Introduction
Intellectual disability (ID) is defined as
substantial impairment of cognitive and adaptive functions that
commonly occur in childhood, and has an estimated prevalence of
1.5~2.0% (1). Most reports conclude
that severe ID (an intellectual quotient of <50) has a
prevalence of 0.3~0.4%, while the estimated prevalence of mild ID
differs between various studies (1).
Related studies have shown that most affected patients exhibited
development delay, speech impediment, feeding difficulties,
gastrointestinal or abdominal wall defects and scoliosis (2,3). In
addition, affected patients exhibited certain facial features,
including abnormal ears, unusually shaped nose and micrognathia. To
date, the application of next generation sequencing technology has
allowed the rapid identification of new gene mutations that lead to
ID (4). The search for de
novo mutations through a trio approach is an efficient strategy
for the identification of the genetic cause of ID in most cases
(5,6). Grozeva et al (2) examined 996 individuals with ID by a
targeted next-generation sequencing approach and discovered 7 de
novo mutations of the SET domain-containing 5 (SETD5)
gene. The clinical phenotype of ID was markedly similar to 3p
deletion syndrome or 3p25 microdeletion syndrome, for which
SETD5 is believed to be the strongest candidate gene
(7,8). Kuechler et al (3) further extended the spectrum of
ID-associated SETD5 mutation by reporting 2 de novo
intragenic variants in two patients. These results suggested that
