Introduction
Hydrogen sulfide (H2S) is a highly
dispersive gasotransmitter that affects cells and organs function
through different mechanisms (1).
H2S is increasingly being considered as an important
signaling molecule in the cardiovascular systems (2,3).
Endogenous production of H2S is primarily catalyzed by
cystathionine β-synthase, cystathionine-γ-lyase (CSE) and
3-mercaptosulphurtransferase (4).
Among them, CSE is the primary H2S-producing enzyme in
cardiovascular tissues. The disordered metabolism and functions of
the CSE/H2S pathway have been associated with several
cardiovascular diseases, including A/R injury, hypertension,
atherosclerosis and oxidative stress (5–9).
Rho-associated protein kinase (ROCK), the
best-characterized effector of the small G protein Rho, has been
proposed to be potential targets in the therapy of cardiovascular
diseases (10,11). Various studies have indicated that
ROCK inhibitors prevent the progress of myocardial infarction by
hemodilution, vascular dilation and inhibition of neutrophil
accumulation (11–13). The useful effects of ROCK inhibition
against A/R damage using the ROCK inhibitors fasudil and Y-27632
have been established (14,15). This suggests that ROCK serves a vital
role in myocardial infarction.
Total flavones of Rhododendra flower (TFR),
an effective compound extracted from the Rhododendra flower,
is comprised of flavones including quercetin, hyperin, rutin and
other flavonoids (16,17). Our previous studies have indicated
that TFR has significant protective effects against myocardial
ischemic injuries in rat and mice models (18,19), and
that the protective mechanism may be engaged with the inhibition of
ROCK1 and ROCK2 and activation of the potassium channel (20). Certain previous studies have
suggested that flavonoid compounds may prevent the RhoA/ROCK signal
pathway by decreasing the contractility of vascular smooth muscle
cells (21–23).
In light of these data, the present study aimed to
evaluate the cardiovascular protective effects of TFR as a ROCK
inhibitor in a mice model of myocardial infarction induced by
isoproterenol. The hearts from wild-type (WT) and CSE knockout (KO)
mice were examined. During the process of myocardial
ischemia-reperfusion injury, the effect of endogenous
H2S on ROCK signaling pathways was explored, and the
effect of TFR on the ROCK and CSE/H2S signaling pathways
was investigated.
Materials and methods
Drugs and reagents
TFR (content of flavones >85%) was provided by
Hefei Heyuan Medical Company Technology Co., Ltd. Isoprenaline
(ISO) was produced by Shanghai Hefeng Pharmaceutical Co. Ltd.
Lactate dehydrogenase (LDH, cat. no. A020-1-2) and creatinine
kinase isoenzyme (CK-MB; cat. no. H197) assay kits were purchased
from Nanjing Jiancheng Bioengineering Institute. Rabbit polyclonal
primary antibodies against ROCK1 and ROCK2 were provided by EnoGene
Biotech Co., Ltd. Membrane protein MLC1 (MLC1) was purchased from
Santa Cruz Biotechnology, Inc.
Animal model
The present study was approved by the Ethics
Committee for Animal Experiments of Anhui Medical University (no.
20160315). The CSE KO and wild-type mice (C57 strain) were produced
by Shanghai Model Organisms Center. Wild-type and CSE KO mice
(n=60; age, 10–16 weeks; weight, 18–24 g; half male and female)
were used for the experiment (Fig.
1).
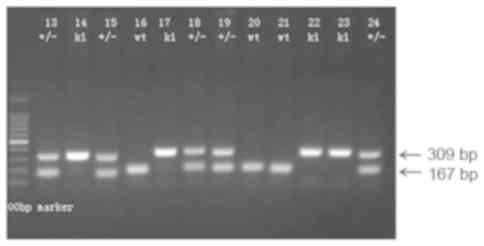 | Figure 1.Polymerase chain reaction
identification of CSE gene expression in mice. Lanes 14, 17, 22 and
23, CSE knockout mice (309 bp). Lanes 16, 20 and 21, wild type mice
(167 bp). Lanes 13, 15, 18, 19 and 24, heterozygous mice (two
bands). CSE, cystathionine γ-lyase. |
The present study was performed in strict accordance
with the recommendations in the Guide for the Care and Use of
Laboratory Animals of the National Institutes of Health (NIH
Publication, 8th edition, 2011) (24). WT and CSE KO mice (6 per group) were
divided to 5 groups. ISO (0.002 mg/kg) was injected in mice. Group
I served as control, with equal dose of normal saline. Myocardial
infarction was induced in groups II–V by subcutaneous
administration of ISO on the first, second and third days
respectively. Group III–V received TFR orally (30, 60 or 120 mg/kg,
respectively) once a day 5 min prior to the ISO for 3 days; group
II received the equal dose of normal saline. The mice were
anaesthetized by intraperitoneal injection of chloral hydrate (350
mg/kg). The mice were confirmed to be fully anaesthetized when the
breathing rate decreased and breathing depth increased, and the
righting reflex, and eyelid and tail-pinch reflexes were lost. At
the end of protocol, mice were sacrificed by anesthesia using 2%
isoflurane, which was then increased to 5%. Then, cardiac puncture
was performed by a qualified technician. Following cardiac
puncture, the mice were observed for respiratory and cardiac
arrest, pupil dilation and disappearance of the pupillary light
reflex. Following these observations, the mice were confirmed to be
dead within 6–10 min.
PCR
Tail tissue was used. DNA polymerase was used (2×
Premix Tag; Takara Bio; cat. no. RR902Q). RNA extraction buffer and
supplier used was TRIzol Reagent from Thermo Fisher Scientific,
Inc. (cat. no. 15596). The RT kit used was iScript gDNA Clear cDNA
Synthesis kit from Bio-rad Laboratories, Inc. (cat. no.
1725035).
DNA polymerase used and supplier sequences of the
forward and reverse primers: P1: CCTGGATATAAGCGCCAAAG, P2:
AGGAACCAGGGCGTATCTCT, P3: CGAGAATTCCATTGCTCAGG. Reverse
transcription protocol was as follows: 94°C for 3 min, 94°C for 30
sec, 57°C for 30 sec, 72°C for 40 sec, 72°C for 10 min then 12°C.
The length of the wild product was 167 bp and the length of the
mutant product was 309 bp.
Determination of ST-segment
elevation
Electrocardiograms (ECGs) recorded ST-segment
elevation at 5, 10, 15, 20 and 60 min following the final injection
of ISO or normal saline. ECGs were recorded under 30 mg/kg
pentobarbital sodium anesthetization administered by
intraperitoneal injection.using needle electrodes and a Biological
Function Experiment System (Chengdu Thaimeng Technology Co. Ltd).
The recorded original data were estimated by the commercial
software included in the acquisition system (AqDAnalysis 7; Lynx
Tecnologia Ltda.).
Measurement of LDH and CK-MB
levels
The supernatant was centrifuged at 3,000 × g for 10
min at 4°C, the LDH and CK-MB levels were detected at 550 and 440
nm, respectively, by spectrophotometry according to the
manufacturer's protocols of the assay kits. The experiment was
repeated 3 times.
Histology
Left ventricular tissues were surgically removed,
fixed in 10% buffered formalin at room temperature for 24 h,
embedded in paraffin and sliced into 5-µm thick sections. The
slides were stained with hematoxylin and eosin (H&E) for 5 min
at room temperature and examined using a confocal microscope
(magnification, ×400; Olympus BX51; Olympus Corporation). The Rona
classification standard (25) was
used to evaluate the degree of myocardial tissue damage.
