R experiments in the bar charts of each figure and in SP,
CGRP release and infarct size experiments were performed by one-way
analysis of variance followed by the Tukey-Kramer multiple
comparison test. To compare two groups in infarct size decrease
(%), a t-test was used. The results were considered statistically
significant at P<0.05.
Results
Protective effects of SLIGRL on
myocardial I/R injury
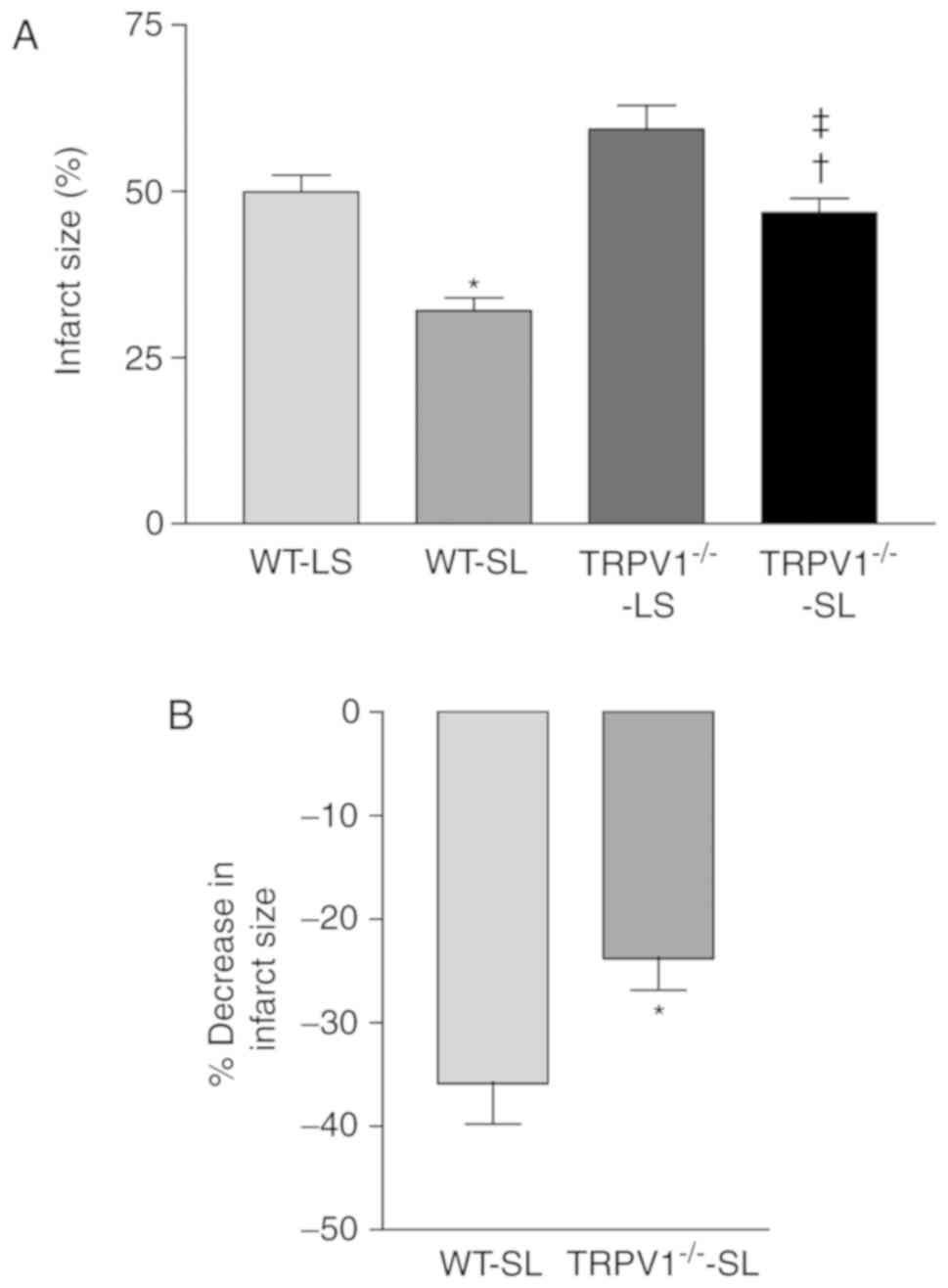
In vivo myocardial I/R experiments showed that
SLIGRL significantly reduced infarct size in both WT and
TRPV1−/− mice (both P<0.05, Fig. 1A), when compared to LSIGRL-treated
control groups. More importantly, the reduction percentage of
infarct size when compared to LSIGRL-treated control group was
greater in WT than that in TRPV1−/− mice (P<0.05;
Fig. 1B).
