Introduction
Cardiovascular diseases (CVDs) are a wide spectrum
of diseases affecting the heart and blood vessels. This spectrum
includes mainly coronary heart disease (CHD) and cerebrovascular
disease (1). Atherosclerosis, the
culprit cause of myocardial and cerebral infarction, is the
principle agent of mortality and morbidity worldwide (2). Atherosclerosis is a cardiovascular
disease marked by the dysfunction of the endothelium, formation of
lipid-laden plaques, and narrowing and hardening of the blood
vessels. Over the centuries, many hypotheses were postulated to
explain the mechanism behind the initiation and development of the
atherosclerotic lesions. The three main theories are the lipid
theory, the oxidation hypothesis of atherogenesis and the
response-to-injury inflammatory hypothesis. The cornerstone focus
of the oxidative hypothesis is that specifically oxidized low
density lipoproteins (LDL) generated by myeloperoxidase pathways
are injurious to the arterial cell wall as reported by Chilsom and
colleagues in 1979. The inflammatory response-to-injury hypothesis
is regarded as the refinement of the previous theory; during the
19th century, the pathologist Rudolf Virchow described
atherosclerosis as a chronic inflammation (3). This hypothesis proposes that injury to
the endothelium and its dysfunction, which leads to fibrin
deposition, are the initiating events along with its increased
permeability to modified lipoproteins (4). Oxidation of low density lipoproteins
have been of major interest since Steinberg et al showed
that native LDL does not accumulate in macrophages whereas modified
lipoproteins do (5).
Oxidized LDL (oxLDL) unlike the native LDL (nLDL)
was shown to initiate and trigger the inflammatory process of the
disease (6). Several candidates were
then proposed to elucidate LDL modification including
myeloperoxidase (MPO). Ample studies showed the responsibility of
MPO in atherogenesis in humans. Immunohistochemical and biochemical
analyses conducted by Daugherty et al (7) co-localized MPO and its products within
human atheromatous plaques (7–9). On the
same note, it has been shown that individuals with a deficiency in
the MPO enzyme are less prone to develop CVDs on the long term.
Another relationship appears in which increased systematic levels
of MPO indicates the presence of coronary artery disease (CAD)
(10). A major function of the
vascular endothelium is the fine-tuning of the delicate components
of the coagulation and fibrinolytic systems. It maintains
anticoagulant and antithrombotic environment by releasing a variety
of molecules that regulate blood hemostasis assuring a
profibrinolytic state (11).
Accordingly, the endothelium secretes major fibrinolytic factors
including tissue plasminogen activator (tPA), urokinase plasminogen
activator (uPA), and plasminogen activator inhibitor-1 (PAI-1) and
express specific receptors that binds these factors supporting a
fibrinolytic environment (12).
Also, as mentioned above, early observations have correlated fibrin
deposition with atheroma plaque formation. This led to the
proposition that a decrease in fibrinolysis in endothelial cells
may negatively influence atherogenesis. In parallel, recent studies
have also revealed that patients with atherosclerosis exhibited a
hypofibrinolytic phenotype (13).
Since it has been previously confirmed that MoxLDL decreases
EA.hy926 profibrinolytic capacity in real-time without delineating
the mechanisms by which this modified LDL can alter pericellular
fibrinolysis (14), we tried in the
present study to perform a preliminary dissection of the molecules
that might be involved in decreasing fibrinolysis by using primary
HAEC as a model. The study also included an ephemeral comparison
regarding the disparate effect of MoxLDL on two different primary
cultures of endothelial cells, bovine aortic endothelial cells and
human aortic endothelial cells, as well as its effect on reactive
oxygen species (ROS) generation in the latter model.
Materials and methods
Cell culture
BAE cells were cultured in Dulbecco's Modified
Eagle's Medium-AQ (DMEM-AQ, Sigma Aldrich) supplemented with 10%
heat inactivated fetal bovine serum (FBS; Sigma Aldrich), and 1%
penicillin/streptomycin mixture. HAEC's were cultured in EBM-2
Basal Medium supplemented with human epidermal growth factor
(hEGF), vascular endothelial growth factor (VEGF), R3-insulin-like
growth factor-1 (R3-IGF-1), ascorbic acid, hydrocortisone, human
fibroblast growth factor-beta (hFGF-β), FBS, and
gentamicin/amphotericin-B (GA) (Lonza). Cells were maintained at
37°C in a humidified 5% CO2 incubator. BAE cells and
HAEC were used between passages 5–9.
In vitro treatment of BAE and
HAEC
BAE cells and HAEC were seeded in 6-well plates at a
density of 5×105 cells/well in triplicates. Cells were
either left untreated or treated with nLDL (100 µg/ml) or with
MoxLDL (25, 50, or 100 µg/ml) (15,16).
Cell morphology was monitored after treatment and images (×200
magnification) were captured using a phase contrast inverted
microscope (Leica).
Recombinant MPO preparation
Recombinant MPO was prepared as described previously
(14). Briefly, in order to express
MPO, a recombinant plasmid that codes for prepromyeloperoxidase was
constructed and named pNIV2703. This plasmid contains an MPO
fragment coding for amino acid 11 in the putative signal sequence
to amino acid 696. The pNIV2703 expression vector was transfected
into CHO cells by electopermeabilization. Cell supernatants were
recovered to assay the production level and the enzymatic activity
of secreted molecules. Each batch solution was characterized by its
activity (U/ml), protein concentration (mg/ml), and specific
activity. Peroxidative activity was determined using
o-dianiside as the substrate. Protein concentration was
measured using the Lowry assay, with ovalbumin as a standard. Each
batch was checked for endotoxin using the endotoxin detection kit
(Lonza). Concentration was always less than 100 pg/ml, which,
taking into account the final dilution of the MPO-treated LDL
fraction, would contribute a final concentration of less than 0.1
pg/ml to the MoxLDL supplemented medium added to the cells.