Western blot analysis
The left ventricular tissues from the mice were
removed and placed in ice-cold RIPA lysis buffer (Beyotime
Institute of Biotechnology). Protein concentrations were determined
using a BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Equal amounts of protein (50 µg) were separated on 10%
polyacrylamide-Tris gels (Beyotime Institute of Biotechnology),
transferred onto polyvinylidene difluoride membranes and blocked
with 5% skim milk in TBST for 1 h at room temperature. Then, the
membranes were incubated at 4°C overnight with rabbit polyclonal
antibodies against ROCK1 (1:1,000; cat. no. E1A7016), ROCK2
(1:1,000; cat. no. E1A6028) or MLC1 (1:1,000; cat. no. SC-86740) or
monoclonal antibody against ß-actin (Bioworld Technology, Inc.).
Following incubation with an anti-rabbit second antibody (OriGene
Technologies, Inc.; 1:10,000 dilution in 5% skim milk) for 1 h at
room temperature, the immunocomplexes were visualized using an
enhanced chemiluminescence detection kit (Thermo Fisher Scientific,
Inc.). The intensity of the immunoreactive bands were quantified by
using the ImageJ analysis software (v.1.8.0; National Institutes of
Health).
Measurement of RhoA activity
To detect the activity of RhoA, left ventricular
tissues from the mice were lysed with radioimmunoprecipitation
assay lysis buffer and incubated with 50 µg of the Rhotekin-RBD
beads, containing a Rho-GTPase binding domain, at 4°C for 1 h. The
samples were then centrifuged at 5,000 × g at 4°C for 1 min and the
supernatant was removed. The beads were removed following washing
with wash buffer. The remaining bead pellets were boiled with 200
µl 2X Laemmli sample buffer (Bio-Rad Laboratories, Inc.) at 85°C
for 5 min. Then, RhoA activity levels were determined using
commercially available absorbance-based G-LISA RhoA activation
assay kits (cat. no. BK 036-S; Cytoskeleton, Inc.). The left
ventricular tissues were homogenized in lysis buffer (Beyotime
Institute of Biotechnology, Haimen, China) and the protein
concentrated according to the manufacturer's protocol. following
indirect immunodetection, RhoA activities were detected by
measuring absorbance at 490 nm using a microplate
spectrophotometer.
Statistical analysis
Data are expressed as means ± standard deviation,
and differences between groups were analyzed by SPSS v15.0 (SPSS,
Inc.). Statistical analyses were performed with one-way analysis of
variance followed by the Duncan post-hoc test to determine the
differences between groups. P<0.05 was considered to indicate a
statistically significant difference.
The statistical analysis of pathology ranking data
was performed using a Kruskal-Wallis H test. To determine the
differences between 5 groups, the Bonferroni method was used. The
difference between the WT and KO groups was analyzed using a
Student's t-test for paired design analysis. P<0.005 was
considered to indicate a statistically significant difference.
Results
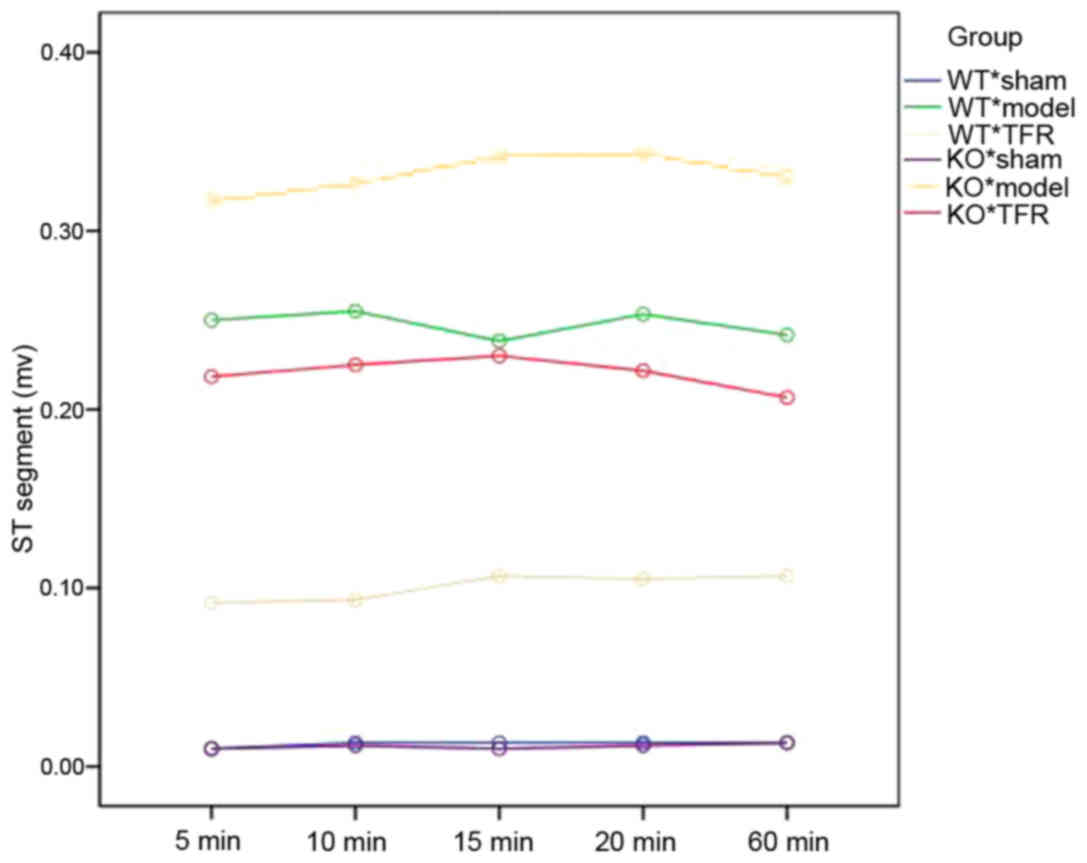
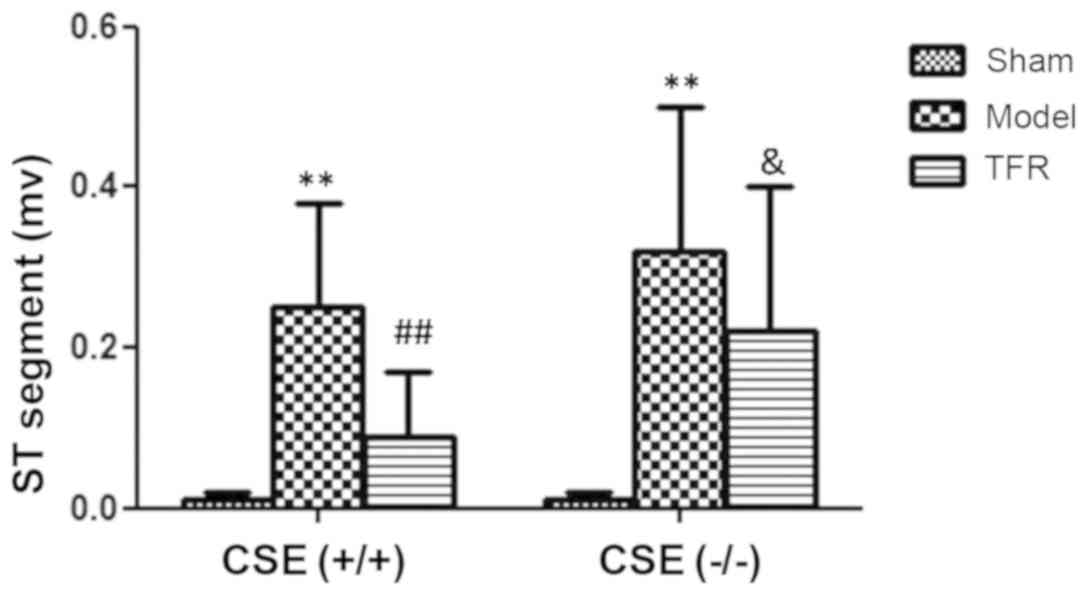
Effect of TFR on ST-segment
elevation
The ST segment of ECG in the model group of the wild
type (WT) mice increased significantly at 5, 10, 15, 20 and 60 min
after the final injection of ISO compared with the sham group
(P<0.01; Figs. 2 and 3). Administration of 60 mg/kg TFR markedly
decreased ST-segment elevation compared with the WT model group
(P<0.05; Table I) and 120 mg/kg
TFR markedly decreased ST-segment elevation compared with the WT
model group (P<0.01; Table
I).
 | Table I.Effect of TFR on the changes of ST
segment (mV) of ECG in the CSE WT and KO mice. |
Table I.