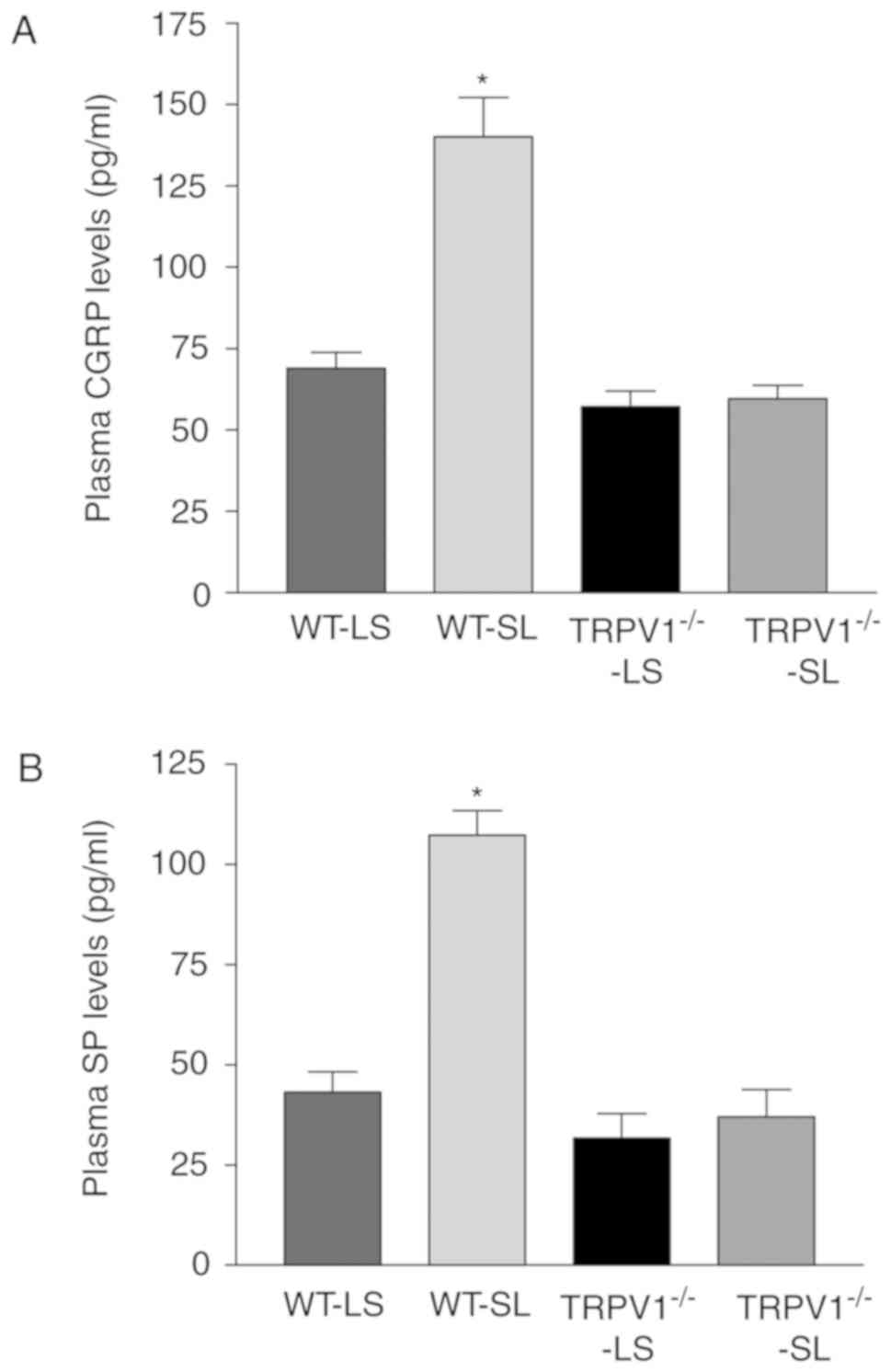
The release of SP and CGRP
Compared to LSIGRL-treated groups, SLIGRL
significantly increased the plasma levels of both CGRP and SP in WT
mice (both P<0.05) but not in TRPV1−/− mice (Fig. 2).
