Introduction
Diabetes mellitus has become the public health
threat with the highest incidence rate. The latest estimated
overall prevalence of total diabetes was 10.9% and the prevalence
of prediabetes was 35.7% in China (1). Overall, diabetes is broadly classified
into 2 major types: Type 1 and type 2. Type 2 diabetes mellitus
(T2DM) is the most common and complex disease, characterised by
insulin resistance, which can lead to a relative state of
hyperinsulinaemia to maintain normal glycaemia, eventually
resulting in β-cell failure (2).
T2DM is considered to be influenced by multifactorial nature,
including genetic, environmental and lifestyle factors (3). However, the molecular mechanisms
underlying insulin dysfunction have not been completely elucidated
yet. Previous studies indicated an association between
sphingosine-1-phosphate (S1P) and T2DM (4,5). S1P was
reported to activate key signalling cascades responsible for the
maintenance of sphingolipid metabolism, which contributed to the
promotion of cell proliferation and suppression of apoptosis
(6). Ceramide and S1P, products of
sphingomyelin metabolism, appeared to represent opposing signalling
cascades, the former signifying cell growth arrest, and the latter
proliferation and survival, the so-called sphingolipid rheostat
(7–10). The sphingolipid rheostat was first
coined in the mid-90′s to describe the repression of
ceramide-mediated programmed cell death through the conversion of
sphingosine to S1P (10–13). Strong evidence suggested that in
balancing the sphingolipid rheostat, S1P signalling was crucial in
the prevention and treatment of diabetes (2). S1P regulates insulin synthesis, insulin
resistance, reduced blood glucose levels, maintenance of blood
vessels and decreased inflammation (2,13).
Therefore, the pharmacological roles of S1P involvement in glucose
metabolism remains unclear, particularly in terms of islet
functions. In the present study, S1P is proposed as a key
regulatory factor of islet β-cell proliferation, potentially
influencing insulin secretion.
S1P is a unique bioactive lipid mediator, which is
generated from sphingomyelin metabolites and has been found to play
an important role in DNA synthesis, Ca2+ mobilization,
MAPK pathway activation (6,14), and, more recently, the proliferation
and survival function of islet β-cell regulation (13). Extracellular S1P was reported to act
as a primary messenger, or ligand receptor, either inside or
outside the cell via the activation of five specific
G-protein-coupled receptors, denoted as S1P receptors 1–5
(S1PR1-5), located at the cell surface and characterized by the
cell type-specific expression pattern (6,14–22).
Each S1PR is involved in distinct and overlapping intracellular
signalling pathways (13,23). S1PRs activate a variety of different
G-proteins, namely Gi (S1PR1-5), Gq (S1PR2/3) and G12/13 (S1PR2-5),
which in turn results in partly diverse functional properties of a
single S1PR (22,24). S1PR1 preferentially couples with Gi
and regulates numerous cell reactions, such as lymphocyte
trafficking and angiogenesis (19,25). In
contrast, S1PR2 and S1PR3 couple with several G proteins, including
Gi, Gq and G12/13, and mediate other pathways (21,26).
S1PR1-3 are widely distributed, whereas S1PR4 is expressed in
lymphoid tissues and lung tissue, and S1PR5 expression is
restricted to tissue of the nervous system. A total of four S1PR
isoforms (S1PR1-4) have been identified in rat and mouse islets and
INS-1 cells (27). This indicates
that S1P, by signalling through S1PR2, plays a key role in pancreas
development, linking lineage allocation and specification, using a
combination of genetic approaches (22,28).
However, the involvement of specific S1PR subtypes for S1P-induced
islet β-cell proliferation remains controversial.
To the best of our knowledge, little is known about
the effects of S1P on islet β-cell proliferation and the role of
S1PR subtypes. Therefore, the aim of the present study was to
investigate the potential effects of S1P on the proliferation and
apoptosis of pancreatic islet β-cell in T2DM mice, established by a
high-fat diet (HFD) with low-dose streptozotocin. The effects of
S1P on blood glucose, the intraperitoneal glucose tolerance test
(IPGTT), homeostatic model assessment of insulin resistance
(HOMA-IR) index and insulin secretion were investigated, and the
expression and localization of S1PR1-3 were observed in diabetic
mice islet cells. This study aimed to explore whether exogenous S1P
induced islet β-cell proliferation and decreased cell apoptosis,
and to evaluate the role of relevant S1PR subtypes in diabetic
mice. The significance of this study was to build further
information on the protective effects of S1P on islet β-cell injury
and the role of S1P in preventing the process of diabetes, and to
provide novel ideas for the prevention and treatment of
diabetes.
Materials and methods
Test materials
Streptozotocin (STZ), S1P and normal rabbit IgG
(cat. no. NI01) were purchased from Sigma-Aldrich; Merck KGaA.
Mouse anti-Ki-67 monoclonal antibody (cat. no. BD550609) was
purchased from BD Biosciences; Becton, Dickinson and Company.
Rabbit anti-insulin (cat. no. sc-9168) and anti-S1P3 (cat. no.
sc-30024) polyclonal antibodies and normal mouse IgG (cat. no.
sc-2025) were purchased from Santa Cruz Biotechnology, Inc. The
TUNEL kit (cat. no. 11684817910) was purchased from Roche
Diagnostics. Rabbit anti-S1P1 polyclonal antibody (cat. no.
ab11424) was purchased from Abcam. Rabbit anti-S1P2 polyclonal
antibody (cat. no. BS2594) was purchased from Bioworld Technology,
Inc. The biotin-streptavidin horseradish peroxidase detection
system include the secondary antibody of 1% biotin-labelled goat
anti-mouse/rabbit IgG (cat. no. SP-9000) and diaminobenzidine (DAB)
kit (cat. no. ZLI-9017) were purchased from Beijing ZSGB-BIO
Technologies, Inc.
Establishment of HFD/STZ diabetic mice
and experimental design
A total of 30 male C57BL/6J mice (age, 4 weeks;
weight, 16–20 g) were obtained from the Experimental Animal Centre
of Xi'an Jiaotong University, Xi'an, China [license no. SCXK (Shan)
2012-003] and were randomly assigned to 2 groups: Normal control
(NC) on a normal diet (n=10), and low-dose-STZ-treated diabetic
group on an HFD, under standard animal housing conditions (n=20).