Isolation of nLDL and MoxLDL
preparation
nLDL was isolated and MoxLDL prepared as previously
described (14); lipoprotein
particles were isolated from plasma from sterile blood pouches
using density-gradient ultracentrifugation. The nLDL fraction
(d=1.019–1.063) was stored under nitrogen at 4°C in the dark
and oxidized according to the procedure described below: Prior to
oxidation, nLDL was gel filtered (PD-10 column, Pharmacia) and 1.6
mg of native LDL was oxidized by 2.1 chlorinating units of
recombinant MPO, to generate MoxLDL in the presence of 1 mM
H2O2 in 2 ml phosphate buffered saline (PBS)
at pH 6.5 for 5 min. LDLs were desalted again after MPO treatment.
Protein concentration was measured by the Lowry assay, using
ovalbumin as protein standard.
RNA extraction from BAE cells and
HAEC
Cells were treated as indicated above and then
harvested for total RNA extraction. RNA was extracted using the
RNeasy plus mini kit (Qiagen) according to manufacturer's
instructions. RNA extracts from different samples were analyzed by
spectrophotometry. Absorbance values (A) were recorded at two
wavelengths, (260 and 280 nm) to assess purity (A260/A280) and
measure the concentration (A260) of extracted RNA.
Reverse transcription of RNA
Extracted RNA was reverse transcribed to a
complementary DNA (cDNA) using QuantiTect reverse transcription kit
according to manufacturer's instructions (Qiagen). Briefly, genomic
DNA (gDNA) contaminants were eliminated by incubating extracted RNA
in gDNA wipeout buffer (Qiagen) at 42°C for 2 min. Then reverse
transcription was performed by incubating samples with master mix
(reverse transcriptase (RT), RT primer mix, and RT buffer; Qiagen)
at 42°C for 15 min and later inactivated at 95°C for 3 min.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was performed using SYBR-Green (Qiagen), on
a Real-time Systems (Bio-Rad). 20 µl of reaction was added in each
well containing: 1 µg cDNA, 1 µl of 10 µM forward and reverse
primers (Table I; Sigma), and 10 µl
Sybr Green. The cycling conditions were: 95°C for 3 min, 40 cycles
of 95°C for 10 s, and 60°C for 30 s. Each reaction was performed in
triplicate and GAPDH was used as a reference gene for
normalization. Relative gene expression levels and resulting fold
changes were calculated using the comparative 2−ΔΔCq
method (17). Primers sequences for
genes of interest were selected from the RTPrimerDB online
real-time PCR primer database (rtprimerdb.org) and verified using the online NCBI
BLAST tool.
 | Table I.Primer sequences for the genes of
interest. |
Table I.
Primer sequences for the genes of
interest.
| Gene | Forward | Reverse |
|---|
| tPA |
5′-CCGGCTACGGCAAGCA-3′ |
5′-TGGATGGGTACAGTCTGACATGA-3′ |
| uPA |
5′-CCGCTTTCTTGCTGGTTGTC-3′ |
5′-TATTGTCGTTCGCCCTGGTG-3′ |
| uPAR |
5′-GGTGACGCCTTCAGCATGA-3′ |
5′-CCCACTGCGGTACTGGACAT-3′ |
| tPAR |
5′-TGGATGGGAGACAATCTGTA-3′ |
5′-TGCCCTCGATTAAAGTCTTG-3′ |
| PAI-1 |
5′-CAGACCAAGAGCCTCTCCAC-3′ |
5′-ATCACTTGGCCCATGAAAAG-3′ |
| FXIII A1 |
5′-CCAGATTGACTTCAACCGTCCC-3′ |
5′-GACACCAGCAAAAACCCAACACTGG-3′ |
| GAPDH |
5′-AAATCCCATCACCATCTTCC-3′ |
5′-TCACACCCATGACGAACA-3′ |
Analysis of BAE cell viability
Propidium iodide (PI) cell viability assay was used
to assess the viability of BAE cells following treatment as
indicated above. BAE cells were washed once with PBS and detached
by incubation with accutase cell detachment solution (Thermo) at
37°C for 2 min. Both detached and floating cells were then
collected and resuspended in 1X binding buffer solution (BD
Biosciences) and stained with PI (BD Biosciences) for 15 min at
room temperature in the dark. Samples were run on a FACSCalibur
flow cytometer (BD Biosciences) and data were analyzed using
CellQuest Pro software version 5.1 (BD Biosciences). BAE cells were
identified by their forward-scatter (FSC) and side-scatter (SSC)
properties. Viable and dead cell populations were identified as PI-
and PI+ cells, respectively (18–20). A
total of 10,000 single cell events were measured for each
sample.
Measurement of ROS generation by
HAEC
ROS production by HAEC was assessed following
treatment as indicated above using the radical-sensitive
fluorescent probe, 2,7-dichlorodihydrofluorescein diacetate
(H2DCFDA; Molecular Probes). HAEC were washed once with
PBS and detached by incubation with accutase cell detachment
solution at 37°C for 2 min. Cells were then incubated with H2DCFDA
(10 µmol/l) for 45 min at 37°C in a humidified 5% CO2
incubator, and washed twice with PBS. Cells were finally
resuspended in PBS and acquired using FACSCalibur flow cytometer.
The experiment was performed in triplicates and
H2O2 (1 and 10 µg/ml) was used as a positive
inducer of ROS. Intracellular ROS levels, reflected by the mean
fluorescence intensity (MFI) of 2,7-dichlorofluorescein
(DCF)-stained HAEC were measured using CellQuest Pro software. HAEC
were identified based on their FSC and SSC characteristics.