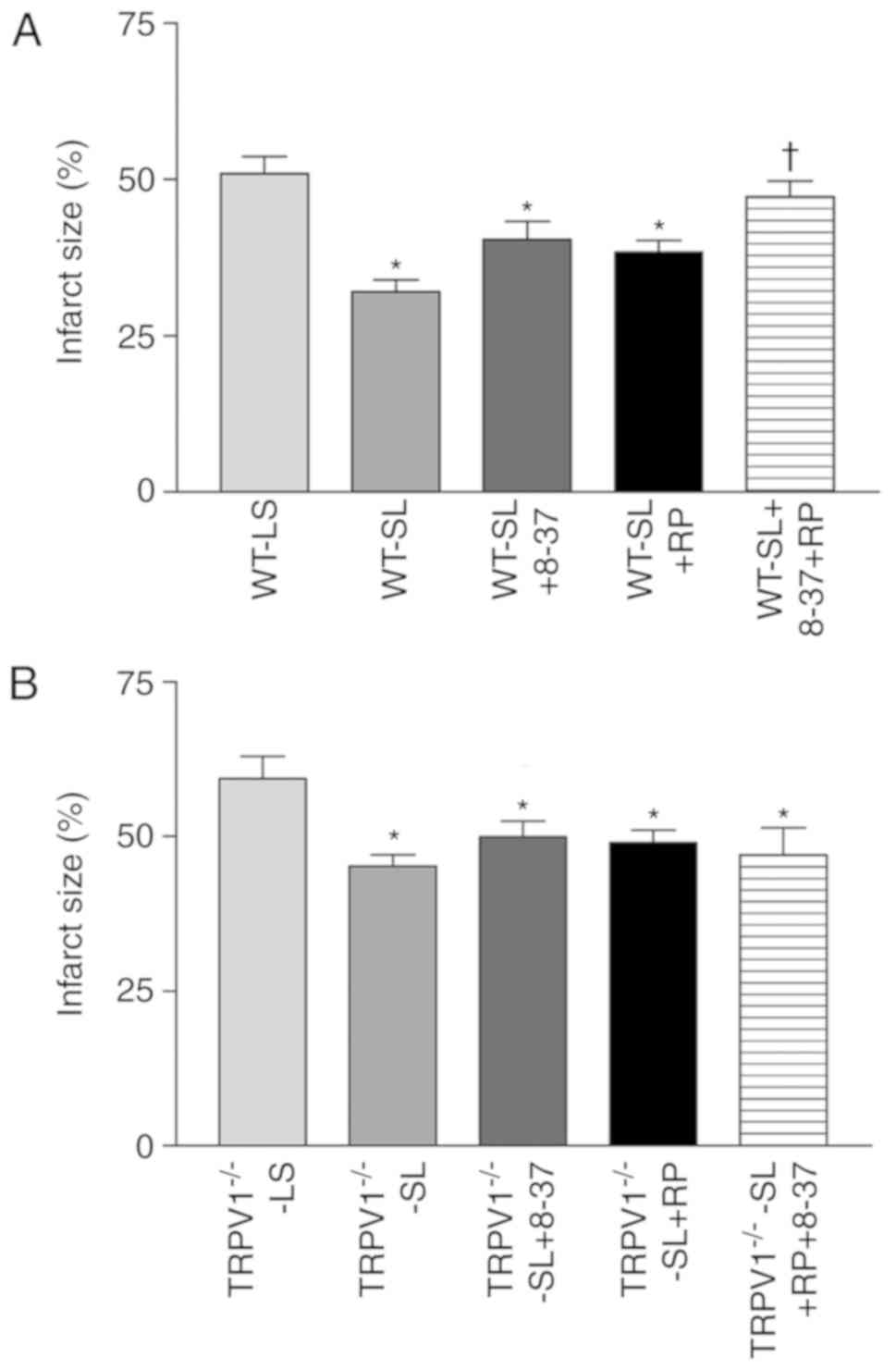
Blockade of the CGRP and SP receptors
on SLIGRL-induced cardioprotection
Pretreatment with CGRP8-37 or RP67580 alone did not
have a significant effect on SLIGRL-induced reduction of infarct
size in both WT and TRPV1−/− mice (Fig. 3). Interestingly, the protective
effects of SLIGRL were abolished by combined treatment with
CGRP8-37 and RP67580 in WT mice but not in TRVR1−/− mice
(Fig. 3).