SETD5 may play a role in the mammalian developmental
process.
In this report, we present an ID patient with a
de novo frameshift mutation in exon 15 of SETD5 that
to the best of our knowledge has never been reported before. We
give a detailed description of the clinical features of the patient
to aid a more comprehensive understanding of the disease.
Case report
Patient
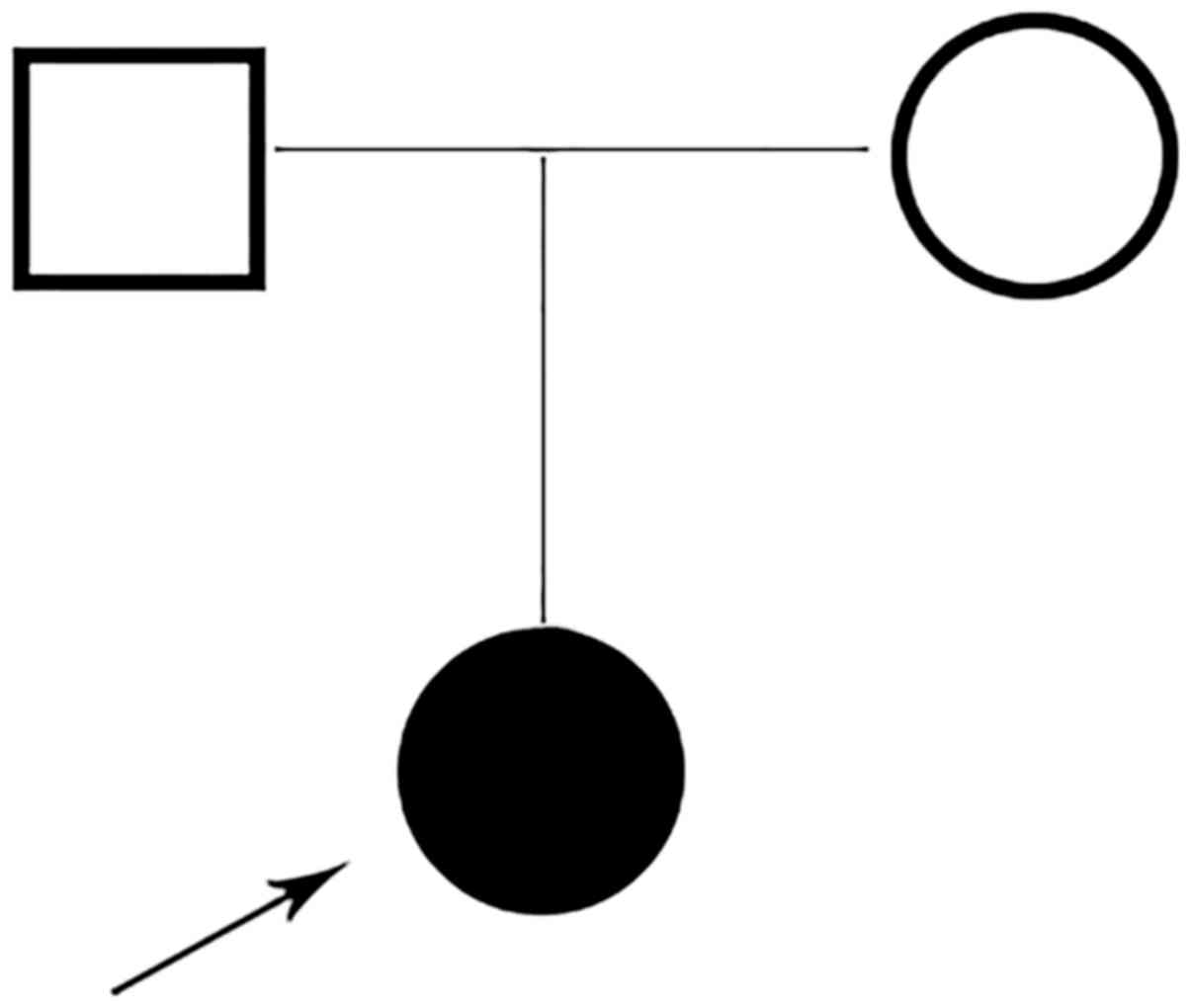
A Chinese family with a child was recruited in this
study (Fig. 1). The proband was a
1.7-year-old girl who was born to a non-consanguineous 32-year
mother and 31-year father. There was no family history of exposure
to teratogens. The proband was born by Caesarean section at
40+5 weeks because of a fetal heartbeat of 68 times per
minute, and Apgar scores were normal. Birthweight was 2,800 g and
length 48 cm. Birth occipito-frontal circumference (OFC) was not
available. The left eyelid showed slight droop not long after
birth. The child's motor development was delayed, especially gross
motor functions. She was unable crawl at 21 months. At the time of
presentation (2 year 5 months), her height was 82 cm with weight
(10 kg) and OFC (46 cm), within normal centile. She was not able to
walk without support or speak fluently. Facial signs comprised
prominent forehead, depressed nasal bridge, abnormal nose, thin
upper lip and prominent philtrum. Moreover, the ears tended to be
large and low set. The eyes were long, narrow and exhibited