Statistics
GraphPad Prism software (version 6) was used to
perform statistical data analysis and drawing of graphs. Data are
presented as mean ± standard error of the mean (SEM). The
non-parametric Kruskal-Wallis test followed by a Dunn's multiple
comparison post-hoc test was performed to determine statistical
differences among the different experimental groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
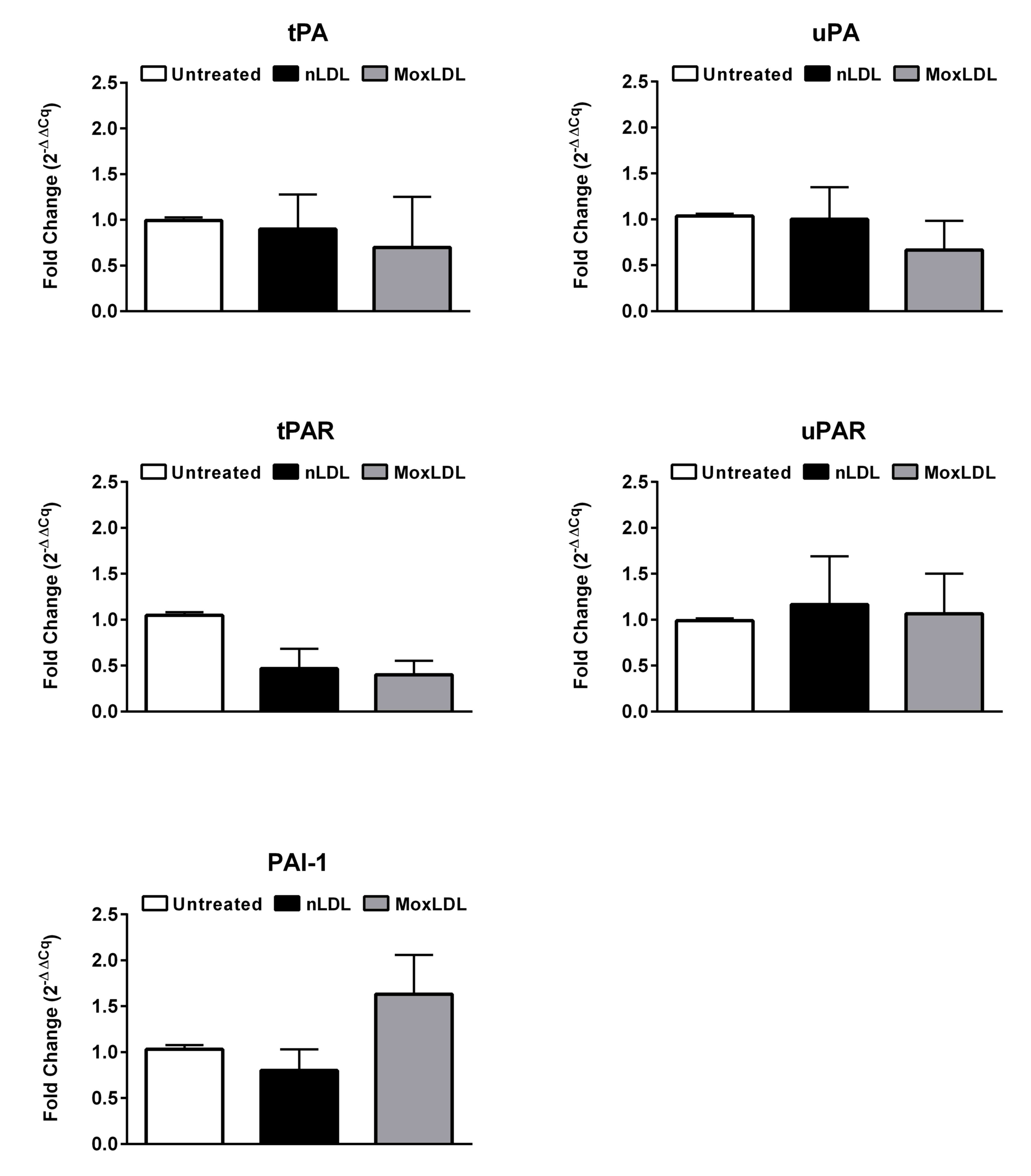
Expression of selected fibrinolytic
genes in HAEC in response to nLDL or MoxLDL treatment
A previous study has demonstrated that MoxLDL delays
fibrinolysis pericellularly in EA.hy926 endothelial cells (14). Several genes such as tPA, uPA, tPA
receptor (tPAR), uPA receptor (uPAR) and PAI-1 are known to be key
players in the process of fibrinolysis (21). However, whether their expression
levels in HAEC are altered upon MoxLDL treatment remains unknown.
Therefore, we have assessed by RT-qPCR the mRNA expression profile
of the above-mentioned genes in HAEC treated with nLDL or MoxLDL
for 24 h. Fibrinolysis activators, tPA and uPA, in addition to
their corresponding receptors, tPAR and uPAR, did not show a
significant variation in their mRNA expression levels following
treatment with nLDL or MoxLDL. PAI-1 also, the major plasminogen
activator inhibitor, did not show a variation in expression
(Fig. 1). Finally, FXIII, a
principal hemostatic factor and the protein responsible for
crosslinking fibrin meshwork was found not to be expressed in HAEC
(data not shown).