Effects of COX inhibition on
SLIGRL-induced cardioprotection
The improvement of hemodynamic parameters after
treated with SLIGRL were reported in our previous publication
(18). The hemodynamic parameters
were impaired in TRPV1−/− mice when compared to WT mice
in ex vivo myocardial I/R model (all P<0.05, Table I). Inhibition of COX with
indomethacin did not affect the hemodynamics of both WT and
TRPV1−/− hearts treated with SLIGRL (Table I).
 | Table I.Effects of cyclooxygenase,
lipoxygenase and epoxygenase inhibitors on SL-induced cardiac
protection. |
Table I.
Effects of cyclooxygenase,
lipoxygenase and epoxygenase inhibitors on SL-induced cardiac
protection.
| Groups | CF, % | +dP/dt, mmHg/s | LVEDP, mmHg | LVDP, mmHg |
|---|
| WT-SL | 81.8±2.2 | 3736.8±137.6 | 10.4±1.5 | 67.0±2.4 |
| TRPV1−/−
-SL |
57.3±5.0a |
2740.6±185.4a |
19.2±1.9a |
52.8±3.4a |
| WT-SL+Indo | 71.4±4.9 | 3479.2±145.4 | 13.0±1.7 | 59.5±3.9 |
|
TRPV1−/−-SL+Indo | 66.8±3.9 | 2732.4±271.9 | 18.8±2.1 | 51.7±2.9 |
| WT-SL+NDGA |
58.9±5.2a |
2961.7±202.0a |
19.0±1.7a |
54.4±3.1a |
|
TRPV1−/−-SL+NDGA |
53.9±4.4a |
2347.8±184.8a |
24.5±3.3a |
47.1±2.3a |
| WT-SL+Bai |
55.0±9.6a |
2680.6±180.5a |
20.4±2.7a |
51.0±1.8a |
|
TRPV1−/−-SL+Bai |
52.5±2.9a |
2431.0±214.3a |
21.0±1.7a |
46.1±3.5a |
| WT-SL+Mico |
57.1±3.3a |
2468.8±159.8a |
21.6±0.7a |
44.9±4.0a |
|
TRPV1−/−-SL+Mico |
44.2±8.7a |
2113.0±255.9a,b |
26.2±2.3a,b |
40.6±4.4a |
| WT-SL+PPOH | 70.5±5.1 | 3063.8±234.7 | 18.1±2.1 | 61.4±4.3 |
|
TRPV1−/−-SL+PPOH |
56.5±7.1a |
2696.2±205.9a |
22.4±2.6a |
52.3±3.4a |
Effects of LOX inhibition on
SLIGRL-induced cardioprotection
The cardioprotective effects of SLIGRL were
attenuated by inhibition of either non-specific LOX inhibitor NDGA
or 12-LOX inhibitor baicalein in WT hearts but not in
TRVR1−/− hearts (Table
I).
Effects of CYP450 inhibition on
SLIGRL-induced cardioprotection
The cardioprotective effects of SLIGRL were
significantly attenuated by inhibition of CYP450 with miconazole in
WT hearts expressed as increased LVED and decreased CF, LVDP, and
+dP/dt, as well as in TRPV1−/− hearts expressed as
increased LVED and decreased +dP/dt (all P<0.05, Table I). However, selective inhibition of
CYP450 epoxygenase with MS-PPOH did not affect SLIGRL-induced
protective effects in WT and TRPV1−/− hearts (Table I).
Discussion
A possible link between PAR2 and TRPV1 in sensory
neurons has been suggested in the literatures (19). Accumulating evidence suggests that
PAR2 activation exerts a cardioprotective effect during myocardial
I/R and produces potent dose-dependent coronary vasodilatation in
NO-dependent or -independent manner (7,9). It is
postulated that PAR2 induces vasodilatation via releasing
lipoxygenase-derived eicosanoid and activating sensory C-fibers
along the coronary arteries (7).