Effect of TFR on the changes of ST
segment (mV) of ECG in the CSE WT and KO mice.
| A, WT group |
|---|
|
|
|
|---|
|
| Time intervals,
min |
|---|
|
|
|
|---|
| Treatment
groups | 5 | 10 | 15 | 20 | 60 |
|---|
| Control (n=6) | 0.01±0.01 | 0.01±0.01 | 0.01±0.01 | 0.01±0.01 | 0.01±0.01 |
| Model (n=6) |
0.25±0.12a |
0.25±0.14a |
0.24±0.14a |
0.25±0.15a |
0.24±0.14a |
| TFR, mg/kg
(n=6) |
| 30 | 0.19±0.14 | 0.19±0.12 | 0.19±0.14 | 0.20±0.14 | 0.21±0.13 |
| 60 |
0.12±0.11b |
0.13±0.11b |
0.13±0.12b |
0.16±0.12b |
0.16±0.13b |
|
120 |
0.09±0.04c |
0.10±0.05c |
0.10±0.05c |
0.11±0.06c |
0.11±0.08c |
|
| B, KO
group |
|
|
| Time intervals,
min |
|
|
|
| Treatment
groups | 5 | 10 | 15 | 20 | 60 |
|
| Control (n=6) | 0.01±0.01 | 0.01±0.01 | 0.01±0.01 | 0.01±0.01 | 0.01±0.01 |
| Model (n=6) |
0.32±0.18a |
0.33±0.19a |
0.34±0.20a |
0.34±0.18a |
0.33±0.19a |
| TFR, mg/kg
(n=6) |
|
|
|
|
|
| 30 | 0.28±0.19 | 0.30±0.19 | 0.27±0.18 | 0.28±0.20 | 0.27±0.18 |
| 60 | 0.26±0.17 | 0.26±0.16 | 0.27±0.18 | 0.26±0.15 | 0.26±0.15 |
|
120 |
0.22±0.10d |
0.23±0.11d |
0.23±0.10d |
0.22±0.09d |
0.21±0.10d |
The ST segment of ECG in the KO mice model group
rose significantly at 5, 10, 15, 20 and 60 min following the final
injection of ISO compared with the sham group (P<0.01; Figs. 2 and 3). However, no significant differences in
the ST-segment elevation were observed following the administration
of 30 and 60 mg/kg TFR compared with the model group in the KO mice
(P>0.05), but the group of 120 mg/kg TFR was significant
decreased compared with WT TFR group (P<0.05; Figs. 2 and 3; Table
I).
In order to compare the elevations of the ST
segments between the WT and KO mice more clearly, ST segment
elevation was recorded at 5 min after the injection of ISO. The
decrease in ST segment elevation in the TFR group in comparison
with the model group was observed to be greater in the WT mice
compared with the KO mice (P<0.05; Fig. 3).
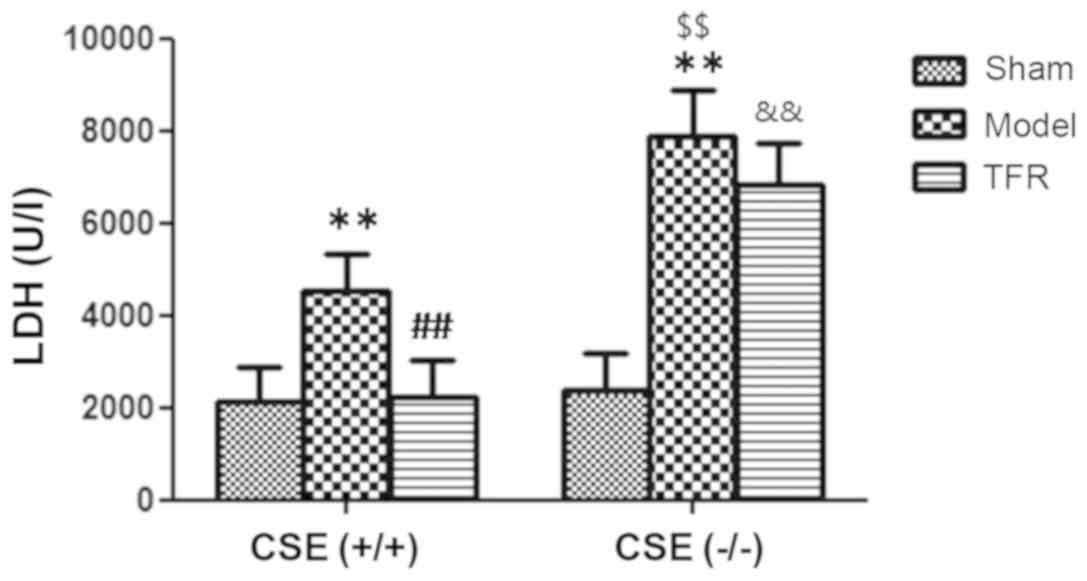
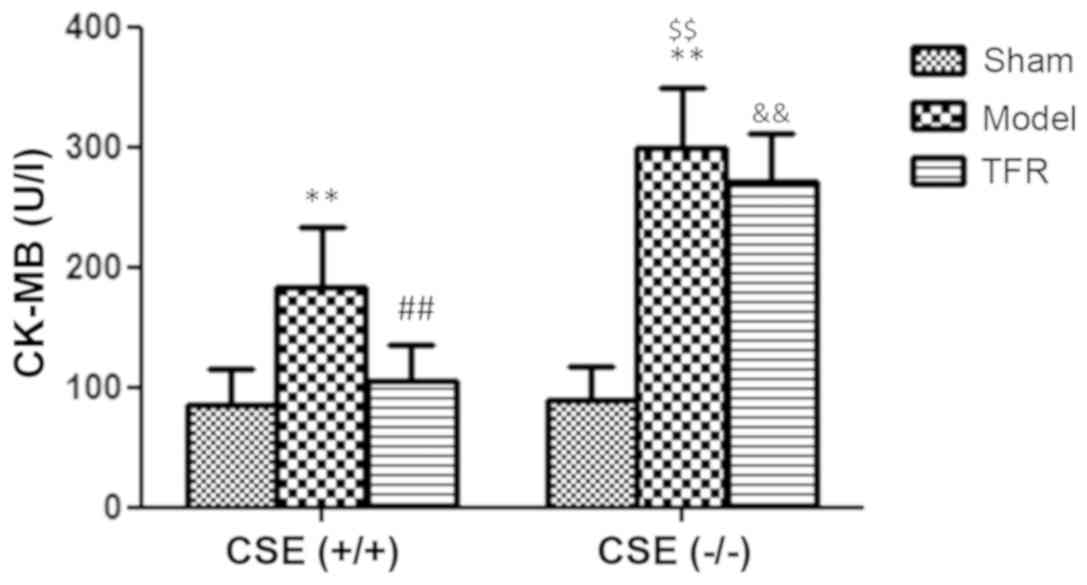
Effect of TFR on the LDH and CK-MB
level
Levels of LDH and CK-MB in the plasma supernatant
are major indicators of myocardial anoxia/reoxygenation (A/R)
injury. A few increases of LDH and CK-MB level were detected in A/R
group of the WT mice (P<0.01). Treatment with 60 mg/kg TFR
markedly inhibited the A/R-induced increases of LDH and CK-MB level
in the plasma supernatant of the WT mice (P<0.01; Figs. 4 and 5).