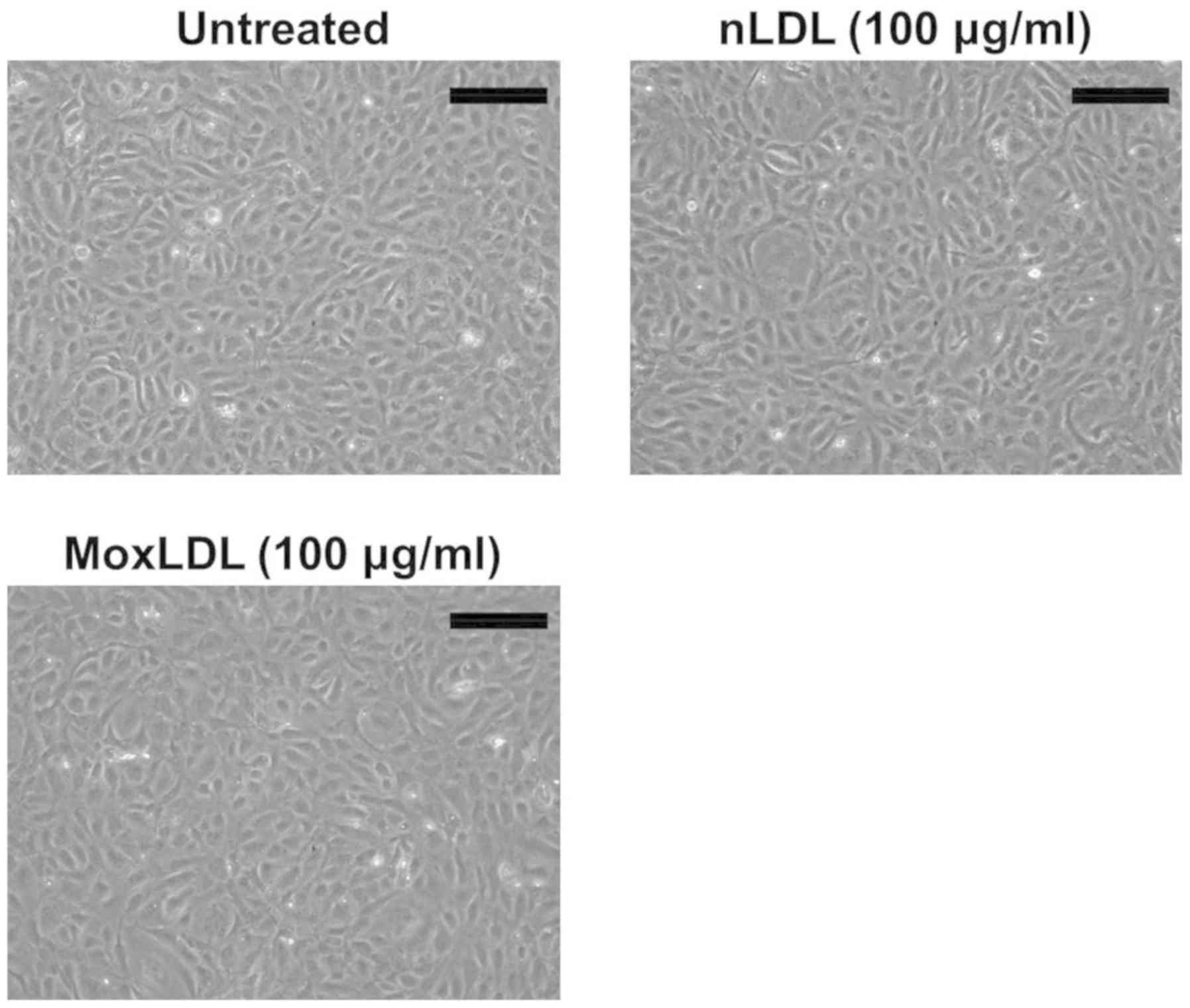
Effect of nLDL or MoxLDL treatment on
the morphology and viability of HAEC
HAEC were cultured in a 6-well plate and were left
untreated or treated with either 100 µg/ml of nLDL or MoxLDL for 24
h. The cells were then visualized under an inverted phase contrast
microscope. HAEC showed a healthy morphology with no or little
detached cells detected (Fig. 2).
Supplementary PI staining and FACS analysis were also carried out
and did not show any significant effect of MoxLDL on HAEC viability
(data not shown).
Effect of nLDL and MoxLDL treatment on
the morphology and adherence of BAE cells
In order to study the effect of nLDL and MoxLDL on
BAE cells, confluent monolayer cells were treated with 100 µg/ml of
nLDL, and 25, 50, and 100 µg/ml of MoxLDL for 24 h. Following
MoxLDL treatment, a significant high percentage of detached and
floating cells was noted when BAE cells were visualized under an
inverted phase contrast microscope. However, nLDL treatment did not
induce BAE cell detachment and cells were maintained as a monolayer
(Fig. 3).