Food intake and body weight were monitored weekly. Following
feeding on an HFD for 4 weeks, a single low dose of STZ (120 mg/kg
in 0.1 M citrate buffer; pH 4.5) was administered via
intraperitoneal injection after a 14-h fasting to induce diabetes,
while the NC group was treated with the same volume of citrate
buffer solution. A total of two weeks after the STZ treatment,
blood glucose levels were measured to confirm diabetes. Values of
>11.1 mmol/l were considered to indicate diabetes in the
animals. A total of two mice did not fulfil the conditions to be
considered diabetic and one mouse died during the aforementioned
process. The remaining 17 diabetic mice were randomly divided into
two groups using a random number table: Vehicle diabetic control
(DC) group (n=8) and S1P-treated group (S1P group; n=9). S1P
storage solution (1 mg S1P dissolved in 1 ml methanol) was
preserved in the dark at −20°C and diluted in PBS buffer to make
the working concentration. The S1P group animals were administered
20 µg/kg working concentration S1P, whereas the vehicle diabetic
control group mice received an equal volume of methanol in PBS by
intraperitoneal injection daily for 3 weeks. The DC and S1P groups
continued to be fed with an HFD, while the non-diabetic NC group
was fed with the normal diet. Fasting blood glucose, body weight,
food intake and drinking volume were measured weekly. All mice were
housed under conditions of a 12-h light/dark cycle at a temperature
of 25°C and a humidity of 55%. All mice were allowed free access to
food and water throughout the entire experiment. All animal
procedures were approved by the Institutional Animal Care and Use
Committee of the Xi'an Jiaotong University of Health Sciences,
Xi'an, China.
IPGTT
Glucose tolerance was evaluated using a conventional
IPGTT and conducted 21 days after S1P administration. Mice were
weighed after fasting for 8 h, before being administered an
intraperitoneal glucose injection (2 g/kg). Blood samples (~0.6 µl)
were collected from the tail before and at 30, 60 and 120 min after
glucose administration. The blood glucose level was determined with
an Accu-Check Active digital glucose meter (Roche Diagnostics). The
areas under the curves (AUCs) of the IPGTT results were calculated
for the blood glucose levels at 0 (BG0), 30
(BG30), 60 (BG60) and 120 (BG120)
min, using the following equation: AUC=1/4 (BG0) + 1/2
(BG30) + 3/4 (BG60) + 1/2 (BG120).
Fasting insulin was determined by radioimmunoassay. The HOMA-IR
index was calculated using Matthew's equation: Fasting insulin
level (µIU/ml) × fasting glucose level (mmol/l)/22.5 (29). A HOMA-IR index value of >2.5 was
considered to indicate the insulin resistance (29).
Histology and
immunohistochemistry
Each mouse had one pancreas and in total the mice in
3 groups had a total of 27 pancreases. The tail of the pancreas was
taken to make paraffin slides. Pancreatic tissues were carefully
excised, fixed in 10% neutral buffered formalin for 24 h at room
temperature, dehydrated, embedded in paraffin and cut into 5-µm
sections. Overall, ≥7 consecutive sections were cut from the
largest cross-sectional area in the middle of each pancreas tissue
specimen and were used for hematoxylin and eosin (HE) staining,
immunohistochemical staining (insulin, Ki-67, S1PR1, S1PR2 and
S1PR3) and TUNEL staining. Each pancreas had one slide for each
protein immunohistochemical staining by strictly following the same
time and conditions in the process of immunohistochemical
staining.
For histological examination, sections were stained
with hematoxylin for 5 min and with eosin for 2 min at room
temperature. For immunohistochemical staining, the samples were
routinely dewaxed, hydrated and incubated with 3%
H2O2 at room temperature for 10 min. Then, a
microwave was used to retrieval the antigen by heating to 95–100°C
for 10 min in 0.01 mol/l sodium citrate buffer (pH, 6.0). The
slices were then blocked with 0.1% goat serum for 15 min at room
temperature. The sections were incubated with anti-insulin
(1:1,400), anti-Ki-67 (1:200), anti-S1PR1 (1:400), anti-S1PR2
(1:150) and anti-S1PR3 (1:40) primary antibodies at 4°C overnight,
followed by incubation with secondary antibody (1% biotin-labelled
goat anti-mouse/rabbit IgG) at 37°C for 15 min. This was followed
by incubation with horseradish peroxidase-labelled
streptomyces ovalbumin at 37°C for 15 min and staining with
DAB at room temperature. Slides were counterstained with
hematoxylin for 1 min at room temperature to identify the cell
nuclei. As a negative control (Fig.
S1A), the insulin, S1PR1, S1PR2 and S1PR3 primary antibodies
were replaced with rabbit non-specific IgG, whereas the Ki-67
primary antibody was replaced with mouse non-specific IgG (Ki-67)
at the same dilution at 4°C overnight. Myocardial (S1PR1-positive),
kidney (S1PR2- and S1PR3-positive) and tumour (Ki-67-positive)
mouse tissue was used for positive controls (Fig. S1B). The staining was observed using
a DP71 fluorescence microscope (magnification, ×400; Olympus
Corporation). The images were analysed using Adobe Photoshop CS4
extended software (Adobe, Inc.). Image pro Plus 6.0 software (Media
Cybernetics, Inc.) was used for semi-quantitative analysis of
insulin, Ki-67 and S1PR expression in pancreatic tissue. A total of
three to five unique fields of view were randomly selected in each
slice (magnification, ×400) and averages were calculated for
analysis. The integrated optical density (IOD) of positively
stained cells and the corresponding islet area in each islet were
measured and calculated automatically, and the average values were
calculated for analysis. The average optical density (IOD/area) was
calculated for the relative amount of protein. After insulin
staining, the number of islets in each slide was counted and the
area of the maximum islet on each slide was measured in order to
assess islet size. Following Ki-67 staining, the proliferating
nuclei were stained dark brown. The cells whose nuclei were dyed
dark brown were considered to be proliferating islet cells and the
proliferation rate in each islet (%) was calculated as the number
of proliferating cells/the total number of cells ×100%. Overall,
three to five islets were counted for each section and average
values were taken.
TUNEL analysis
The formalin-fixed and paraffin-embedded pancreas
tissue sections were stained with HE and immunohistochemical
staining of insulin. Following the HE and insulin staining, images
of at least 5 fields on each slide were captured using a
fluorescence microscope (magnification, ×400; Olympus Corporation).
Apoptosis was demonstrated using a TUNEL assay kit (Roche
Diagnostics), according to the manufacturer's protocols. After
TUNEL staining, apoptotic nuclei were stained dark brown and cells
with dark brown nuclei were considered to be apoptotic islet cells.