Significant increases in LDH and CK-MB levels were
detected in the A/R model group of the KO mice (P<0.01), and the
LDH and CK-MB levels were increased significantly in A/R model
group of the WT mice compared with the KO mice (P<0.01).
Treatment with 60 mg/kg TFR had no effect of the A/R-induced
increases in LDH and CK-MB level in the plasma supernatant of the
KO mice (P>0.05). However, the LDH and CK-MB levels of the TFR
group of the KO mice were significantly increased compared with
those in the TFR group of the WT mice (Figs. 4 and 5).
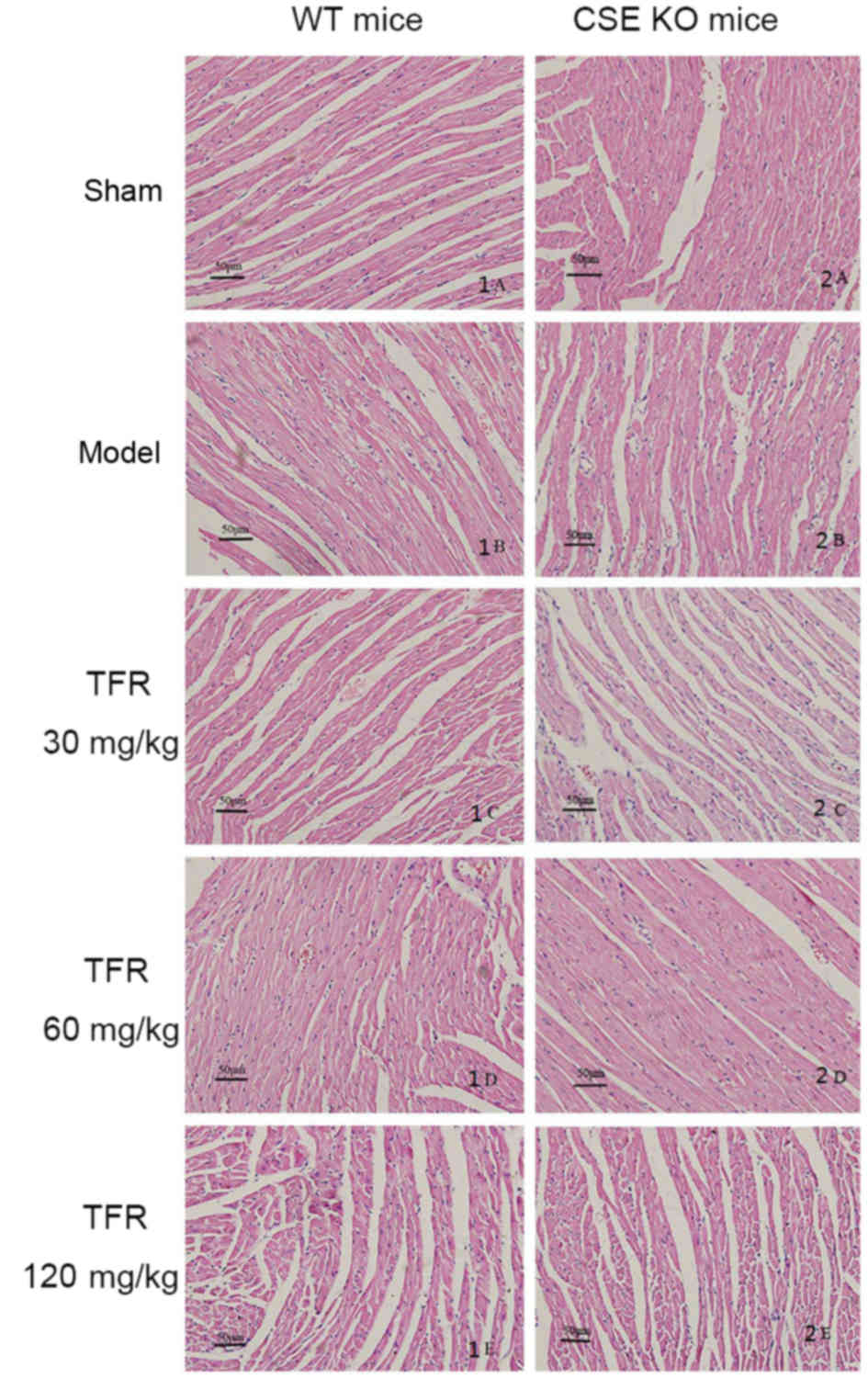
Pathological observations
Analysis of the myocardium in the sham group in the
WT mice population revealed a normal myofibrillar structure with
stripes, branched appearance, and connections with adjacent
myofibrils. In the mice treated with A/R, disorganized myocardium
structure and loss of attachment between cardiomyocytes was
observed. Tissues from the A/R mice exhibited obvious myocardial
cell hypertrophy, cytopathy, loss of transverse striations and
occasional cytoplasmic vacuolization. The TFR groups exhibited less
severe histological damage, normal myocardial arrangement, clear
transverse striations and fewer inflammatory cells.
The architecture of the myocardium was intact with
erratic myofiber array in the sham group of the KO mice. Tissue
from the A/R group of the KO mice revealed severely focal necrosis,
myocardial cytopathy, loss of striations, severe infiltration of
inflammatory cells and cytoplasmic vacuolization. Compared with
this group, tissues from the A/R group of the WT mice population
revealed less severe histological damage. In addition, the tissue
sections from the TFR group of the KO mice demonstrated myocardial
cell swelling, indistinct transverse striations and inflammatory
cell infiltration. There were no marked differences in the
pathological changes of 120 mg/kg TFR group of the KO mice compared
with the A/R group of the same KO mice population (Fig. 6).
The pathological grades of myocardium from each
group are presented in Table II.
The level of significance was corrected as P-value of comparisons
between different groups, when several comparisons were performed
between groups. Analysis of the data demonstrated that there were
significantly improvements in the pathological grades between the
TFR and sham groups in the WT mice population (P<0.001). In
addition, there were no significant changes between the TFR and
sham groups in the KO mice population. Using a Z-test, it was
demonstrated that there were significant improvements in the
pathological changes of the 60 or 120 mg/kg TFR groups of the WT
mice than the 60 or 120 mg/kg TFR groups of the KO mice
(P<0.005). The results of Kruskal-Wallis H test in the WT and KO
groups were χ2=24.310 (P<0.001) and
χ2=21.858 (P<0.001), respectively (Table II).
| Table II.Pathological grades of cardiomyocytes
in CSE WT and KO mice. |
Table II.