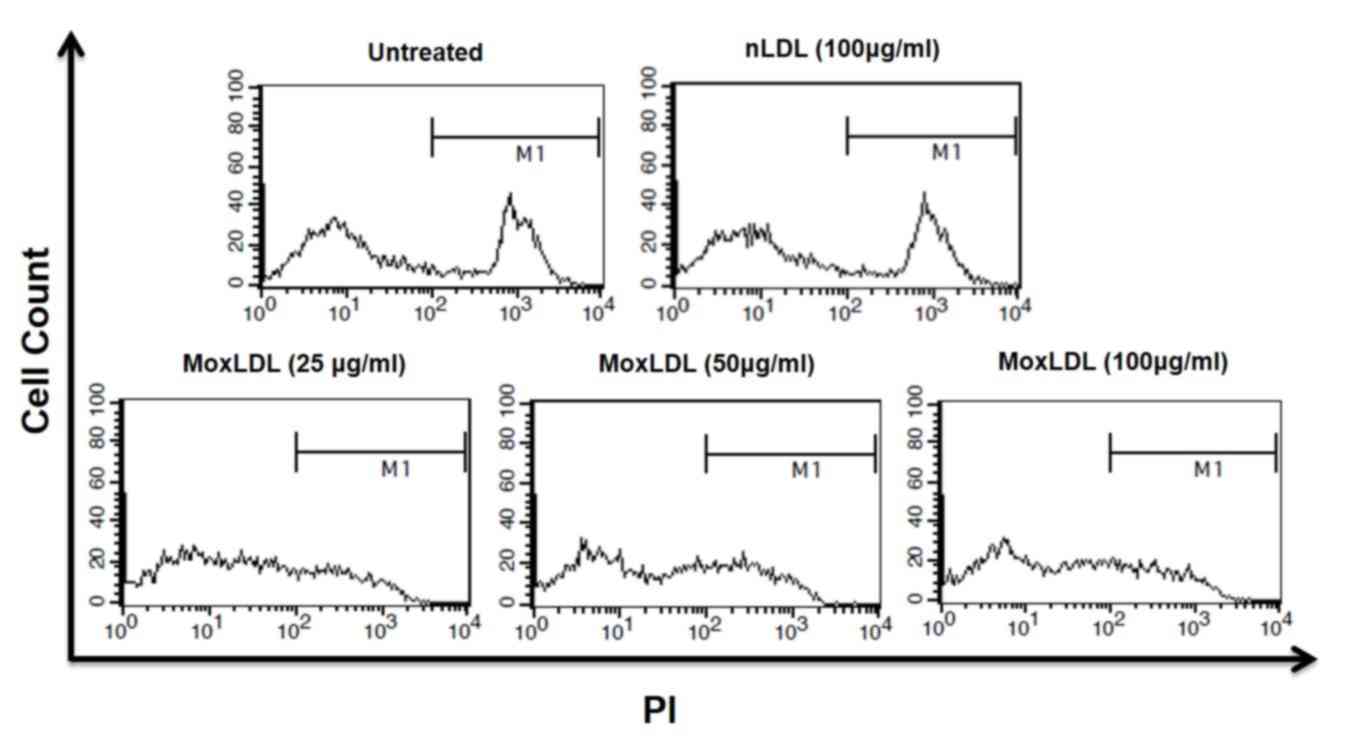
Effect of nLDL and MoxLDL treatment on
the viability of BAE cells
We assumed that the detachment of BAE cells might be
a consequence of a cytotoxic effect induced by MoxLDL treatment.
Therefore, PI viability assay was performed in order to assess the
cytotoxic effect of MoxLDL as well as the effect of nLDL on BAE
cells. Treated BAE cells were harvested, stained with PI, and
analyzed by flow cytometry. As expected, untreated cells and
nLDL-treated cells showed some little extent of spontaneous cell
death. However, adequate cell viability analysis of MoxLDL-treated
BAE cells was not possible due to that fact that cell fragments and
debris compromised the bulk of the culture (Fig. 4).
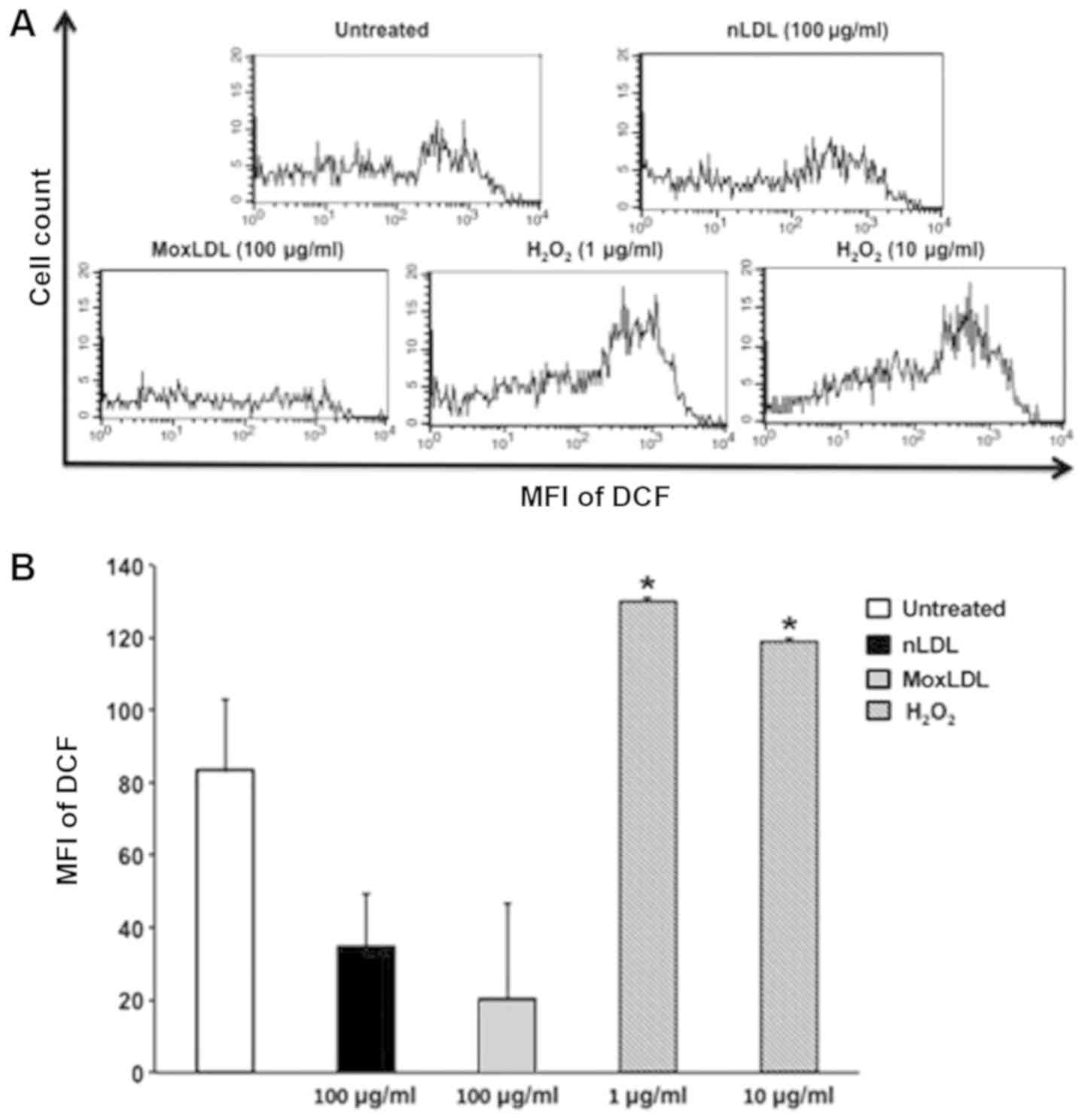
Effect of nLDL or MoxLDL treatment on
ROS production by HAEC
OxLDL has been previously reported, to increase ROS
production by endothelial cells (22). Therefore, the ability of MoxLDL to
induce endothelial cells to generate ROS was assessed by H2DCFDA
staining combined with flow cytometry analysis. MoxLDL treatment
resulted with no increase but in a decrease in the production of
ROS as compared to untreated cells; however, this decrease did not
attain statistical significance (P>0.05; Fig. 5). Likewise, nLDL treatment showed a
non-significant (P>0.05) decrease in ROS production (Fig. 5). H2O2 (1 and
10 µg/ml), used as a positive control, induced high and significant
levels of ROS by HAEC (Fig. 5).