unilateral mild ptosis. Both hands were able to hold items and
muscle tension was normal. At presentation, her anterior fontanelle
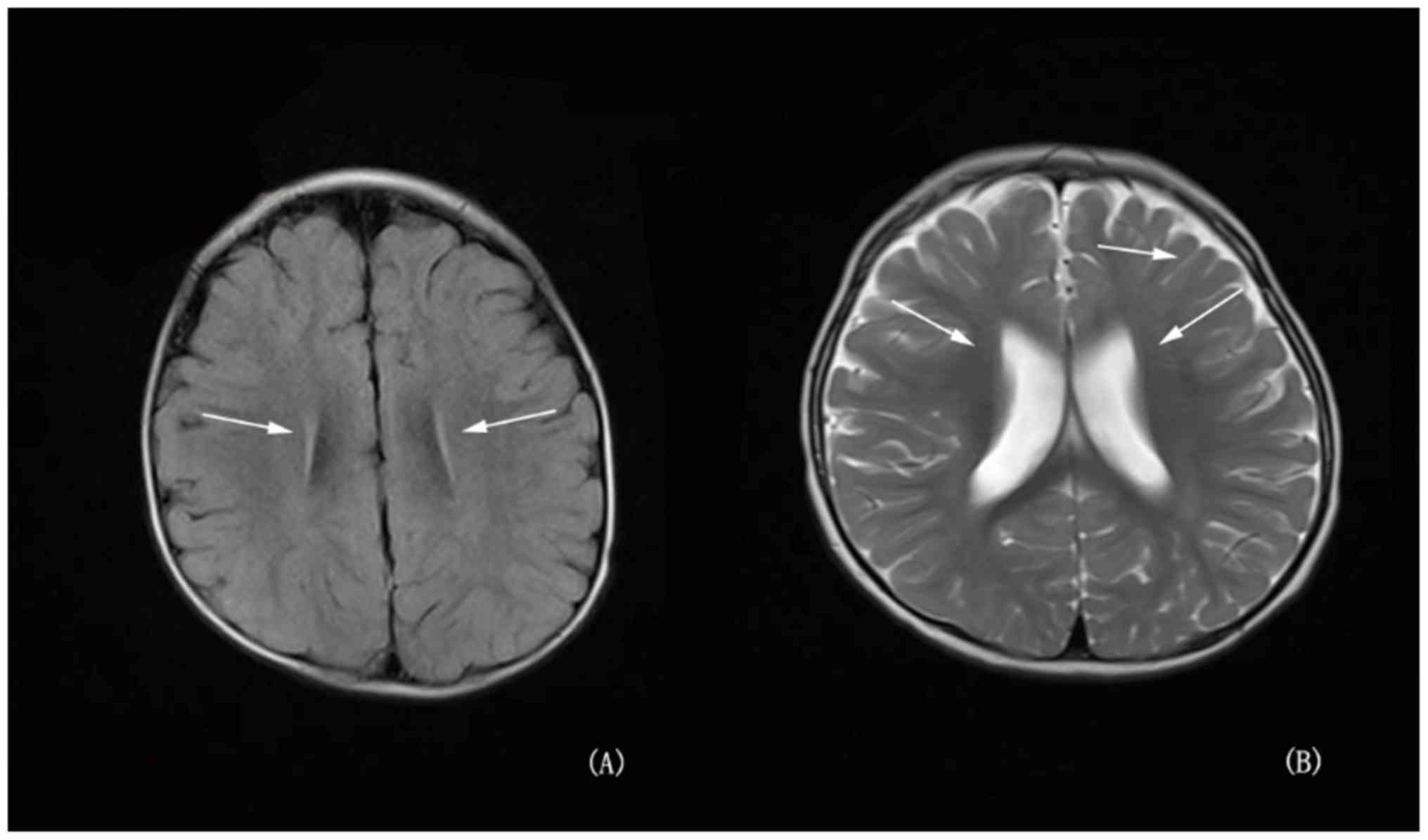
had not been closed. Brain magnetic resonance imaging (MRI) showed
high signal in bilateral parietal white matter area about
T2W1 and fluid attenuated inversion recovery (FLAIR); in
addition to a widened ventricle, outside the brain interval
(Fig. 2). Routine chromosome
analysis showed a normal female 46, XX karyotype. Screening for
metabolic disorders and electromyography examinations detected no
pathological results. The clinical phenotype of the parents was
normal.
Methods
Full informed consent was obtained from the guardian
of the patient, after which peripheral blood samples were collected
from the 3 family members, and genomic DNA was extracted from blood
samples using a DNA Extraction Kit (Tiangen Biotech Co., Ltd.,
Beijing, China) according to the manufacturer's instructions. Whole
exome sequencing was performed with trios including the proband and
her parents using the Illumina HiSeq 2500 platform (Illumina, Inc.,
San Diego, CA, USA). All sequencing fragments were aligned to the
reference human genome (hg19) using Burrows-Wheeler Alignment (BWA)
(9), and duplicated reads were
removed. Single nucleotide variants (SNVs) and small indels were
identified using the SAMtools (http://samtools.sourceforge.net/). For each sample,
variants of indels and SNVs were called using the Genome Analysis
Tool kit v3.2 (GATK) program (10).