The apoptosis rate in each islet (%) was calculated as the number
of apoptotic cells/the total number of cells ×100% in each islet.
Overall, three to five islets were counted for each section and
average values were taken.
Statistical analysis
Statistical analysis was performed using SPSS
version 21.0 software (IBM Corp.). Continuous variables are
expressed as the mean ± standard deviation or median
(interquartile) from at least three independent experiments.
Intervention and control groups were compared with one-way analysis
of variance followed by Least Significant Difference post hoc test
for parametric analysis and Kruskal-Wallis H test for nonparametric
analysis. P<0.05 was considered to indicate statistically
significant differences.
Results
Islet β-cell morphology and structural
changes
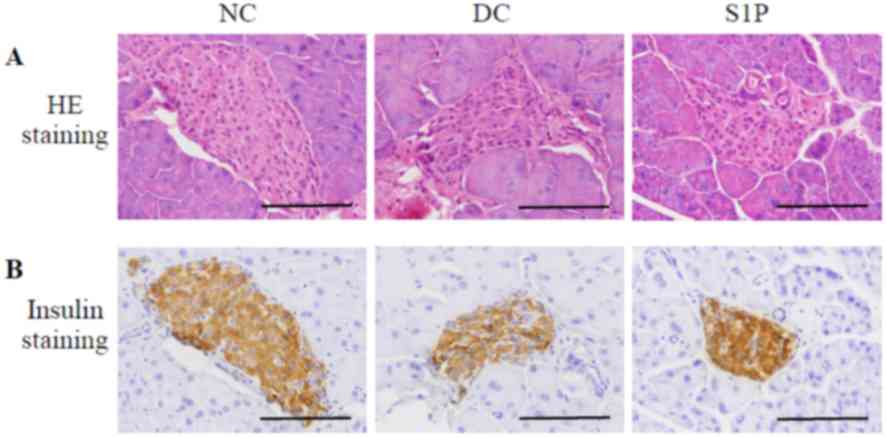
The islets of the NC mice were round or oval, with
clear boundaries and complete film. In addition, the islets were
evenly distributed between the pancreatic acinus; the islet cells
were round or oval, of uniform size, strong staining in the
cytoplasm and a visible centred nucleus. The number of islets in
the DC group was decreased and they were unevenly distributed, with
different degrees of atrophy, and vacuoles appeared within them. A
further loss of β-cells was noted in the diabetic group within
irregularly shaped islets. Ill-defined β-cells were loosely
arranged, the cell volume was increased and the nucleus exhibited
an abnormal distribution (Fig. 1A).
The number of positively stained cells decreased and the insulin
staining was decreased compared with in the NC group (Fig. 1B), suggesting that islet β-cells
showed marked damage after establishment of the animal model. Due
to the limitations of tissue size and the specific parts of
sections, the number of the islets was not identical on different
slides, and each slide could count ~three to ten islets. A number
of the small islets only appear to be small as they were produced
by cutting through the edge of an islet, the maximum islet area was
used as an indicator in each slide to assess the islet area.
Compared with that in the NC group, the islet area in the DC group
was significantly smaller (P<0.01; Table I). The S1P administration group
exhibited less damage; the number and volume of islet cells was
increased, and the islet cells had a clearer structure, and a more
regular shape and arrangement compared with the DC group (Fig. 1A). Immunohistochemistry on the
pancreas of the NC mice revealed intense insulin staining, whereas
weak staining was observed in the core of the islets in the DC
group. The islets in the S1P administration group displayed
stronger insulin immunostaining intensity compared with the DC
group (Fig. 1B). The islet area in
the S1P group was significantly decreased compared with that in the
NC group (P<0.05) and appeared larger than that in the DC group,
but the difference compare with the NC group was not statistically
significant (Table I). An increase
in positive insulin staining granules suggested that S1P could
decrease islet damage and improve β-cell morphology. The average
optical density of the insulin immunohistochemistry (IOD/area) and
maximum islet area were calculated using Image Pro Plus 6.0 image
analysis system (Table I).
 | Table I.The average optical density and the
maximum islet area of insulin immunohistochemistry in all
groups. |
Table I.
The average optical density and the
maximum islet area of insulin immunohistochemistry in all
groups.
| Group | n | IOD/area | Area
(µm2) |
|---|
| NC | 10 | 0.2246±0.0414 | 1,4236±7906 |
| DC | 8 | 0.1896±0.0376 |
8,404±5217a |
| S1P | 9 | 0.2251±0.0366 |
9,992±4955b |
Effect of S1P on fasting blood
glucose
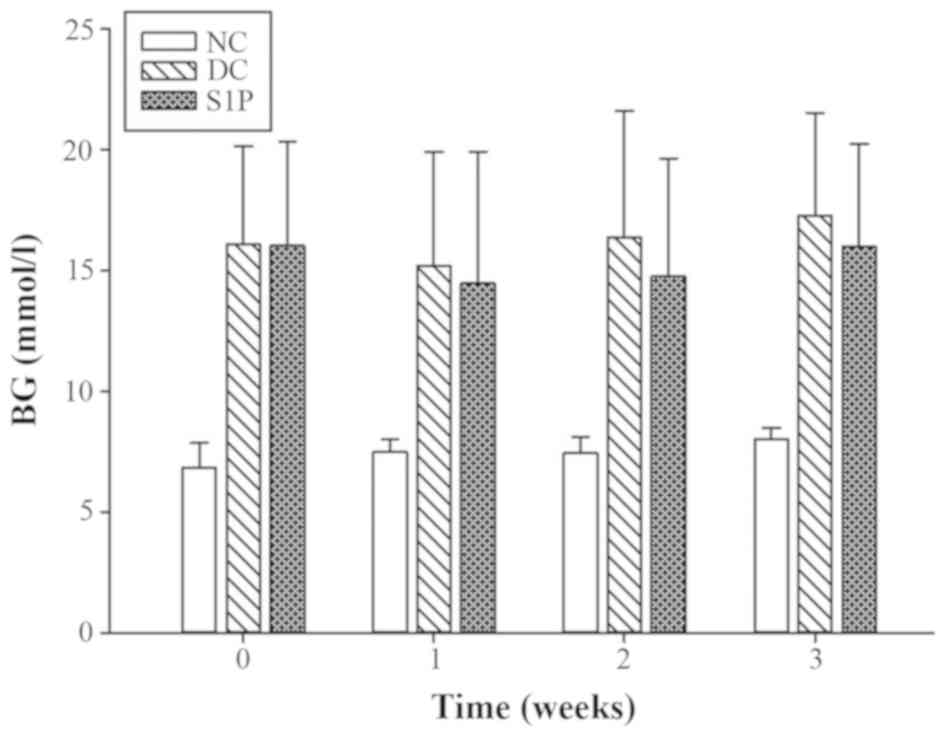
The blood glucose level was measured before and 1, 2
and 3 weeks after S1P administration to observe the effect of S1P
on the fasting blood glucose levels of T2DM mice. The fasting blood
glucose in the DC mice was significantly increased compared with
the NC group (P<0.01; Table II);
1 week after S1P administration, the fasting levels in the S1P
group were decreased slightly compared with the diabetic model
group, but the difference was not significant (P>0.05). The same
trend was observed at weeks 2 and 3 after the administration of
S1P. No statistically significant difference was observed in the
fasting blood glucose between the DC and S1P group (Fig. 2; Table
SI).
| Table II.IPGTT in all groups (mmol/l). |
Table II.