Pathological grades of cardiomyocytes
in CSE WT and KO mice.
| A, (WT group), CSE
(+/+) |
|---|
|
|---|
|
| Pathological
grades, n |
|
|---|
|
|
|
|
|---|
| Treatment
group | 0 | I | II | III | IV | P-value |
|---|
| Sham | 6 | 0 | 0 | 0 | 0 |
|
| Model | 0 | 0 | 0 | 3 | 3 |
<0.005a |
| TFR, mg/kg |
|
|
|
|
|
|
| 30 | 0 | 0 | 2 | 3 | 1 |
<0.005a |
| 60 | 0 | 3 | 2 | 1 | 0 |
<0.005a,<0.005b |
|
120 | 0 | 4 | 2 | 0 | 0 |
<0.005a,<0.005b |
|
| B, (KO group),
CSE (−/-) |
|
|
| Pathological
grades |
|
|
|
|
|
| Treatment
group | 0 | I | II | III | IV | P-value |
|
| Sham | 6 | 0 | 0 | 0 | 0 |
|
| Model | 0 | 0 | 0 | 2 | 4 |
<0.005a |
| TFR, mg/kg |
| 30 | 0 | 0 | 1 | 3 | 2 |
<0.005a |
| 60 | 0 | 1 | 2 | 3 | 0 |
<0.005a |
|
120 | 0 | 2 | 2 | 2 | 0 |
<0.005a |
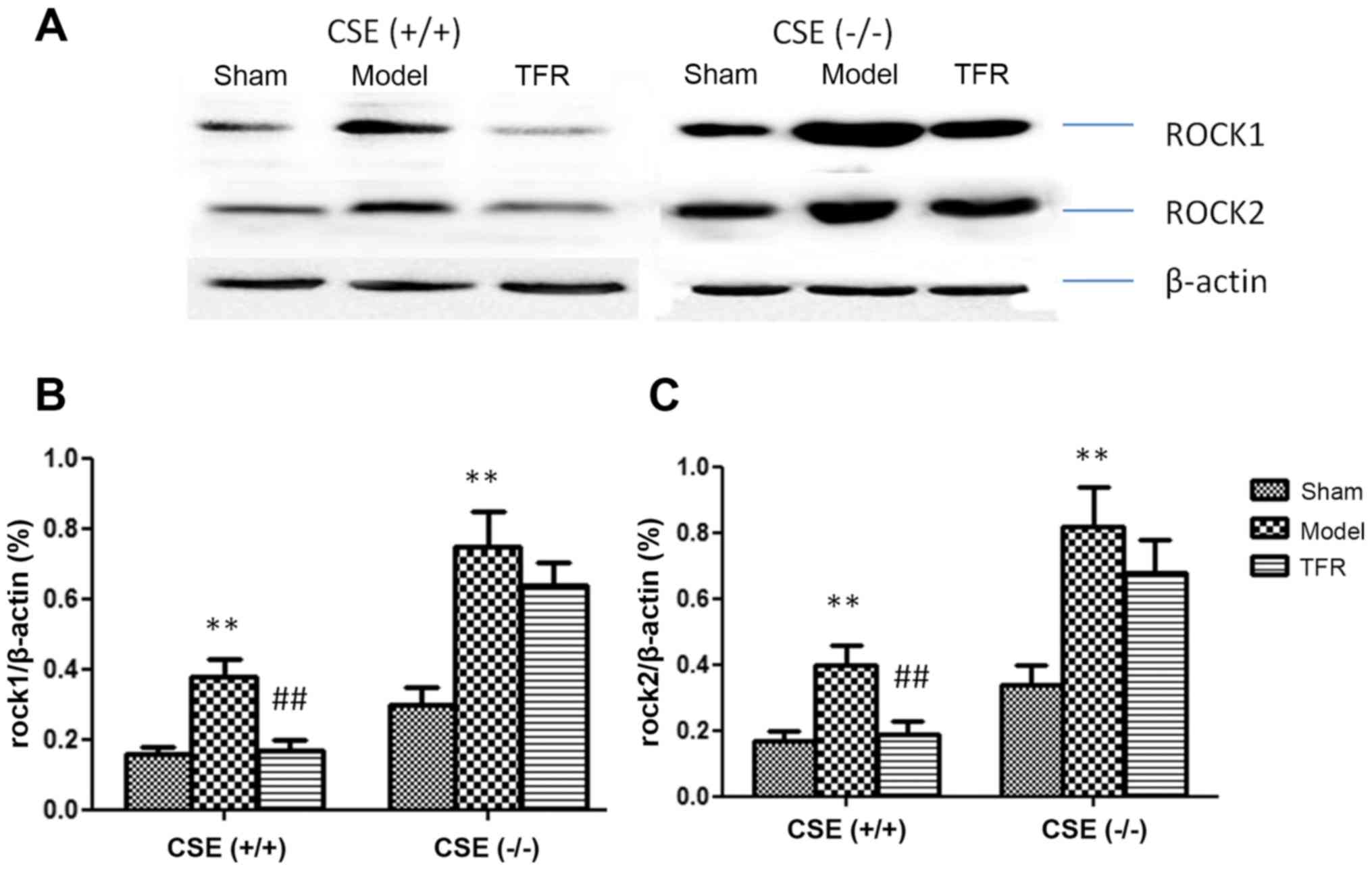
Effect of TFR on ROCKs protein
expression
The expression levels of ROCK1 and ROCK2 proteins
were examined in each group (Fig.
7A), and were quantified by using densitometric analysis
(Fig. 7B and C). Exposure to A/R
markedly increased both ROCK1 and ROCK2 protein levels in the WT
mice (P<0.01). The increases of ROCK1 and ROCK2 were markedly
inhibited by treatment with 60 mg/kg TFR (P<0.01). Exposure to
A/R significantly increased ROCK1 and ROCK2 protein levels in the
KO mice (P<0.01). The increases of ROCK1 and ROCK2 were not
markedly altered by treatment with 60 mg/kg TFR group of the KO
mice compared with the A/R group of the KO mice. The results
indicated that TFR treatment inhibited the expression of the ROCK
proteins associated with the CSE/H2S pathway.