Activation of PAR2 led to sensitization or activation of TRPV1 and
increased CGRP and SP release (3),
and these neuropeptides may induce coronary vasodilatation,
(20) which are expected to protect
heart from I/R injury. Pretreatment with capsaicin, leading to CGRP
and SP release, significantly improved the recovery of hemodynamics
after I/R injury (21). Our previous
study has also shown that treatment with exogenous CGRP and SP
before ischemia protected heart from I/R injury in both WT and
TRPV1−/− hearts (22). In
addition, PAR2 agonists-induced vasodilatation was profoundly
attenuated by the TRPV1 selective antagonist capsazepine (7). However, capsazepine inhibits not only
TRPV1 but also mitochondrial function to induce cellular apoptosis
and necrosis via non-receptor mediated mechanisms (23). To further examine whether TRPV1 is
involved in PAR2-mediated cardioprotection during I/R injury in
vivo, TRPV1−/− and WT mice were used, and we
demonstrated that WT mice showed greater improvement of heart
function and larger reduction of infarct size after treated with
SLIGRL when compared with TRPV1−/− mice, suggesting that
TRPV1 plays a critical role in PAR2-induced cardioprotection.
Reperfusion of ischemic tissue results in increased
production of oxidants and radicals that can initiate inflammatory
response at reflow. It was reported that ROS participate in
mediating TRPV1-dependent and neuropeptide-dependent vasodilatation
(20). Moreover, SP and CGRP are not
only related to vasodilatation but also to inflammatory response.
There is increasing evidence that both TRPV1 and PAR2 play an
important role in inflammation acting as either a pro-inflammatory
or anti-inflammatory mediator, which depends on the activated cells
and the type of inflammation (2,24–26).
Those pro-inflammatory and anti-inflammatory effects involve
neurogenic mechanisms, because it is prevented by ablation of
sensory nerves or using TRPV1−/− mice (25,26).
CGRP is one of the most potent vasodilators identified to date, and
has been shown particularly sensitive to coronary vasculature
(27). In addition to vasodilation,
CGRP exerts positive chronotropic and inotropic effects (28). CGRP also regulates inflammatory
processes, including inhibiting NF-κB activation and lowering ROS,
interleukin-2, and monocyte chemoattractant protein-1, suggesting
that CGRP-induced cardioprotection might also be mediated by its
anti-inflammatory effects (29,30). SP
can act on inflammatory cell in vivo and has
pro-inflammatory effects (31).
Studies showed that pretreatment with a tachykinin NK1
receptor antagonist markedly inhibited I/R injury (32,33).
However, in intestinal I/R model, tissue damage was significantly
reduced by SLIGRL and this effect was abolished by pretreatment
with RP67580, indicating that NK1 receptor mediates
PAR2-induced protective effects. In the present study, SLIGRL
induced higher plasma levels of SP and CGRP in WT than
TRPV1−/− mice. The protective effect of SLIGRL on
myocardial I/R injury was not able to be abolished by pretreatment
with CGRP8-37 or RP67580 alone. However, pretreatment with CGRP8-37
and RP67580 together can significantly increase infarct size in
SLIGRL-treated WT mice but not in TRPV1−/− mice. The
results indicate that SP and CGRP may have a cooperative effect on
cardioprotection, and PAR2 activator-induced myocardial protection
is, at least partially, mediated by SP and CGRP. PAR2 activator
sensitizes not only TRPV1 but also TRPV4, the latter is also
co-expressed with PAR2 in DRG (34).
Thus, the TRPV4 channel may also contribute to the cardioprotective
effect of PAR2 activation. This is probably a mechanism through
which the infarct size was slightly reduced by PAR2 activation in
TRPV1−/− mice. Interestingly, the protective effects of
PAR2 on TRPV1−/− mice was not attenuated by combination
treatment with CGRP8-37 and RP67580, suggesting a different
mechanism between WT and TRPV1−/− mice. One of the
possible reasons could be that TRPV4 might compensate the function
of TRPV1 in the knockout mice.
Evidence demonstrates that various metabolic
products of AA such as LOX products and PGs can activate or
sensitize TRPV1 (11,35). Studies showed that PAR2 activators
stimulated the release of PGs. PGs can enhance release of SP and
CGRP (35). Moreover,
PGI2 protects the heart from myocardial I/R injury and
decreases oxidative stress (36).