All the identified variations were annotated and classified by
Variant Effect Predictor software (11). The variants were filtered to excluded
those with <1% minor allele frequency in all the following
databases: ExAC (http://exac.broadinstitute.org), 1,000 Genomes
(http://www.1000genomes.org/), ESP6500
(http://evs.gs.washington.edu/EVS), and
an in-house database of ~500 Polish exomes. Then, known
disease-causing variants were screened based on ClinVar, OMIM and
HGMD. Finally, the pathogenic variant was determined in combination
with clinical manifestation.
The variant of interest was confirmed by Sanger
sequencing of exon 15 of SETD5. DNA was amplified by
polymerase chain reaction (forward primer: GACCAGTTCAGCCCAAAGAC,
reverse primer: CAGCCTTTTGTGTCAAAGCA). The PCR products were
identified by agarose gel electrophoresis and sequenced directly
using an ABI3730 automated sequencer (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). SETD5 variants were
annotated based on the transcript number NM_001080517.2.
Results
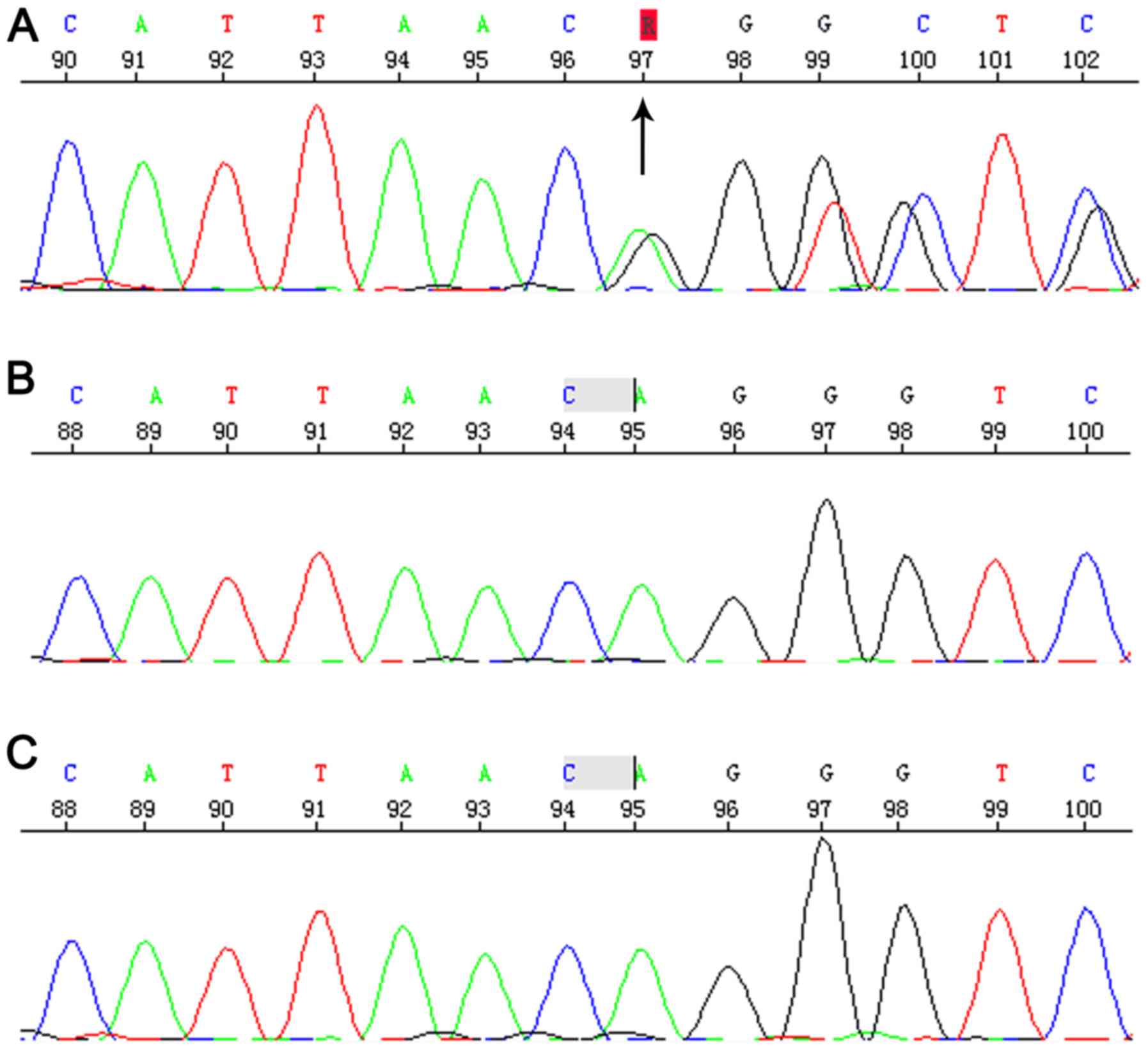
Sequencing revealed the Chr3:9489612-9489613
c.2025_2026del AG that located in 15 exon of SETD5 gene.