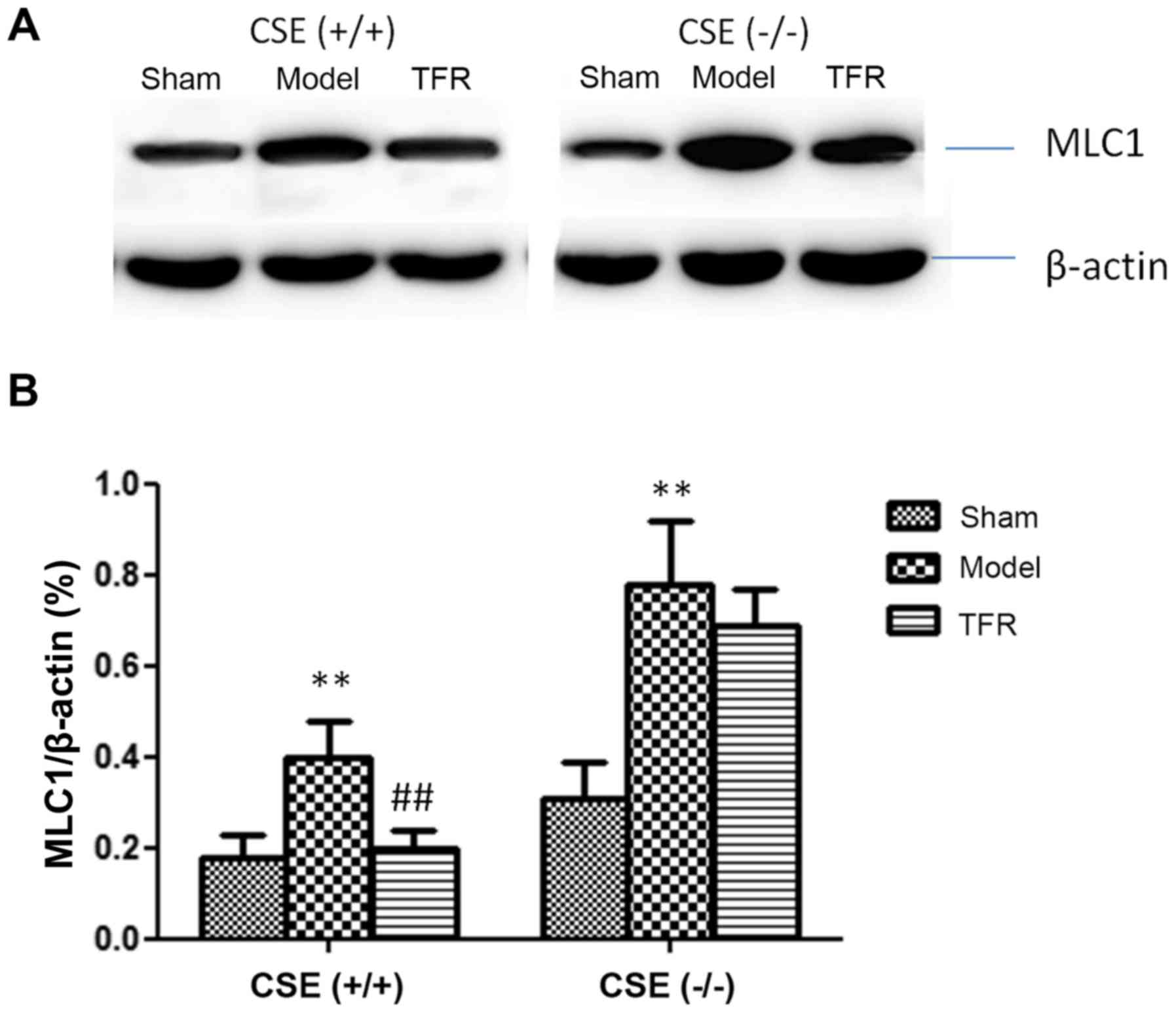
The expression levels of MLC1 proteins were
determined in each group (Fig. 8A),
and levels of MLC1 proteins were quantified using densitometry
(Fig. 8B). Exposure to A/R markedly
increased MLC1 protein levels compared with the sham group in the
WT mice (P<0.01). In addition, the increases of MLC1 were
markedly inhibited by 60 mg/kg TFR compared with the A/R group of
the WT mice (P<0.01). Exposure to A/R apparently increased MLC1
protein levels compared with the sham group of the KO mice
(P<0.01). In the KO mice population, the inhibitory effect of
TFR on the increased expression of MLC1 protein was significantly
decreased compared with the TFR group of the WT mice population.
These data demonstrated the TFR inhibited the expression of the
MLC1 protein associated with the CSE/H2S pathway.
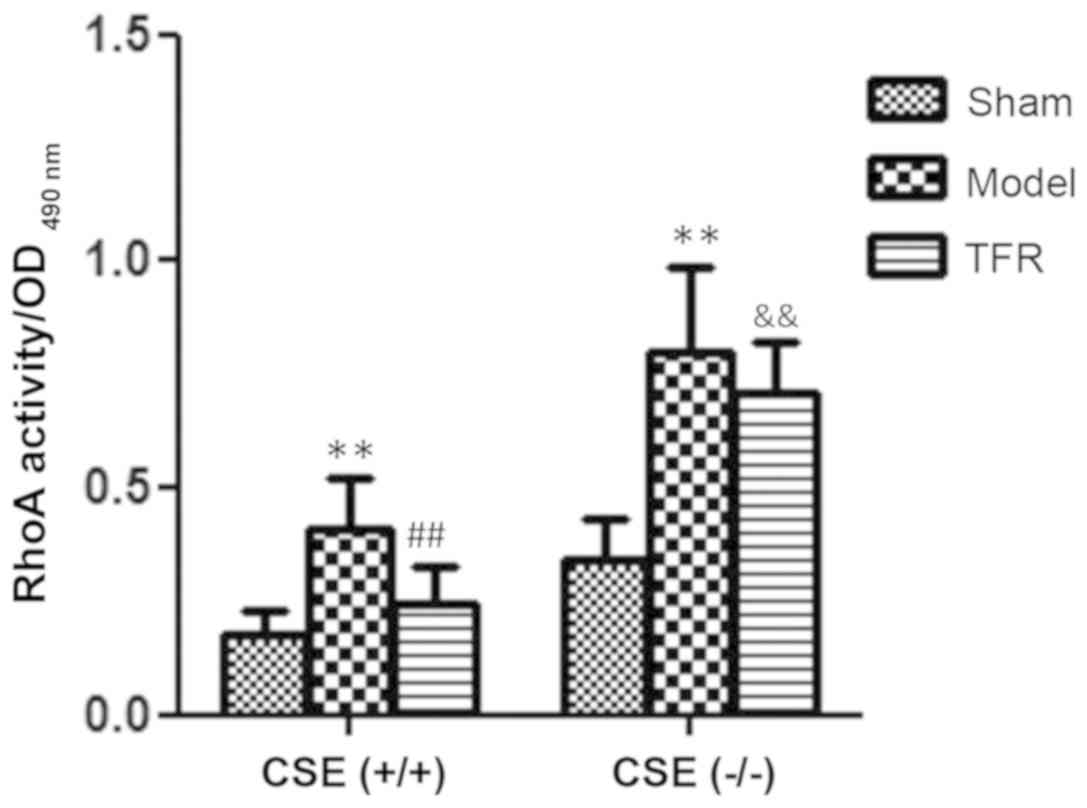
Effect of TFR on RhoA activity
RhoA activity in the left ventricular tissues was
detected using an absorbance-based G-LISA RhoA activation assay. As
demonstrated in Fig. 9, RhoA
activity in the model group (0.41±0.11) was significantly increased
compared with that in the sham group (0.18±0.05) (P<0.01). In
comparison with the model group, treatment with 60 mg/kg TFR
markedly inhibited the increase in RhoA activity, which was
decreased to 0.25±0.08 (P<0.01; Fig.
9).
Significant increases of RhoA activity were detected
in the model group of KO mice (0.8±0.09) compared with the sham
group (0.34±0.09) (P<0.01). In addition, the RhoA activity was
increased significantly in the model group of KO mice compared with
that in the WT mice (P<0.01). Compared with the model group,
treatment with 60 mg/kg TFR had no effect on A/R-induced increases
in RhoA activity of the KO mice (0.71±0.11) (P>0.05). However,
in the KO mice, the RhoA activity of the TFR group was
significantly increased compared with the TFR group of the WT mice
(P<0.01; Fig. 9).
Discussion
ISO is a synthetic β-adrenergic agonist that may
cause serious stress in the cardiac muscle and necrosis of
myocardium. Therefore, in the present study, ISO was used to induce
acute myocardial ischemia. The acute myocardial ischemia caused by
ISO was confirmed by loss of integrity of myocardial membranes on
histological changes, increased ST segment elevation, and increased
serum levels of LDH and CK-MB. In the present study, TFR treatment
decreased the ST-segment elevation induced by ISO; TFR also
decreased LDH and CK-MB levels in the serum. TFR treatment resulted
in significant improvements in the pathological changes caused by
hypoxia injury. The increases in expression levels of ROCK1, ROCK2
and MLC1 induced by the ISO were markedly inhibited by TFR
treatment. These results suggested that TFR had cardioprotective
effects in myocardial ischemia that may be attributed to the
inhibition of the RhoA/ROCK signal pathway.