However, in present study, COX inhibition with indomethacin did not
affect SLIGRL-induced protection in both WT and TRPV1−/−
hearts. It is consistent with other studies showing that perfusion
with indomethacin before ischemia did not significantly alter LVDP
recovery (37). These results
indicate that metabolic products of COX pathway might not be
related to PAR2-induced myocardial protection.
Products of LOX have been implicated in mediating
inflammatory responses and can function as potent vasodilator,
particularly in the setting of oxidative stress. LOX products, such
as hydroperoxyeicosatetraenoic acids, hydroxyeicosatetraenoic
acids, and leukotriene B4 directly activate TRPV1 in
sensory neurons (11). Moreover,
12-LOX-derived eicosanoids protect against myocardial I/R injury
via activation of neuronal TRPV1 (38). The present study showed that the
protective effects of SLIGRL were suppressed in the presence of
NDGA and baicalein in WT mice but there were no significant
differences in TRVR1−/− hearts. These results are in
line with previous study demonstrated that baicalein suppressed
SLIGRL-induced coronary vasodilatation (7), indicating that the 12-LOX/TRPV1 pathway
is involved in PAR2 activation-induced myocardial protection.
AA can also be metabolized by the CYP450 pathway. A
group of products of CYP450 metabolites of the epoxidation,
including epoxyeicosatrienoic acid (EETs), have protective effects
in I/R injury in the heart and vasculature, and EETs possess
anti-inflammatory properties (39).
EETs induce an endothelium-independent relaxation of coronary
arteries and cause vasodilation when NO synthesis is impaired
40). In this study, the protective effects of SLIGRL
were suppressed in the presence of miconazole not only in WT but
also in TRVR1−/− hearts. The effects of miconazole in
hearts may be due to inhibition of either CYP450 or adenylyl
cyclase.
This study provides direct evidence that the TRPV1
receptor plays a role in mediating PAR2 activator SLIGRL-induced
cardiac protection via at least in part, increased endogenous CGRP
and SP release. The 12-LOX/TRPV1 pathway is involved in PAR2
activator-induced myocardial protection.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by grants
from The National Institutes of Health (Bethesda, MD, USA; grant
nos. HL-57853 and HL-73287) and a grant from Michigan Economic
Development Corporation (Lansing, USA).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BZ performed experiments, contributed to the
acquisition, analysis and interpretation of the data, and drafted
the manuscript. SM contributed to the interpretation of the data
and revision of the manuscript. DHW was responsible for the
conception and design of the experiments, interpretation of the
data, and revision of the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All experimental procedures involving animals were
approved by the Michigan State University Animal Care and Use
Committee (East Lansing, USA) and conform to The National
Institutes of Health Guidelines (Bethesda, MD, USA).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ossovskaya VS and Bunnett NW:
Protease-activated receptors: Contribution to physiology and
disease. Physiol Rev. 84:579–621. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steinhoff M, Buddenkotte J, Shpacovitch V,
Rattenholl A, Moormann C, Vergnolle N, Luger TA and Hollenberg MD:
Proteinase-activated receptors: Transducers of proteinase-mediated
signaling in inflammation and immune response. Endocr Rev. 26:1–43.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Steinhoff M, Vergnolle N, Young SH,
Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD,
Wallace JL, Caughey GH, et al: Agonists of proteinase-activated
receptor 2 induce inflammation by a neurogenic mechanism. Nat Med.
6:151–158. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gunthorpe MJ, Benham CD, Randall A and
Davis JB: The diversity in the vanilloid (TRPV) receptor family of
ion channels. Trends Pharmacol Sci. 23:183–191. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Caterina MJ, Leffler A, Malmberg AB,
Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI
and Julius D: Impaired nociception and pain sensation in mice
lacking the capsaicin receptor. Science. 288:306–313. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai Y, Moriyama T, Higashi T, Togashi K,
Kobayashi K, Yamanaka H, Tominaga M and Noguchi K:
Proteinase-activated receptor 2-mediated potentiation of transient
receptor potential vanilloid subfamily 1 activity reveals a
mechanism for proteinase-induced inflammatory pain. J Neurosci.