IPGTT in all groups (mmol/l).
| Group | n | 0 min | 30 min | 60 min | 120 min |
|---|
| NC | 10 | 8.03±0.47 | 16.54±1.96 | 11.56±1.29 | 8.61±0.93 |
| DC | 8 |
17.26±4.27a |
29.76±4.14a |
29.59±2.84a |
24.35±3.52a |
| S1P | 9 |
16.01±4.26a |
29.70±3.09a |
28.51±4.49a |
22.93±4.44a |
Effect of S1P on mouse IPGTT
results
IPGTT was conducted 3 weeks after S1P administration
to observe the effect of S1P on pancreatic β-cell function and
metabolic regulation in T2DM mice. Compared with that in the NC
group, the blood glucose in the DC mice reached its peak 30 min
after glucose injection and maintained a high level throughout the
whole experiment. The blood glucose in the S1P mice appeared to
decrease 1 and 2 h after glucose injection, compared with in the DC
group, but the difference was not significant (P>0.05; Table II).
Effect of S1P on fasting serum insulin
levels and the HOMA-IR index
Fasting serum insulin levels were not significantly
different among groups (all P>0.05; Table III) and were consistent with
insulin levels in T2DM. The AUC and HOMA-IR index were calculated
in all groups. Compared with that measured in the NC group, the
HOMA-IR index in the DC mice was significantly increased
(P<0.01), whereas that in S1P group decreased slightly compared
with that in the DC group, but the difference was not statistically
significant (P>0.05; Table
III). The HOMA-IR exhibited a trend similar to that observed
with the AUC. Compared with that calculated in the NC group, the
AUC was significantly increased in the DC and S1P groups (both
P<0.01). The AUC calculated for the S1P group appeared to
decrease compared with the DC group, but with no statistical
significance (P>0.05; Table
III).
| Table III.FINS (mIU/L), HOMA-IR and AUC in all
groups. |
Table III.
FINS (mIU/L), HOMA-IR and AUC in all
groups.
| Group | n | FINS (mIU/l) | HOMA-IR | AUC |
|---|
| NC | 10 | 7.51±2.60 | 2.68±0.98 | 23.25±1.68 |
| DC | 8 | 8.37±2.31 |
6.56±2.80a |
53.56±5.76a |
| S1P | 9 | 8.39±1.93 |
5.82±1.44a |
51.70±6.45a |
Mouse pancreatic β-cell proliferation
assay
Next, an investigation into whether exogenous S1P
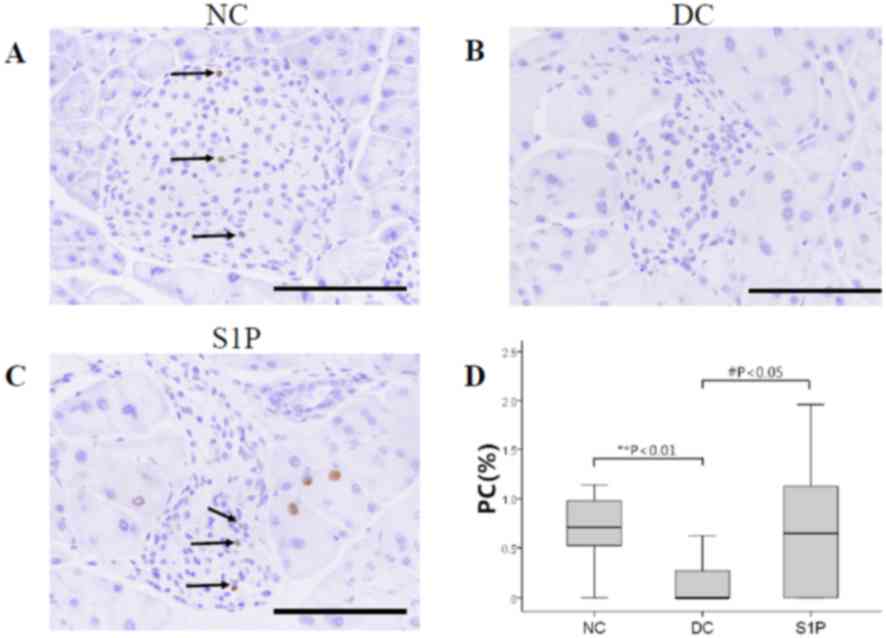
promoted β-cell proliferation was carried out. Ki-67-positive
staining appeared as a brownish yellow colour in the nucleus,
whereas the cell membrane and cytoplasm displayed no staining
(Fig. 3A and C). A total of three to
five unique fields of view of the islets were selected randomly and
the cell proliferation rate of each islet was calculated
separately, then the mean value was taken for analysis. The rates
of Ki-67 expression were <3% in all groups of mouse islets
(Fig. 3D). Fewer Ki-67-positive
cells were observed within the S1P administration group of mouse
islets compared with the NC group (Fig.
3A and C), whereas almost no Ki-67-positive cells were noted in
the diabetic model control group (Fig.
3B). Ki-67-positive cells increased significantly in the S1P
group compared with the DC group (P<0.05; Fig. 3D; Table
SII).