In the CSE KO mice, the ST-segment elevation induced
by ISO was significantly increased compared with the WT mice. CSE
KO mice also demonstrated increased LDH and CK-MB levels in the
serum compared with the WT mice. CSE KO mice exhibited more severe
pathological changes as a result of hypoxia injury compared with
the WT mice, suggesting that H2S was involved in the
pathological process of myocardial ischemic injury. It was also
observed that the expression levels of ROCK1, ROCK2 and MLC1
induced by ISO in the KO mice were markedly increased compared with
the WT mice. These results suggested that CSE KO led to the
decrease in H2S expression and activation of the
RhoA/ROCK signal pathway, which may have aggravated the myocardial
ischemic injury. H2S has protective effects against A/R
injury in mice heart tissues by preventing the RhoA/ROCK signal
pathway (20,26). TFR inhibition of the RhoA/ROCK signal
pathway may be mediated by the CSE-H2S pathway.
The study by Zhang et al (20) indicated that the cardioprotection
afforded by TFR treatment involved the stimulation of nitric oxide
release and the inhibition of lipid peroxidation. Increasing
evidence has suggested that the RhoA/ROCK pathway serves an
important role in the A/R damage, vascular smooth muscle cell
proliferation, cardiac hypertrophy, heart failure and ventricular
remodeling (27,28). Development of hypertension and
myocardial infarction (MI); the two primary drivers of
cardiovascular disease are associated with cardiac ROCK activation
and phosphorylation of ROCK target proteins (29). ROCK inhibitors have a beneficial
effect in attenuating hypertension and MI associated with ROCK
activation (30). In addition,
inhibition of the ROCK pathway may have a protective effect on
cardiovascular function; the inhibitory agents Y27632 or fasudil
were demonstrated to limit infarct size, alleviate the A/R damage,
decrease the release of the MDA and LDH and promote the recovery of
myocardial function following ischemia (31,32).
During agonist-induced vascular smooth muscle cell
(VSMC) contraction, MLC phosphorylation is a crucial step for force
development. ROCK, when activated by the small GTPase RhoA,
inhibits MLC phosphatase (MLCP) activity by phosphorylating its
myosin-binding subunit, thereby serving a key role in
agonist-induced Ca2+ sensitization and VSMC hypercontraction
(33).
The major regulatory mechanism of smooth muscle
contraction is the phosphorylation/dephosphorylation of MLC
(34). MLC is phosphorylated by the
Ca2+-calmodulin-activated MLC kinase (MLCK) and
dephosphorylated by the Ca2+-independent MLCP.
Therefore, a rise in cytosolic Ca2+ concentration
produces smooth muscle contraction via the activation of MLCK and
consequent phosphorylation of MLC (10). Hyperin is the primary active
ingredient of TFR; it inhibits the contraction of the rabbit
cardiac papillary muscle (35). In
the present study, the increases of ROCK1, ROCK2 and MLC1 induced
by ISO were markedly inhibited by TFR treatment. These results
suggested that TFR had cardioprotective effects in myocardial
ischemia that may be attributed to the inhibition of the RhoA/ROCK
signal pathway.
Endogenous H2S has been suggested as a
novel signal transmitter and neuromodulator (36). In recent years, growing evidence has
demonstrated that H2S is a critical mediator of heart
functions and serves a protective function in the pathogenesis and
progress of heart diseases. Geng et al (37) identified that the CSE/H2S
pathway exists in the heart and has physiological effects such as
negative inotropy and reduced central venous pressure. NaHS
significantly decreased the infarct size of the left ventricle and
mortality after acute MI in rats (38). In an additional study, sulfur dioxide
(SO2) preconditioning significantly decreased A/R
induced myocardial injury in vivo, which is associated with
increased myocardial antioxidative capacity and upregulated H2S/CSE
pathway (39). It also has been
revealed to protect against hyperglycemia-induced ROS-mediated
apoptosis by upregulating the PI3K/AKT/nuclear factor erythroid
2-related factor 2 (Nrf2) pathway, which subsequently activates
Nrf2-regulated antioxidant enzymes in cardiomyocytes exposed to
high glucose (40). However, the
association between H2S and the RhoA/ROCK signaling
pathway remains unknown.
In the present study, it was identified that
TFR-mediated inhibition of the RhoA/ROCK signal pathway may have
been mediated by the CSE-H2S axis. These results
suggested that TFR exhibited cardioprotective effects in myocardial
ischemia that may be attributed to an inhibition of the RhoA/ROCK
signal pathway. The expression levels of ROCK1, ROCK2 and MLC1
induced by ISO in the KO mice were markedly increased compared with
the WT mice. These results suggested that CSE KO led to decreased
H2S expression and activation of the RhoA/ROCK signal
pathway and that H2S had protective effects against A/R
injury in mice hearts by inhibiting the RhoA/ROCK signal
pathway.
In the present study, accompanying the
pathophysiological process of ISO-induced myocardial injury was the
impaired endogenous CSE/H2S pathway. Administering
exogenous H2S resulted in effective protection of the
myocytes and contractile activity by directly scavenging oxygen
free radicals and decreasing the accumulation of lipid
peroxidations. These results suggest that H2S not only
alleviated the pathological process of ischemic heart disease but
may also serve as a cardiovascular protective regulator, and as a
novel target in the prevention or treatment of cardiovascular
diseases. TFR exhibited protective effects against A/R injury in
mice hearts by inhibiting the RhoA/ROCK signal pathway and may have
been mediated by the CSE-H2S.
Acknowledgements
The authors thank Dr. Yin-Guang Fan (Department of
Epidemiology and Biostatistics, School of Public Health, Anhui
Medical University) for the guidance on statistical methods.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81374002 and
81173596), the Natural Science Research in Universities of Anhui
Province (grant no KJ2016A326) and Doctor Research Foundation of
Anhui Medical University (grant no XJ201502).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZC designed the experiment. YJ, YG and YL performed
the experiment. YJ analyzed the data. YJ prepared the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee for Animal Experiments of Anhui Medical University (no.
20160315).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
H2S
|
hydrogen sulfide
|
|
ROCK
|
Rho-associated protein kinase
|
|
A/R
|
anoxia/reoxygenation
|
|
TFR
|
Total flavones of Rhododendra
flower
|
References
|
1
|
Zhao K, Li H, Li S and Yang G: Regulation
of cystathionine gamma-lyase/H2S system and its
pathological implication. Front Biosci (Landmark Ed). 19:1355–1369.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang L, Geng B, Yu F, Zhao J, Jiang H, Du
J and Tang C: Hydrogen sulfide inhibits myocardial injury induced
by homocysteine in rats. Amino Acids. 34:573–585. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sivarajah A, Collino M, Yasin M, Benetti
E, Gallicchio M, Mazzon E, Cuzzocrea S, Fantozzi R and Thiemermann
C: Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide
in a mice model of regional myocardial I/R. Shock. 31:267–274.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang G, Li H, Tang G, Wu L, Zhao K, Cao Q,
Xu C and Wang R: Increased neointimal formation in cystathionine
gamma-lyase deficient mice: Role of hydrogen sulfide in
α5β1-integrin and matrix metalloproteinase-2 expression in smooth
muscle cells. J Mol Cell Cardiol. 52:677–688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mani S, Li H, Untereiner A, Wu L, Yang G,
Austin RC, Dickhout JG, Lhoták Š, Meng QH and Wang R: Decreased
endogenous production of hydrogen sulfide accelerates
atherosclerosis. Circulation. 127:2523–2534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang R: Two's company, three's a crowd:
can H2S be the third endogenous gaseous transmitter? FASEB J.
16:1792–1798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang R: The gasotransmitter role of
hydrogen sulfide. Antioxid Redox Signal. 5:493–501. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao
K, Meng Q, Mustafa AK, Mu W, Zhang S, et al: H2S as a physiologic
vasorelaxant: hypertension in mice with deletion of cystathionine
gamma-lyase. Science. 322:587–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li H, Mani S, Cao W, Yang G, Lai C, Wu L
and Wang R: Interaction of hydrogen sulfide and estrogen on the
proliferation of vascular smooth muscle cells. PLoS One.