24:4293–4299. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McLean PG, Aston D, Sarkar D and Ahluwalia
A: Protease-activated receptor-2 activation causes EDHF-like
coronary vasodilation: Selective preservation in
ischemia/reperfusion injury: Involvement of lipoxygenase products,
VR1 receptors, and C-fibers. Circ Res. 90:465–472. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong B and Wang DH: TRPV1 gene knockout
impairs preconditioning protection against myocardial injury in
isolated perfused hearts in mice. Am J Physiol Heart Circ Physiol.
293:H1791–H1798. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Napoli C, Cicala C, Wallace JL, de Nigris
F, Santagada V, Caliendo G, Franconi F, Ignarro LJ and Cirino G:
Protease-activated receptor-2 modulates myocardial
ischemia-reperfusion injury in the rat heart. Proc Natl Acad Sci
USA. 97:3678–3683. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Robin J, Kharbanda R, McLean P, Campbell R
and Vallance P: Protease-activated receptor 2-mediated
vasodilatation in humans in vivo: Role of nitric oxide and
prostanoids. Circulation. 107:954–959. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ,
Jung J, Cho S, Min KH, Suh YG, Kim D and Oh U: Direct activation of
capsaicin receptors by products of lipoxygenases: Endogenous
capsaicin-like substances. Proc Natl Acad Sci USA. 97:6155–6160.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Starkopf J, Andreasen TV, Bugge E and
Ytrehus K: Lipid peroxidation, arachidonic acid and products of the
lipoxygenase pathway in ischaemic preconditioning of rat heart.
Cardiovasc Res. 37:66–75. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yet SF, Tian R, Layne MD, Wang ZY, Maemura
K, Solovyeva M, Ith B, Melo LG, Zhang L, Ingwall JS, et al:
Cardiac-specific expression of heme oxygenase-1 protects against
ischemia and reperfusion injury in transgenic mice. Circ Res.
89:168–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cattaruzza F, Cenac N, Barocelli E,
Impicciatore M, Hyun E, Vergnolle N and Sternini C: Protective
effect of proteinase-activated receptor 2 activation on motility
impairment and tissue damage induced by intestinal
ischemia/reperfusion in rodents. Am J Pathol. 169:177–188. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhong B and Wang DH: N-oleoyldopamine, a
novel endogenous capsaicin-like lipid, protects the heart against
ischemia-reperfusion injury via activation of TRPV1. Am J Physiol
Heart Circ Physiol. 295:H728–H735. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hochhauser E, Alterman I, Weinbroum A,
Barak Y, Harell D, Raz A, Erman A and Vidne B: Effects of
vasoactive substances released from ischemic reperfused liver on
the isolated rat heart. Exp Clin Cardiol. 6:29–34. 2001.PubMed/NCBI
|
|
17
|
Nayeem MA, Ponnoth DS, Boegehold MA,
Zeldin DC, Falck JR and Mustafa SJ: High-salt diet enhances mouse
aortic relaxation through adenosine A2A receptor via CYP
epoxygenases. Am J Physiol Regul Integr Comp Physiol.
296:R567–R574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhong B and Wang DH: Protease-activated
receptor 2-mediated protection of myocardial ischemia-reperfusion
injury: Role of transient receptor potential vanilloid receptors.
Am J Physiol Regul Integr Comp Physiol. 297:R1681–R1690. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amadesi S, Cottrell GS, Divino L, Chapman
K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S,
Vergnolle N and Bunnett NW: Protease-activated receptor 2
sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent
mechanisms in rats and mice. J Physiol. 575:555–571. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Starr A, Graepel R, Keeble J, Schmidhuber
S, Clark N, Grant A, Shah AM and Brain SD: A reactive oxygen
species-mediated component in neurogenic vasodilatation. Cardiovasc
Res. 78:139–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang DH: The vanilloid receptor and
hypertension. Acta Pharmacol Sin. 26:286–294. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang L and Wang DH: TRPV1 gene knockout
impairs postischemic recovery in isolated perfused heart in mice.
Circulation. 112:3617–3623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Athanasiou A, Smith PA, Vakilpour S,
Kumaran NM, Turner AE, Bagiokou D, Layfield R, Ray DE, Westwell AD,
Alexander SP, et al: Vanilloid receptor agonists and antagonists
are mitochondrial inhibitors: How vanilloids cause non-vanilloid
receptor mediated cell death. Biochem Biophys Res Commun.