S1P inhibits apoptosis in islet
β-cell
Islet β-cell dysfunction and insulin resistance are
multifaceted with their interdependence for triggering the
pathogenesis of T2DM. TUNEL staining of mouse islet β-cell was used
to determine the percentage of β-cells that were undergoing
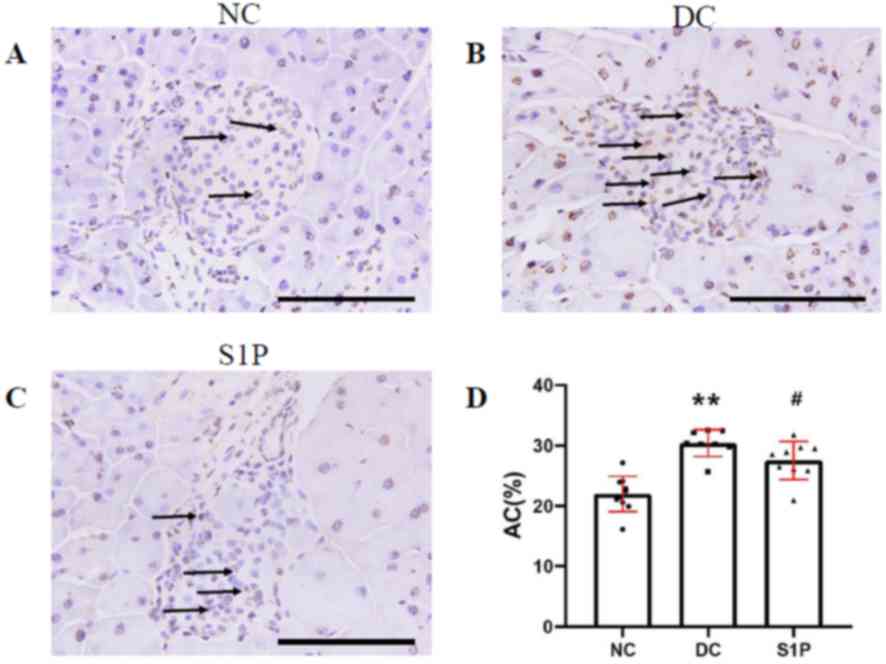
apoptosis (Fig. 4). The apoptotic
islet cell nuclei were stained brown, whereas normal nuclei were
blue (Fig. 4A-C). A significantly
increased number of apoptotic cells were observed in the islets of
the DC mice, compared with those counted in the NC mouse islets
(P<0.01; Fig. 4D; Table SIII). A significantly increased
number of TUNEL-positive cells was observed in the diabetic model
group compared with the S1P administration group (P<0.05;
Fig. 4D; Table SIII), suggesting that S1P serves a
positive role in protecting islet cells against apoptosis.
S1P may enhance islet β-cell
proliferation and survival via S1PR1 and S1PR2
As most of the S1P-induced effects were mediated via
S1PR, it was of interest to investigate the differences of protein
expression of the S1PR subtypes in the mouse pancreas. S1P
signalling is mediated via the activation of specific S1PRs
(S1PR1-5), of which mouse islet β-cell express mainly S1PR1-3
(22). To determine which S1PR
subtypes mediate the S1P-induced protective response in islet
cells, normal and diabetic mouse islets were incubated with
anti-S1PR1-3 antibodies. The positive S1PR1, S1PR2 and S1PR3
protein staining was localised in the cytoplasm and membrane of the
islet cells, with almost no expression in the exocrine pancreas,
consistent with the insulin staining results (Fig. 5A). Compared with that in the NC
group, the positive staining of the S1PR1 protein in the diabetic
mice was stronger and exhibited more uneven distribution (Fig. 5A). The Image Pro Plus 6.0 software
image analysis system was used to calculate the average density of
immunohistochemical S1PR1 protein staining (IOD/area) and
significant differences were observed between the two diabetic
groups, and the NC group (P<0.01; Fig. 5B; Table
SIV). S1PR2 exhibited a similar expression trend to S1PR1
(Fig. 5C; Table SIV). No statistical difference was
observed in the expression of S1PR3 protein between the diabetic
and NC group (P>0.05; Fig. 5D;
Table SIV). These results indicate
that S1PR1, S1PR2 and S1PR3 proteins were expressed in the
pancreatic β-cells. The protein expression of S1PR1 and S1PR2 in
the diabetic mice was significantly increased compared with the NC
group (P<0.01 and P<0.05, respectively; Fig. 5B and C), whereas no significant
difference was observed in the expression of S1PR3 (Fig. 5D). Overall, the findings of the
present study indicate that extracellular S1P induces islet β-cell
proliferation and inhibits cell apoptosis, possibly via S1PR1 and
S1PR2 activation, leading to enhanced islet β-cell protection.
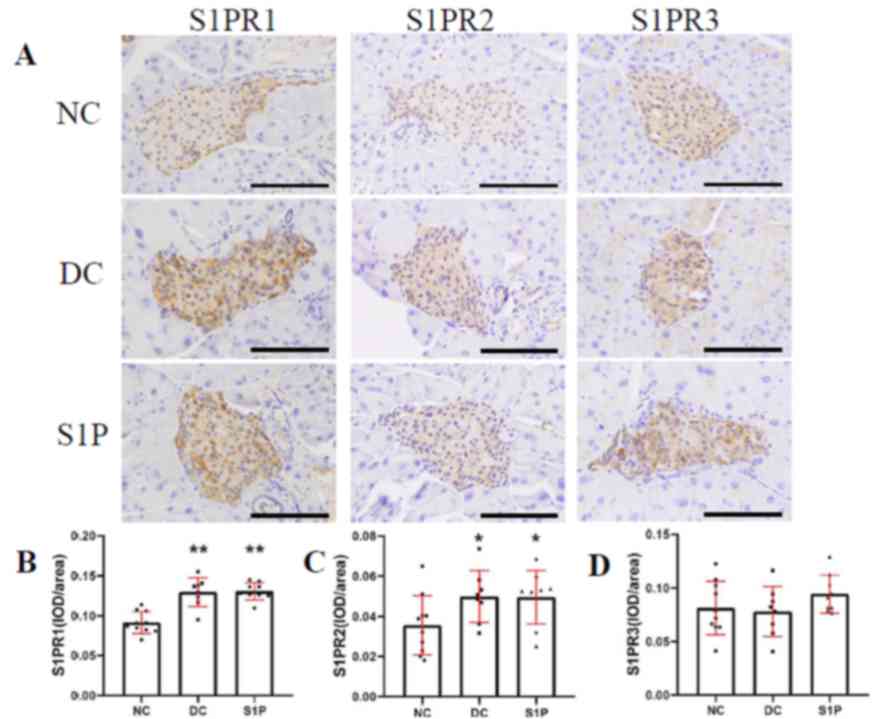 | Figure 5.Immunohistochemical staining of
S1PR1-3 subtypes in the mouse islets. (A) Representative images of
immunohistochemical staining of S1PR1, S1PR2 and S1PR3 in the NC
(n=10), DC (n=8), and S1P (n=9) groups. S1PR1-3 expression is
indicated by the brown staining in the cells. Magnification, ×400;
scale bar, 100 µm. IOD/area of (B) S1PR1, (C) S1PR2 and (D) S1PR3
in all groups. Values are expressed as the mean ± standard
deviation. Parametric one-way analysis of variance followed by
Least Significant Difference post hoc test was performed for the
comparison of the groups. *P<0.05 and **P<0.01 vs. NC. NC,
normal control; DC, diabetic control; S1P, sphingosine-1-phosphate;
S1PR, S1P receptor; IOD, integrated optical density. |
Discussion
T2DM is characterized by insulin resistance in
target tissues and deficiency in the production of insulin from
pancreatic β-cells, caused by decreased β-cell mass (30). Multiple signalling mechanisms have
been demonstrated to influence the islet β-cell mass, of which
β-cell apoptosis emerged as a key mechanism causing decompensation
of β-cells and the development of diabetes (31,32).