7:e416142012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Loirand G, Guérin P and Pacaud P: Rho
kinases in cardiovascular physiology and pathophysiology. Circ Res.
98:322–334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong M, Yan BP, Liao JK, Lam YY, Yip GW
and Yu CM: Rho-kinase inhibition: A novel therapeutic target for
the treatment of cardiovascular diseases. Drug Discov Today.
15:622–629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satoh K, Fukumoto Y and Shimokawa H:
Rho-kinase: Important new therapeutic target in cardiovascular
diseases. Am J Physiol Heart Circ Physiol. 301:H287–H296. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimokawa H and Satoh K: 2015 ATVB plenary
lecture translational research on rho-kinase in cardiovascular
medicine. Arterioscler Thromb Vasc Boil. 35:1756–1769. 2015.
View Article : Google Scholar
|
|
14
|
Zhang J, Li XX, Bian HJ, Liu XB, Ji XP and
Zhang Y: Inhibition of the activity of Rho kinase reduces
cardiomyocyte apoptosis in heart ischemia/reperfusion via
suppressing JNK-mediated AIF translocation. Clin Chim Acta.
401:76–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Zhu W, Tao J, Xin P, Liu M, Li J and
Wei M: Fasudil protects the heart against ischemia-reperfusion
injury by attenuating endoplasmic reticulum stress and modulating
SERCA activity: The differential role for PI3K/Akt and JAK2/STAT3
signaling pathways. PLoS One. 7:e481152012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dai SJ, Chen RY and Yu DQ: Studies on the
flavonoid compounds of Rhododendron anthopogonoides. Zhongguo Zhong
Yao Za Zhi. 29:44–47. 2004.(In Chinese). PubMed/NCBI
|
|
17
|
Huang Y, Yin P, Jiang DF, Wang CY, Tan R,
Wan L, Zhang Y and Fan G: Quality standard of rhododendron flos.
World Sci Technol. 16:151–155. 2014.
|
|
18
|
Yuan LP, Chen ZW, Li F, Dong LY and Chen
FH: Protective effect of total flavones of Rhododendra on ischemic
myocardial injury in rabbits. Am J Chin Med. 34:483–492. 2006.(In
Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang JH, Chen ZW and Wu Z: Late
protective effect of pharmacological preconditioning with total
flavones of Rhododendra against myocardial ischemia-reperfusion
injury. Can J Physiol Pharmacol. 86:131–138. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiao Y, Fan YF, Wang YL, Zhang JY, Chen S
and Chen ZW: Protective effect and mechanism of total flavones from
rhododendron simsii Planch flower on cultured Rat cardiomyocytes
with anoxia and reoxygenation. Evid Based Complement Alternat Med.
2015:8635312015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hausenloy DJ, Tsang A and Yellon DM: The
reperfusion injury salvage kinase pathway: A common target for both
ischemic preconditioning and postconditioning. Trends Cardiovasc
Med. 15:69–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Terrell AM, Crisostomo PR, Wairiuko GM,
Wang M, Morrell ED and Meldrum DR: Jak/STAT/SOCS signaling circuits
and associated cytokine-mediated inflammation and hypertrophy in
the heart. Shock. 26:226–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Demirynrek S, Kara AF, Celik A, Babül A,
Tarakçioglu M and Demiryürek AT: Effects of fasudil. A Rho-kinase
inhibitor.on myocardial preconditioning in anesthetized mice. Eur J
Pharmacol. 527:129–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janet C, Garber R, Wayne B, Joseph T,
Bielitzki, Leigh AC, John C, Donovan, et al: Guide for the Care and
Use of Laboratory Animals of the National Institutes of HealthNIH
Publication; 85-23. revised. 2011
|
|
25
|
Rona G, Chappel CI, Balazs T and Gaudry R:
An infarct-like myocardial lesion and other toxic manifestations
produced by isoproterenol in the rat. AMA Arch Pathol. 67:443–455.
1959.PubMed/NCBI
|
|
26
|
Xu X, Li H, Gong Y, Zheng H and Zhao D:
Hydrogen sulfide ameliorated lipopolysaccharide-induced acute lung
injury by inhibiting autophagy through PI3K/Akt/mTOR pathway in
mice. Biochem Biophys Res Commun. 507:514–518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chau VQ, Salloum FN, Hoke NN, Abbate A and
Kukreja RC: Mitigation of the progression of heart failure with
sildenafil involves inhibition of RhoA/Rho-kinase pathway. Am J
Physiol Heart Circ Physiol. 300:H2272–H2279. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yatani A, Irie K, Otani T, Abdellatif M
and Wei L: RhoA GTPase regulates L-type Ca2+ currents in cardiac
myocytes. Am J Physiol Heart Circ Physiol. 288:H650–H659. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shimokawa H, Hiramori K, Iinuma H, Hosoda
S, Kishida H, Osada H, Katagiri T, Yamauchi K, Yui Y, Minamino T,
et al: Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in
patients with stable effort angina: A multicenter study. J
Cardiovasc Pharmacol. 40:751–761. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito Y, Kondo H and Hojo Y: Granzyme B as
a novel factor involved in cardiovascular diseases. J Cardiol.
57:141–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hamid SA, Bower HS and Baxter GF: Rho
kinase activation plays a major role as a mediator of irreversible
injury in reperfused myocardium. Am J Physiol Heart Circ Physiol.
292:2598–2606. 2007. View Article : Google Scholar
|
|
32
|
Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin
ES, Moore PK and Bian JS: Cardioprotection induced by hydrogen
sulfide preconditioning involves activation of ERK and PI3K/Akt
pathways. Pflugers Arch. 455:607–616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uehata M, Ishizaki T, Satoh H, Ono T,
Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M
and Narumiya S: Calcium sensitization of smooth muscle mediated by
a Rho-associated protein kinase in hypertension. Nature.
389:990–994. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abe K, Shimokawa H, Morikawa K, Uwatoku T,
Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K
and Takeshit A: Long-term treatment with a Rho-kinase inhibitor
improves monocrotaline-induced fatal pulmonary hypertension in
rats. Circ Res. 94:385–393. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen ZW, Ma CG, Fang M and Xu SY: The
blocking effect of hyperin on the inward flow of calcium ion. Yao
Xue Xue Bao. 29:15–19. 1994.(In Chinese). PubMed/NCBI
|
|
36
|
Kimura H: Hydrogen sulfide as a
neuromodulator. Mol Neurobiol. 26:13–19. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J
and Tang C: H2S generated by heart in rat and its effects on
cardiac function. Biochem Biophys Res Commun. 313:362–368. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC,
Huang SH, Tan CS, Whiteman M, Lu J and Moore PK: Hydrogen sulfide
and its possible roles in myocardial ischemia in experimental rats.
J Appl Physiol (1985). 102:261–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin HF, Wang Y, Wang XB, Sun Y, Tang CS
and Du JB: Sulfur dioxide preconditioning increases antioxidative
capacity in rat with myocardial ischemia reperfusion (I/R) injury.
Nitric Oxide. 32:56–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai CY, Wang CC, Lai TY, Tsu HN, Wang CH,
Liang HY and Kuo WW: Antioxidant effects of diallyl trisulfide on
high glucose-induced apoptosis are mediated by the
PI3K/Akt-dependent activation of Nrf2 in cardiomyocytes. Int J
Cardiol. 168:1286–1297. 2013. View Article : Google Scholar : PubMed/NCBI
|