This is a frameshift mutation, causing termination of the protein
in advance (p.Gly676Vlfs*2). This variant had not previously
reported in the literature and was absent from the ExAC and 1,000
Genomes databases. Parents tested negative for this particular
variant, suggesting that this was a de novo mutation
(Fig. 3) and autosomal dominant
transmission of ID (MIM:615761). Therefore, this variant was
considered as probably pathogenic in the proband.
Discussion
ID patients can be characterized by specific
features, including speech and motor deficits, growth retardation,
cardiovascular and renal defects, and behavioral problems (2,12). These
features can vary depending on the main underlying genetic cause.
Previous papers have suggested that the ID caused by SETD5 de
novo mutations might be part of a more complex clinical
phenotype with variable expressivity. These observations are in
conformity with the previously emphasized value of familial cases
of newly recognized genetic syndromes in expanding knowledge of
their phenotype (13).
The phenotypic features of individuals with the
SETD5 de novo mutations in our family were similar to
previously described patients, although there were some obvious
differences (2). As to the reason
for these differences, it should be noted that the previous studies
reporting SETD5 mutations analyzed hundreds of ID patients
for de novo variants (2,3).
SETD5 is located on chromosome 3q3.25 and
includes 49 exons, which code a 1,442 amino acid protein, SET
domain-containing protein 5. The protein belongs to the SET
methyltransferase family, which catalyzes methylation of histone H3
and H4 lysine residues (14). It is
highly expressed in the cerebral cortex, the intestine and the eye
(3). Related studies have confirmed
that SET-containing-domain protein could possess histone
H3-specific methyltransferase activity (15).
Although rare, patients with ID might exhibit large
deletions (up to 12 Mb), small deletions, substitutions, or
insertions (16). Our results
indicated that a new de novo SETD5 gene variant [c.2025_2026
delAG (p.Gly676Vlfs*2)] as the genetic cause of ID in the patient
under study, but her parents did not carry the mutation.
Fortunately, related studies have shown that rare de novo
loss of function (LoF) variants of SETD5 have recently been
proposed to be a relatively frequent (0.7%) cause of ID (3). In addition, Rauch et al
described a new one de novo nonsense mutation resulting in
the deletion of 7 genes (5).
Kuechler et al (3) revealed 2
de novo intragenic variants; one was a nonsense variant and
the other an 81 bp deletion located across a splice-donor site.
Szczałuba et al (17)
prioritized a potential LoF mutation. Related studies have
confirmed that ID could be caused by de novo variants in
45~55% of patients (5). These data
demonstrated that mutations were commonly found in patients with
ID. Surprisingly, these studies further showed that de novo
mutations might be the main cause of most ID (18).
In conclusion, we described a novel variant in
SETD5 gene as the putative genetic cause of a patient with
ID. The findings may aid deeper understanding of this disease.
Acknowledgements
Not applicable.
Funding
This work was supported by the Key Project of
Tianjin Health Care Professionals (grant no.16KG166), the National
Natural Science Foundation of China (grant no.81771589), and the
Program of Tianjin Science and Technology Plan (grant no.
18ZXDBSY00170).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YLF conceived and designed this study, and drafted
the manuscript. RPZ performed the data analysis and interpretation.