Apoptosis of β-cell plays an important role in the occurrence and
development of diabetes, mediated by glucose, free fatty acids,
sulfonylurea, amylin and ceramide (33). The sphingolipid rheostat signalling
pathway is a highly conserved balanced system comprising ceramide
and pro-apoptotic functions on the one hand, and S1P-induced cell
proliferation and survival on the other (7–10,34).
Until now, there are only few studies on S1P signaling pathway in
diabetes which are in the initial stage and not fully understood,
and not much is known about the effect of S1P on islet β-cell
proliferation. In the present study, the effects of S1P on the
proliferation and apoptosis of pancreatic islet β-cells in type 2
diabetic mice were focused on, and the expression and localization
of S1P receptors S1PR1-3 in the pancreatic of type 2 diabetic mice
was observed. On the one hand, the present study tried to indicate
that S1P promotes proliferation and decreases apoptosis of islet
β-cell in diabetes. On the other hand, this study first observed
the expression and localization of S1P receptors S1PR1-3 from
histomorphology in situ in the pancreatic islets of T2DM
mice. The present study demonstrated that compared with in the DC
group, the islets in the S1P administration group showed a greater
insulin immunostaining intensity, higher proliferation rate and
increased numbers of Ki-67-positive cells, whereas the apoptosis
rates were lower. Currently, apoptosis is categorized into two
pathways: Extrinsic and intrinsic pathways, while intrinsic
pathways includes the mitochondrial pathway and endoplasmic
reticulum pathway, extrinsic pathways includes the death receptor
pathway (35). Apoptosis can be
induced through apoptosis inducing factor without relying on
caspases in the mitochondrial pathway. Therefore, caspase 3
staining was not chosen but TUNEL staining to determine apoptosis
of β-cells (36). These results
suggest that S1P leads to an improvement in the morphology of islet
β-cells, the promotion of their proliferation and inhibition of
apoptosis in diabetic mice, indicating that this compound serves a
role in protecting islet cells via S1PRs. Previous studies
investigated the role of S1P on islet β-cell proliferation, insulin
secretion and apoptosis, and the subsequent prevention of diabetes
development in obese mice (37) as
well as its effect on other cell types (10,38–42).
Furthermore, addition of exogenous S1P at nanomolar levels
significantly protected β-cell against cytokine-induced cell death
(43), as well as MIN6 cells against
palmitate-induced apoptosis (37).
A previous study indicated that S1P was able to
significantly stimulate glucose-independent insulin secretion
through the activation of phospholipase C in the clonal hamster
β-cell HIT-T15 line, as well as in isolated mouse islets (44). The present study demonstrated that
S1P had an effect on improving blood glucose control and IPGTT
outcomes, suggesting the protective effect of S1P on β-cells in
diabetes. Although the results of immunohistochemical staining with
Ki67 showed that the proliferation of β-cells in islets was
increased after S1P treatment compared with the DC group, the
number of proliferating β-cells was still small and the
proliferation rate of Ki67 in islets was <3%, which may still
not be enough to secrete enough insulin to reduce blood glucose
significantly. So even though the result of blood glucose and some
other results showed no statistical significance, the therapeutic
and protective effects of S1P on diabetes cannot be denied. In
agreement with the results of the present study, S1P has been shown
to be important for insulin synthesis and secretion in a rat
insulinoma cell line (45). Strong
evidence exists supporting the critical roles of S1P on the
progression of diabetes mellitus, including insulin sensitivity and
secretion, pancreatic β-cell apoptosis, and the development of
diabetic inflammatory state (46).
The results of the present study suggested that S1P
lead to an improvement in the morphology of islet β-cells, the
promotion of their proliferation and inhibition of apoptosis in
diabetic mice, indicating that this compound serves a role in
protecting islet cells via S1PRs. S1P has been reported to regulate
insulin resistance through receptor-mediated pathways in pancreatic
β-cells, but the specific S1PR subtypes involved remain unknown.
Among the five cognate receptors (S1PR1-5), a previous study
reported that islet and INS-1 cells expressed S1PR1, S1PR2, S1PR3
and S1PR4 subtypes (27). S1P
signalling is mediated via activation of specific S1PRs (S1PR1-5),
of which mouse islet β-cell express mainly S1PR1-3 (22). Mechanistically, S1P decreased and
inhibited adipocyte proliferation and differentiation via the
downregulation of S1PR1, and decreased the activity of the
peroxisome proliferator activator receptor γ in the adipose tissues
of a serine palmitoyltransferase 2 knockout mouse model (47). In addition, S1PR1, S1P seemed to
counteract insulin signalling and confer insulin resistance via
S1PR2 in pancreatic β-cells (4,48).
Specific S1PR2 antagonist JTE-013-treatment in S1PR2-deficient mice
attenuated β-cell apoptosis, ameliorated blood glucose elevation,
rescued β-cell damage and decreased the incidence of diabetes in
STZ-induced wild-type models (49,50). In
agreement with these findings, the present study demonstrated that
extracellular S1P induced proliferation and decreased apoptosis in
pancreatic β-cells. The present study also indicated that the
expression of S1PR1 and S1PR2 proteins in diabetic mice group were
increased compared with the normal control group, while S1PR3
showed no difference. S1PR1 and S1PR2 may play a certain role in
the pathogenesis and pathophysiological changes in T2DM, whereas
the S1PR3 subtype may not be involved in diabetes. All these
results confirmed that the S1P signaling pathway was involved in
the development of T2DM.