354:50–55. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nilius B, Owsianik G, Voets T and Peters
JA: Transient receptor potential cation channels in disease.
Physiol Rev. 87:165–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Babánková D, Huang J, Swain GM and
Wang DH: Deletion of transient receptor potential vanilloid type 1
receptors exaggerates renal damage in deoxycorticosterone
acetate-salt hypertension. Hypertension. 52:264–270. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Helyes Z, Elekes K, Németh J, Pozsgai G,
Sándor K, Kereskai L, Börzsei R, Pintér E, Szabó A and Szolcsányi
J: Role of transient receptor potential vanilloid 1 receptors in
endotoxin-induced airway inflammation in the mouse. Am J Physiol
Lung Cell Mol Physiol. 292:L1173–L1181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brain SD, Williams TJ, Tippins JR, Morris
HR and MacIntyre I: Calcitonin gene-related peptide is a potent
vasodilator. Nature. 313:54–56. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Asimakis GK, DiPette DJ, Conti VR, Holland
OB and Zwischenberger JB: Hemodynamic action of calcitonin
gene-related peptide in the isolated rat heart. Life Sci.
41:597–603. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li W, Wang T, Ma C, Xiong T, Zhu Y and
Wang X: Calcitonin gene-related peptide inhibits
interleukin-1beta-induced endogenous monocyte chemoattractant
protein-1 secretion in type II alveolar epithelial cells. Am J
Physiol Cell Physiol. 291:C456–C465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harzenetter MD, Novotny AR, Gais P, Molina
CA, Altmayr F and Holzmann B: Negative regulation of TLR responses
by the neuropeptide CGRP is mediated by the transcriptional
repressor ICER. J Immunol. 179:607–615. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O'Connor TM, O'Connell J, O'Brien DI,
Goode T, Bredin CP and Shanahan F: The role of substance P in
inflammatory disease. J Cell Physiol. 201:167–180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kramer JH, Phillips TM and Weglicki WB:
Magnesium-deficiency-enhanced post-ischemic myocardial injury is
reduced by substance P receptor blockade. J Mol Cell Cardiol.
29:97–110. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Souza DG, Mendonça VA, de A Castro MS,
Poole S and Teixeira MM: Role of tachykinin NK receptors on the
local and remote injuries following ischaemia and reperfusion of
the superior mesenteric artery in the rat. Br J Pharmacol.
135:303–312. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grant AD, Cottrell GS, Amadesi S,
Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi
GW, Bautista-Cruz F, et al: Protease-activated receptor 2
sensitizes the transient receptor potential vanilloid 4 ion channel
to cause mechanical hyperalgesia in mice. J Physiol. 578:715–733.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moriyama T, Higashi T, Togashi K, Iida T,
Segi E, Sugimoto Y, Tominaga T, Narumiya S and Tominaga M:
Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive
mechanism of prostaglandins. Mol Pain. 1:32005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shinmura K, Tamaki K, Sato T, Ishida H and
Bolli R: Prostacyclin attenuates oxidative damage of myocytes by
opening mitochondrial ATP-sensitive K+ channels via the EP3
receptor. Am J Physiol Heart Circ Physiol. 288:H2093–H2101. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Camitta MG, Gabel SA, Chulada P, Bradbury
JA, Langenbach R, Zeldin DC and Murphy E: Cyclooxygenase-1 and −2
knockout mice demonstrate increased cardiac ischemia/reperfusion
injury but are protected by acute preconditioning. Circulation.
104:2453–2458. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sexton A, McDonald M, Cayla C, Thiemermann
C and Ahluwalia A: 12-Lipoxygenase-derived eicosanoids protect
against myocardial ischemia/reperfusion injury via activation of
neuronal TRPV1. FASEB J. 21:2695–2703. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gross GJ, Falck JR, Gross ER, Isbell M,
Moore J and Nithipatikom K: Cytochrome P450 and arachidonic acid
metabolites: Role in myocardial ischemia/reperfusion injury
revisited. Cardiovasc Res. 68:18–25. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ding Z, Gödecke A and Schrader J:
Contribution of cytochrome P450 metabolites to bradykinin-induced
vasodilation in endothelial NO synthase deficient mouse hearts. Br
J Pharmacol. 135:631–638. 2002. View Article : Google Scholar : PubMed/NCBI
|