Discussion
In our study, the interaction between MoxLDL,
endothelial cells, and mainly fibrinolysis was investigated. All
previous research documented and explored the signaling pathway by
which CuoxLDL initiate inflammation and subsequent atherogenesis
(23). On the contrary, very little
is known about the MoxLDL. MPO modified LDL is the more
physiologically relevant model of LDL oxidation due to the fact
that immunohistochemical analyses conducted by Daugherty et
al (7) and others co-localize
MPO and some modified amino acids in the ApoB100 moiety of LDL,
such as chlorotyrosine or nitrotyrosine, within human atheromatous
plaques (7–9,24).
Therefore, in our study, we aimed to examine the effect of MoxLDL
on different cell models: BAE and HAEC.
BAE are primary cells derived from the aorta of
cows. Previous research performed on these cells showed that 150
µg/ml of MoxLDL treatment for 24 h was markedly cytotoxic as judged
by MTT assay (25). However, this
study was performed using another oxidation product of MPO, which
is peroxinitrite modified LDL. Similarly, our results showed
tremendous death and fragmentation of BAE cells following MoxLDL
treatment (LDL modified by hypochlorus acid), even at low to normal
physiological concentrations (25, 50 and 100 µg/ml) (15). By recurring to PI staining and FACS
analysis, we unsuccessfully tried to assess the exact magnitude of
MoxLDL's effect in our experimental model and that was due to the
remarkable fragmentation of the cells that were difficultly sorted
and in a very bad shape. Hence, our results pave the way for future
investigation at this level that should be carried out using maybe
lower concentrations of modified LDL in order to better
characterize the mechanism of cell death. On the same note and in
the context of atherosclerosis, little in vivo
experimentation was conducted in animals. It was not documented
that animals especially bovine develop atherosclerosis naturally.
This may be due to the fact of their short life span, in comparison
to humans, unique digestive and metabolic characteristics,
different diet and cholesterol intake. This raises again an
interesting question regarding the modification of LDL molecules in
their system and whether it involves similar mechanisms that are
already seen and documented in humans.
Contradictory to the effect of MoxLDL on cell death
in BAE cells, previous reports confirmed that MoxLDL treatment (up
to 100 µg/ml) does not reduce viability or induce cell death in
human endothelial cells, human umbilical Vein Endothelial Cells
(HUVEC) (26). Preponderance of
studies reported that uptake by LOX-1 receptor, a type of scavenger
receptors minimally expressed on inactivated vascular endothelium,
is involved in endothelial activation, dysfunction and subsequent
initiation of atherosclerosis. It has been shown that CuoxLDL binds
to LOX-1 receptor increasing transcriptional activation of LOX-1
mRNA synthesis, entering in a positive feedback loop that
exacerbate the vascular dysfunction (27,28).
Intracellularly, this binding activates membrane bound NADPH
oxidase that rapidly elevates the level of ROS by generating super
oxide ion, exacerbating the inflammatory signal (29). Enough evidence had been accumulated
to state that CuoxLDL stimulates ROS generation; however, very
limited research was conducted on endothelial cells using MoxLDL to
check the levels of ROS production. As a matter of fact, CuoxLDL is
not physiologically relevant. In particular, CuoxLDL is not closely
related to the oxidized LDL present in vivo. High
concentrations of Cu or Fe used in vitro to oxidized LDL are
never met physiologically. Several enzymes were proposed as a
physiological alternative to the modification of LDL such as MPO
and peroxidasin (16). Additionally,
immunohistochemical and biochemical analyses conducted by Delporte
et al (8) co-localize MPO and
its modified amino acids products, such as chlorotyrosine or
nitrotyrosine within human atheromatous plaques (7,9,13,24).
Also, high serum levels of MPO are regarded as a risk factor in
CADs (30).
Data in the literature show MoxLDL's key involvement
in the interplay and fine-tuning between the coagulation and
fibrinolytic systems on the endothelial cell surface; yet little is
known about the mechanism by which MoxLDL binds to the endothelium
initiating the dysfunction. The endothelium secretes tPA and uPA,
and expresses specific receptors that bind these factors supporting
a fibrinolytic environment (13).