However, the study has certain limitations. The
sample size of the animals included was relatively small and the
course, and observation time of S1P administration was short. The
protein expression of the S1PR subtypes was detected in pancreatic
islets using immunohistochemical staining, but not further
confirmed by isolation of the mouse islet β-cells and detection
using reverse transcription PCR and western blot analysis. Each
step in the process of immunohistochemical staining may affect the
final result of staining. The influencing factors include the fixed
time of tissue samples, the thickness of the slide, the time and
method of antigen retrieval process, the time of antibody
incubation, the time of DAB color rendering and the time of
hematoxylin redyeing. Therefore, the present study strictly
followed the same time and conditions in the process of
immunohistochemical staining, so as to minimize the differences in
staining conditions. Although immunohistochemistry is only a
semi-quantitative analysis, the results can explain certain
problems under strict experimental operation. Finally, further
animal and cell experiments to investigate the mechanism of S1P
signalling in diabetes are required.
In conclusion, the present study demonstrated that
S1P serves a positive role in protecting islet β-cells against
apoptosis, suggesting the physiological significance of S1P in
preserving β-cell mass in diet-induced diabetic mice, indicating a
potential novel therapeutic strategy for delaying the occurrence
and development of diabetes. However, to date, the role of S1P on
β-cell function and diabetes mellitus is still not fully
understood, this study has laid a foundation for the further
research on the relationship between S1P and diabetes. Further
studies are required to address the function of S1P and S1PR
subtypes in islet β-cells.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank Dr Gang Tian, The First Affiliated
Hospital of Xi'an Jiaotong University, for assisting with helpful
suggestions and a review of the manuscript.
Funding
The present study was supported by The First
Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China
(grant no. 2013YK20), the Clinical Research Award of the First
Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China
(grant no. XJTU1AF-CRF-2016-016) and the Chinese Medical
Association (grant no. 13040470432).
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JS designed and conceived the study. YH designed and
performed the experiments. BS and XZ analysed the data. JS and YH
wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the
Institutional Animal Care and Use Committee of the Xi'an Jiaotong
University of Health Sciences, Xi'an, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang L, Gao P, Zhang M, Huang Z, Zhang D,
Deng Q, Li Y, Zhao Z, Qin X, Jin D, et al: Prevalence and ethnic
pattern of diabetes and prediabetes in China in 2013. JAMA.
317:2515–2523. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haass NK, Nassif N and McGowan EM:
Switching the sphingolipid rheostat in the treatment of diabetes
and cancer comorbidity from a problem to an advantage. Biomed Res
Int. 2015:1651052015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Billings LK and Florez JC: The genetics of
type 2 diabetes: What have we learned from GWAS? Ann N Y Acad Sci.
1212:59–77. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fayyaz S, Japtok L and Kleuser B:
Divergent role of sphingosine 1-phosphate on insulin resistance.
Cell Physiol Biochem. 34:134–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Silva VR, Micheletti TO, Pimentel GD,
Katashima CK, Lenhare L, Morari J, Mendes MC, Razolli DS, Rocha GZ,
de Souza CT, et al: Hypothalamic S1P/S1PR1 axis controls energy
homeostasis. Nat Commun. 5:48592014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spiegel S and Milstien S: Functions of a
new family of sphingosine-1-phosphate receptors. Biochim Biophys
Acta. 1484:107–116. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pulkoski-Gross MJ, Donaldson JC and Obeid
LM: Sphingosine-1-phosphate metabolism: A structural perspective.
Crit Rev Biochem Mol Biol. 50:298–313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maceyka M, Harikumar KB, Milstien S and
Spiegel S: Sphingosine-1-phosphate signaling and its role in
disease. Trends Cell Biol. 22:50–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pyne S and Pyne NJ: New perspectives on
the role of sphingosine 1-phosphate in cancer. Handb Exp Pharmacol.
55–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cuvillier O, Pirianov G, Kleuser B, Vanek
PG, Coso OA, Gutkind S and Spiegel S: Suppression of
ceramide-mediated programmed cell death by sphingosine-1-phosphate.
Nature. 381:800–803. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gómez-Muñoz A, Waggoner DW, O'Brien L and
Brindley DN: Interaction of ceramides, sphingosine, and sphingosine
1-phosphate in regulating DNA synthesis and phospholipase D
activity. J Biol Chem. 270:26318–26325. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pyne S and Pyne NJ: The differential
regulation of cyclic AMP by sphingomyelin-derived lipids and the
modulation of sphingolipid-stimulated extracellular signal
regulated kinase-2 in airway smooth muscle. Biochem J. 315:917–923.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hatoum D, Haddadi N, Lin Y, Nassif NT and
McGowan EM: Mammalian sphingosine kinase (SphK) isoenzymes and
isoform expression: Challenges for SphK as an oncotarget.
Oncotarget. 8:36898–36929. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spiegel S and Milstien S:
Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett.
476:55–57. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Strub GM, Maceyka M, Hait NC, Milstien S
and Spiegel S: Extracellular and intracellular actions of
sphingosine-1-phosphate. Adv Exp Med Biol. 688:141–155. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Obinata H and Hla T: Sphingosine
1-phosphate in coagulation and inflammation. Semin Immunopathol.
34:73–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Blaho VA and Hla T: An update on the
biology of sphingosine 1-phosphate receptors. J Lipid Res.
55:1596–1608. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kihara Y, Maceyka M, Spiegel S and Chun J:
Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br
J Pharmacol. 171:3575–3594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang F, Van Brocklyn JR, Hobson JP,
Movafagh S, Zukowska-Grojec Z, Milstien S and Spiegel S:
Sphingosine 1-phosphate stimulates cell migration through a
G(i)-coupled cell surface receptor. Potential involvement in
angiogenesis. J Biol Chem. 274:35343–35350. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ishii I, Fukushima N, Ye X and Chun J:
Lysophospholipid receptors: Signaling and biology. Annu Rev
Biochem. 73:321–354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takuwa Y, Okamoto Y, Yoshioka K and Takuwa
N: Sphingosine- 1-phosphate signaling in physiology and diseases.
Biofactors. 38:329–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Völzke A, Koch A, Meyer ZHD, Huwiler A and
Pfeilschifter J: Sphingosine 1-phosphate (S1P) induces COX-2
expression and PGE2 formation via S1P receptor 2 in renal mesangial
cells. Biochim Biophys Acta. 1841:11–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takuwa Y, Takuwa N and Sugimoto N: The Edg
family G protein-coupled receptors for lysophospholipids: Their
signaling properties and biological activities. J Biochem.