YZW and LRC performed the Sanger confirmation and analysis. YQZ and
CQC participated in the design and coordination of this study in
addition to revising and critiquing the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Tianjin Children's Hospital. Informed consent for participation
in the study or use of their medical data was obtained from all
participants or their legal guardian.
Patient consent for publication
Informed consent was obtained from the guardian
(parents), who agreed to join this study, and for the use of
medical information and images for scientific research and
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Leonard H and Wen X: The epidemiology of
mental retardation: Challenges and opportunities in the new
millennium. Ment Retard Dev Disabil Res Rev. 8:117–134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grozeva D, Carss K, Spasic-Boskovic O,
Parker MJ, Archer H, Firth HV, Park SM, Canham N, Holder SE, Wilson
M, et al: De novo loss-of-function mutations in SETD5, encoding a
methyltransferase in a 3p25 microdeletion syndrome critical region,
cause intellectual disability. Am J Hum Genet. 94:618–624. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuechler A, Zink AM, Wieland T, Lüdecke
HJ, Cremer K, Salviati L, Magini P, Najafi K, Zweier C, Czeschik
JC, et al: Loss-of-function variants of SETD5 cause intellectual
disability and the core phenotype of microdeletion 3p25.3 syndrome.
Eur J Hum Genet. 23:753–760. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 42:30–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rauch A, Wieczorek D, Graf E, Wieland T,
Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di
Donato N, et al: Range of genetic mutations associated with severe
non-syndromic sporadic intellectual disability: An exome sequencing
study. Lancet. 380:1674–1682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Ligt J, Willemsen MH, van Bon BW,
Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA,
de Vries P, Gilissen C, et al: Diagnostic exome sequencing in
persons with severe intellectual disability. N Engl J Med.
367:1921–1929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shuib S, McMullan D, Rattenberry E, Barber
RM, Rahman F, Zatyka M, Chapman C, Macdonald F, Latif F, Davison V
and Maher ER: Microarray based analysis of 3p25-p26 deletions
(3p-syndrome). Am J Med Genet A 149A. 2099–2105. 2009. View Article : Google Scholar
|
|
8
|
Peltekova IT, Macdonald A and Armour CM:
Microdeletion on 3p25 in a patient with features of 3p deletion
syndrome. Am J Med Genet A 158A. 2583–2586. 2012. View Article : Google Scholar
|
|
9
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A mapreduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pinto D, Delaby E, Merico D, Barbosa M,
Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z,
et al: Convergence of genes and cellular pathways dysregulated in
autism spectrum disorders. Am J Hum Genet. 94:677–694. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carey JC: Significance of case reports in
the advancement of medical scientific knowledge. Am J Med Genet
Part A. 140:2131–2134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martin C and Zhang Y: The diverse
functions of histone lysine methylation. Nat Rev Mol Cell Biol.
6:838–849. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rea S, Eisenhaber F, O'Carroll D, Strahl
BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD
and Jenuwein T: Regulation of chromatin structure by site-specific
histone H3 methyltransferases. Nature. 406:593–599. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Riess A, Grasshoff U, Schäferhoff K, Bonin
M, Riess O, Horber V and Tzschach A: Interstitial 3p25.3-p26.1
deletion in a patient with intellectual disability. Am J Med Genet
158A. 2587–2590. 2012. View Article : Google Scholar
|
|
17
|
Szczałuba K, Brzezinska M, Kot J,
Rydzanicz M, Walczak A, Stawiński P, Werner B and Płoski R: SETD5
loss-of-function mutation as a likely cause of a familial syndromic
intellectual disability with variable phenotypic expression. Am J
Med Genet A. 170:2322–2327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grozeva D, Carss K, Spasic-Boskovic O,
Tejada MI, Gecz J, Shaw M, Corbett M, Haan E, Thompson E, Friend K,
et al: Targeted next-generation sequencing analysis of 1,000
individuals with intellectual disability. Hum Mutat. 36:1197–1204.
2015. View Article : Google Scholar : PubMed/NCBI
|