Moreover, the binding of plasminogen and tPA to fibrin or their
respective receptors ensures the protection from
α2-antiplasmin and α2-macroglobulin
inhibition. This means that endothelial cells and their receptors
aid and promote pericellular fibrinolysis (31). Early evidence correlates fibrin
deposition and plaque formation. Consistent with that, studies
documented that fibrin deposition on endothelial cells alters their
cobblestone morphology, induce the production of IL-8 and
inflammatory and chemotactic molecules, and most importantly
renders them more permeable to LDL infiltration (32). Due to previous technical limitations,
studying the effect of MoxLDL on pericellular fibrinolysis was a
challenge. However, more recently, a technical device that detects
fibrinolysis in real-time was successfully created (14). In the latter model, the authors
associated MoxLDL with a delayed fibrinolytic capacity of the
endothelium, but no effect on PAI-1, tPA, uPAR, tPAR or plasmin
inhibitors: α2-antiplasmin and
α2-macroglobulin was recorded. These cells that were
used in the study were hybrid cells that arise from a fusion
between HUVECs and thioguanine-resistant clone of A549 human
carcinoma cell line (33). In
accordance with this previous finding suggesting the null
transcriptional effect of MoxLDL on the fribrinolytic key players,
our results verified so while using the role model of cells to
study atherosclerosis: Human aortic endothelial cells (HAEC). We
additionally studied another potential effector that might be the
cause behind this delay: Factor XIII. Factor XIII, also known as
fibrin stabilizing factor, crosslinks every E-unit with a D-unit of
fibrin monomers further stabilizing the fibrin meshwork.
Contradictory observations are published on the fact whether
endothelial cells secrete FXIII or not (34). Our results showed that HAEC that were
analyzed by RT-qPCR do not express it.
As for the negative effect of MoxLDL on pericellular
fibrinolysis in endothelial cells, and since we've shown that it
was not related to a change in gene expression, one potential
mechanism that remains effective is the possibility of a physical
interaction between MoxLDL and specific cell membrane receptors
that are expressed on endothelial cells, more specifically tPAR and
uPAR receptors, which can explain this delay as a competitive
inhibition on the receptor itself by MoxLDL. This was seen with
other pro-atherogenic molecules such as apolipoprotein (a) which
was shown to bind tPAR receptors with high affinity (35).
Finally, preceding reports stated that MoxLDL lacked
an effect on the expression of LOX-1 gene expression as opposed to
CuoxLDL that was reported to increase LOX-1 expression (14,36).
This was further validated by our experiments that did not show an
increase in ROS production intracellulary in HAEC (15). Therefore, it is likely that MoxLDL
acts through a still undetermined receptor(s), eliciting signaling
transduction pathways that are dissimilar to CuoxLDL.
This study gave interesting insights onto the effect
of MoxLDL on the gene expression of central players in fibrinolysis
in HAEC as well as a potentially different mechanism of action for
MoxLDL as compared to CuoxLDL. In hope that our results will pave
the way for more experiments, some prospective research work can be
proposed including the investigation of the potential receptor(s)
that is/are responsible for binding to MoxLDL. Accordingly, a
series of knockdown experiments (tackling potential
receptors/signaling proteins) can be conducted in order to study
the signaling transduction pathway(s) promoted by MoxLDL in HAEC
thus helping us reveal the key players that are responsible for the
phenotype that is being uncovered after subjecting the cells to
physiological levels of MPO-modified LDL.
Acknowledgements
The authors would like to thank Dr Marwan El-Sabban
(American University of Beirut, Beirut, Lebanon) for his valuable
help and contribution to the study by providing HAEC and BAE
cell.
Funding
This present study was funded by the National
Council for Scientific Research in Lebanon CNRS-L and the
University of Balamand (grant no. 1849-18).
Availability of data and materials
Previously reported [expression of Selected
Fibrinolytic Genes in HAEC in Response to nLDL or MoxLDL Treatment]
data were used to support this study and are available at [doi:
10.1155/2014/134635-doi: 10.1371/journal.pone.0038810]. These prior
studies (and datasets) are cited at relevant places within the text
as references [Daher et al, 2014-Zouaoui Boudjeltia et
al, 2012].
Authors' contributions
LV, KB, MK and JD conceived the current study and
designed the experiments. GS and JD wrote the manuscript. JD and SB
edited the manuscript. GS and SB performed data analysis.
Ethics approval and consent to
participate
The CHU Charleroi Hospital Ethics Committee (Comité
d'Ethique I.S.P.PC: OM008) has approved blood sampling and has
specifically approved this study. The studies conform to the
principles outlined in the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3:e4422006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the global
burden of disease study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Virchow R: Gesammelte Abhandlungen zur
Wissenschaftlichen Medicin (Collected treatises on scientific
medicine)Meidinger Sohn & Comp.; 1856
|
|
4
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis (*). Ann Rev Immunol.
27:165–197. 2009. View Article : Google Scholar
|
|
5
|
Steinberg D, Parthasarathy S, Carew TE,
Khoo JC and Witztum JL: Beyond cholesterol. Modifications of
low-density lipoprotein that increase its atherogenicity. N Engl J
Med. 320:915–924. 1989.PubMed/NCBI
|
|
6
|
Hansson GK: Regulation of immune
mechanisms in atherosclerosis. Ann N Y Acad Sci. 947:157–166. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Daugherty A, Dunn JL, Rateri DL and
Heinecke JW: Myeloperoxidase, a catalyst for lipoprotein oxidation,
is expressed in human atherosclerotic lesions. J Clin Invest.
94:437–444. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Delporte C, Boudjeltia KZ, Noyon C,
Furtmuller PG, Nuyens V, Slomianny MC, Madhoun P, Desmet JM, Raynal
P, Dufour D, et al: Impact of myeloperoxidase-LDL interactions on
enzyme activity and subsequent posttranslational oxidative
modifications of apoB-100. J Lipid Res. 55:747–757. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hazell LJ, Baernthaler G and Stocker R:
Correlation between intima-to-media ratio, apolipoprotein B-100,
myeloperoxidase and hypochlorite-oxidized proteins in human
atherosclerosis. Free Radic Biol Med. 31:1254–1262. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang R, Brennan ML, Fu X, Aviles RJ,
Pearce GL, Penn MS, Topol EJ, Sprecher DL and Hazen SL: Association
between myeloperoxidase levels and risk of coronary artery disease.