131:767–771. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brinkmann V: Sphingosine 1-phosphate
receptors in health and disease: Mechanistic insights from gene
deletion studies and reverse pharmacology. Pharmacol Ther.
115:84–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mandala S, Hajdu R, Bergstrom J,
Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D,
Keohane C, et al: Alteration of lymphocyte trafficking by
sphingosine-1-phosphate receptor agonists. Science. 296:346–349.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taha TA, Argraves KM and Obeid LM:
Sphingosine-1-phosphate receptors: Receptor specificity versus
functional redundancy. Biochim Biophys Acta. 1682:48–55. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Laychock SG, Tian Y and Sessanna SM:
Endothelial differentiation gene receptors in pancreatic islets and
INS-1 cells. Diabetes. 52:1986–1993. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Serafimidis I, Rodriguez-Aznar E, Lesche
M, Yoshioka K, Takuwa Y, Dahl A, Pan D and Gavalas A: Pancreas
lineage allocation and specification are regulated by
sphingosine-1-phosphate signalling. PLoS Biol. 15:e20009492017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matthews DR, Hosker JP, Rudenski AS,
Naylor BA, Treacher DF and Turner RC: Homeostasis model assessment:
Insulin resistance and beta-cell function from fasting plasma
glucose and insulin concentrations in man. Diabetologia.
28:412–419. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Butler AE, Janson J, Bonner-Weir S, Ritzel
R, Rizza RA and Butler PC: Beta-cell deficit and increased
beta-cell apoptosis in humans with type 2 diabetes. Diabetes.
52:102–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ashcroft FM and Rorsman P: Diabetes
mellitus and the β cell: The last ten years. Cell. 148:1160–1171.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bonner-Weir S, Li WC, Ouziel-Yahalom L,
Guo L, Weir GC and Sharma A: Beta-cell growth and regeneration:
Replication is only part of the story. Diabetes. 59:2340–2348.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mandrup-Poulsen T: beta-cell apoptosis:
Stimuli and signaling. Diabetes. 50 (Suppl 1):S58–S63. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim RH, Takabe K, Milstien S and Spiegel
S: Export and functions of sphingosine-1-phosphate. Biochim Biophys
Acta. 1791:692–696. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei Q, Deng H, Cui H, Fang J, Zuo Z, Deng
J, Li Y, Wang X and Zhao L: A mini review of fluoride-induced
apoptotic pathways. Environ Sci Pollut Res Int. 25:33926–33935.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo H, Chen L, Cui H, Peng X, Fang J, Zuo
Z, Deng J, Wang X and Wu B: Research advances on pathways of
nickel-induced apoptosis. Int J Mol Sci. 17:E102015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qi Y, Chen J, Lay A, Don A, Vadas M and
Xia P: Loss of sphingosine kinase 1 predisposes to the onset of
diabetes via promoting pancreatic beta-cell death in diet-induced
obese mice. FASEB J. 27:4294–4304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xia P, Wang L, Gamble JR and Vadas MA:
Activation of sphingosine kinase by tumor necrosis factor-alpha
inhibits apoptosis in human endothelial cells. J Biol Chem.
274:34499–34505. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Olivera A, Kohama T, Edsall L, Nava V,
Cuvillier O, Poulton S and Spiegel S: Sphingosine kinase expression
increases intracellular sphingosine-1-phosphate and promotes cell
growth and survival. J Cell Biol. 147:545–558. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sarkar S, Maceyka M, Hait NC, Paugh SW,
Sankala H, Milstien S and Spiegel S: Sphingosine kinase 1 is
required for migration, proliferation and survival of MCF-7 human
breast cancer cells. FEBS Lett. 579:5313–5317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sukocheva O, Wadham C, Holmes A, Albanese
N, Verrier E, Feng F, Bernal A, Derian CK, Ullrich A, Vadas MA and
Xia P: Estrogen transactivates EGFR via the sphingosine 1-phosphate
receptor Edg-3: The role of sphingosine kinase-1. J Cell Biol.
173:301–310. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Morales A, Lee H, Goñi FM, Kolesnick R and
Fernandez-Checa JC: Sphingolipids and cell death. Apoptosis.
12:923–939. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Laychock SG, Sessanna SM, Lin MH and
Mastrandrea LD: Sphingosine 1-phosphate affects cytokine-induced
apoptosis in rat pancreatic islet beta-cells. Endocrinology.
147:4705–4712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shimizu H, Okajima F, Kimura T, Ohtani K,
Tsuchiya T, Takahashi H, Kuwabara A, Tomura H, Sato K and Mori M:
Sphingosine 1-phosphate stimulates insulin secretion in HIT-T 15
cells and mouse islets. Endocr J. 47:261–269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hasan NM, Longacre MJ, Stoker SW, Kendrick
MA, Druckenbrod NR, Laychock SG, Mastrandrea LD and MacDonald MJ:
Sphingosine kinase 1 knockdown reduces insulin synthesis and
secretion in a rat insulinoma cell line. Arch Biochem Biophys.
518:23–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ng ML, Wadham C and Sukocheva OA: The role
of sphingolipid signalling in diabetesassociated pathologies
(Review). Int J Mol Med. 39:243–252. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee SY, Lee HY, Song JH, Kim GT, Jeon S,
Song YJ, Lee JS, Hur JH, Oh HH, Park SY, et al: Adipocyte-specific
deficiency of de novo sphingolipid biosynthesis leads to
lipodystrophy and insulin resistance. Diabetes. 66:2596–2609. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kitada Y, Kajita K, Taguchi K, Mori I,
Yamauchi M, Ikeda T, Kawashima M, Asano M, Kajita T, Ishizuka T, et
al: Blockade of sphingosine 1-phosphate receptor 2 signaling
attenuates high-fat diet-induced adipocyte hypertrophy and systemic
glucose intolerance in mice. Endocrinology. 157:1839–1851. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Japtok L, Schmitz EI, Fayyaz S, Krämer S,
Hsu LJ and Kleuser B: Sphingosine 1-phosphate counteracts insulin
signaling in pancreatic beta-cells via the sphingosine 1-phosphate
receptor subtype 2. FASEB J. 29:3357–3369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Imasawa T, Koike K, Ishii I, Chun J and
Yatomi Y: Blockade of sphingosine 1-phosphate receptor 2 signaling
attenuates streptozotocin-induced apoptosis of pancreatic
beta-cells. Biochem Biophys Res Commun. 392:207–211. 2010.
View Article : Google Scholar : PubMed/NCBI
|