JAMA. 286:2136–2142. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wobst J, Kessler T, Dang TA, Erdmann J and
Schunkert H: Role of sGC-dependent NO signalling and myocardial
infarction risk. J Mol Med (Berl). 93:383–394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cesarman-Maus G and Hajjar KA: Molecular
mechanisms of fibrinolysis. Br J Haematol. 129:307–321. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Delporte C, Van Antwerpen P, Vanhamme L,
Roumeguere T and Zouaoui Boudjeltia K: Low-density lipoprotein
modified by myeloperoxidase in inflammatory pathways and clinical
studies. Mediators Inflamm. 2013:9715792013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zouaoui Boudjeltia K, Daher J, Van
Antwerpen P, Moguilevsky N, Delree P, Ducobu J, Raes M, Badran B,
Vanhaeverbeek M, Brohee D, et al: Exposure of endothelial cells to
physiological levels of myeloperoxidase-modified LDL delays
pericellular fibrinolysis. PLoS One. 7:e388102012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
El Samad G: Effect of myeloperoxidase
modified LDL on bovine and human aortic endothelial cells
(unpublished PhD thesis)University of Balamand; El Koura: 2018
|
|
16
|
Colon S, Page-McCaw P and Bhave G: Role of
hypohalous acids in basement membrane homeostasis. Antioxid Redox
Signal. 27:839–854. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eisenberg T, Carmona-Gutierrez D, Büttner
S, Tavernarakis N and Madeo F: Necrosis in yeast. Apoptosis.
15:257–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ocampo A and Barrientos A: Quick and
reliable assessment of chronological life span in yeast cell
populations by flow cytometry. Mech Ageing Dev. 132:315–323. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deere D, Shen J, Vesey G, Bell P,
Bissinger P and Veal D: Flow cytometry and cell sorting for yeast
viability assessment and cell selection. Yeast. 14:147–160. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chapin JC and Hajjar KA: Fibrinolysis and
the control of blood coagulation. Blood Rev. 29:17–24. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eruslanov E and Kusmartsev S:
Identification of ROS using oxidized DCFDA and flow-cytometry.
Methods Mol Biol. 594:57–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barter P: Lipoprotein metabolism and CKD:
Overview. Clin Exp Nephrol. 18:243–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sugiyama S, Okada Y, Sukhova GK, Virmani
R, Heinecke JW and Libby P: Macrophage myeloperoxidase regulation
by granulocyte macrophage colony-stimulating factor in human
atherosclerosis and implications in acute coronary syndromes. Am J
Pathol. 158:879–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Steffen Y, Schewe T and Sies H:
Epicatechin protects endothelial cells against oxidized LDL and
maintains NO synthase. Biochem Biophys Res Commun. 331:1277–1283.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Daher J, Martin M, Rousseau A, Nuyens V,
Fayyad-Kazan H, Van Antwerpen P, Courbebaisse G, Martiat P, Badran
B, Dequiedt F, et al: Myeloperoxidase oxidized LDL interferes with
endothelial cell motility through miR-22 and heme oxygenase 1
induction: Possible involvement in reendothelialization of vascular
injuries. Mediators Inflamm. 2014:1346352014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen XP, Xun KL, Wu Q, Zhang TT, Shi JS
and Du GH: Oxidized low density lipoprotein receptor-1 mediates
oxidized low density lipoprotein-induced apoptosis in human
umbilical vein endothelial cells: Role of reactive oxygen species.
Vascul Pharmacol. 47:1–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sawamura T, Kume N, Aoyama T, Moriwaki H,
Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T and Masaki
T: An endothelial receptor for oxidized low-density lipoprotein.
Nature. 386:73–77. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Neri Serneri GG, Coppo M, Bandinelli M,
Paoletti P, Toscano T, Micalizzi E, Chiostri M and Boddi M:
Exaggerated myocardial oxLDL amount and LOX-1 receptor
over-expression associated with coronary microvessel inflammation
in unstable angina. Atherosclerosis. 226:476–482. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meuwese MC, Stroes ES, Hazen SL, van Miert
JN, Kuivenhoven JA, Schaub RG, Wareham NJ, Luben R, Kastelein JJ,
Khaw KT and Boekholdt SM: Serum myeloperoxidase levels are
associated with the future risk of coronary artery disease in
apparently healthy individuals: The EPIC-Norfolk prospective
population study. J Am Coll Cardiol. 50:159–165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sueishi K, Ichikawa K, Kato K, Nakagawa K
and Chen YX: Atherosclerosis: Coagulation and fibrinolysis. Semin
Thromb Hemost. 24:255–260. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qi J, Goralnick S and Kreutzer DL: Fibrin
regulation of interleukin-8 gene expression in human vascular
endothelial cells. Blood. 90:3595–3602. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ahn K, Pan S, Beningo K and Hupe D: A
permanent human cell line (EA.hy926) preserves the characteristics
of endothelin converting enzyme from primary human umbilical vein
endothelial cells. Life Sci. 56:2331–2341. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koch M and Zernecke A: The hemostatic
system as a regulator of inflammation in atherosclerosis. IUBMB
Life. 66:735–744. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Romagnuolo R, Marcovina SM, Boffa MB and
Koschinsky ML: Inhibition of plasminogen activation by apo(a): Role
of carboxyl-terminal lysines and identification of inhibitory
domains in apo(a). J Lipid Res. 55:625–634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aoyama T, Fujiwara H, Masaki T and
Sawamura T: Induction of lectin-like oxidized LDL receptor by
oxidized LDL and lysophosphatidylcholine in cultured endothelial
cells. J Mol Cell Cardiol. 31:2101–2114. 1999. View Article : Google Scholar : PubMed/NCBI
|