Introduction
Despite clinical progress in the past few decades,
acute kidney injury (AKI) remains to be major medical concern. Each
year, ~13.3 million are afflicted with AKI worldwide (1). In general, AKI is temporary disease and
does not usually result in mortality directly; however, this injury
is associated with high morbidity and mortality, with ~1.7 million
deaths each year worldwide (1,2). In
developed countries, AKI is mainly observed in elderly patients
within the intensive care unit, whereas in lower- and middle-income
countries young adults are likely to be affected by AKI and be at
risk of mortality (3–5). One-third of individuals that experience
AKI may develop chronic kidney disease and end-stage renal disease,
drastically reducing the quality of life and incurring high
long-term medical costs (6).
Despite the health problems that are caused by AKI,
effective therapeutic strategies other than dialysis are not yet
available to increase the survival rate, reduce injury or
accelerate recovery (7). Potential
tubular, vascular and inflammatory processes have been reported to
be associated with AKI pathogenesis (8). Among these factors, inflammation is
generally believed to serve a predominant role in the
pathophysiology of AKI, especially in septic AKI (7–9).
Therefore, developing novel strategies to reduce inflammation at
injured sites may represent a feasible and practical way to treat
or prevent septic AKI, and has previously been investigated on
murine models with promising results (9,10). By
using a lipopolysaccharide (LPS)-induced septic AKI mouse as a
model, Hu et al (11)
demonstrated that the systemic delivery of a plasmid expressing an
immunosuppressive cytokine interleukin-35 (IL-35) effectively
prevented LPS-induced AKI by inhibiting NF-κB activation.
Additionally, another study has revealed that the inhibition of
leukocyte infiltration into the kidneys could reduce renal injury
and protect renal function (12),
whilst the activation of the cholinergic anti-inflammatory pathway,
which is mediated by α7-nicotinic acetylcholine receptor on
CD4+ T cells, has also been demonstrated to exhibit
renal protection (13).
As a key component of the innate immune response,
Toll-like receptors (TLRs) serve to recognize pathogen-associated
molecular patterns (PAMPs) that are present on pathogens, and
initiate the innate immune response by producing inflammatory
cytokines (14,15). In particular, previous studies have
shown that TLR2 could be activated in mice using LPS stimulation,
which contribute to the development of septic AKI by enhancing
inflammatory cytokine production via the NF-κB signaling pathway
(14–19). Histological evaluation has
demonstrated that TLR2 overactivation in AKI is mainly identified
in podocytes, which may be indicative of the important roles
podocytes serve in septic AKI pathogenesis (17).
Since preventing inflammation has been demonstrated
to be an effective approach for the treatment and prevention of
septic AKI, it was hypothesized that TLR2 inhibition may serve as a
potentially valuable target for inhibiting inflammation,
consequently reducing the risk of AKI. To assess this, the
potential protective effects of ortho-vanillin (OV), a small
molecule inhibitor against TLR2, were investigated on LPS-induced
septic AKI in vitro and in vivo. OV is an organic
solid that is present in a number of plants and has been reported
to inhibit TLR2 signaling without inducing cytotoxicity (20,21).
Therefore, the results of the present study revealed that treatment
with OV effectively alleviated LPS-induced septic AKI in a podocyte
cell line and in a mouse AKI model.
Materials and methods
Cell line culture, LPS and OV
treatment
The mouse podocyte cell line MPC5 was a kind gift
from Professor Mundel, Department of Neuro-Ophthalmology, Mount
Sinai School of Medicine and was cultured at 33°C with 5%
CO2 in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin (Sigma-Aldrich; Merck KGaA),
100 µg/ml streptomycin (Sigma-Aldrich; Merck KGaA) and 10 U/ml
recombinant mouse interferon-γ (Sigma-Aldrich; Merck KGaA). To
initiate differentiation, cells were transferred to RPMI-1640
medium without interferon-γ and cultured at 37°C with 5%
CO2 for 10 days (18).
After differentiation, podocytes were treated at 37°C, 5%
CO2 with LPS to a final concentration of 10 µg/ml for 24
h. For OV treatment at 37°C, 5% CO2, OV was added to a
final concentration of 50 µM 2 h prior to LPS treatment and
remained for the duration of the LPS treatment. Podocytes treated
with vehicle (saline solution for LPS and H2O for OV)
were used as negative controls.
Cell viability
Differentiated MPC5 cells were treated with 0, 10,
50 or 150 µM OV at 37°C and 5% CO2 for 24 h before cell
viability was assessed using a MTT assay kit (Abcam) according to
the manufacturer's protocol. Briefly, following OV treatment,
medium was removed and cells were resuspended in serum-free
RPMI-1640 medium at 1×106 cells/ml. A total of 50 µl of
the cell suspension was mixed with 50 µl MTT reagent and incubated
at 37°C for 3 h. Following incubation, MTT solvent was added and
incubated at room temperature for an additional 15 min on a shaker.
Finally, optical absorbance was read at OD590 nm and
cell viability was calculated with cells without OV treatment
considered to be 100%.
Animals and ethical statement
A total of 60 male BALB/c mice (age, 6–8-weeks;
weight, 20–22 g; Shanghai SLAC Laboratory Animal Co., Ltd.) were
used in the current study. All mice were housed in a specific
pathogen-free environment (18°C, 50% humidity and light from 04.00
to 17.00) with freely available food and water supplied. All
protocols involving animals in the present study were reviewed and
approved by the Bioethics Committee of the First People's Hospital
of Kunshan (Kunshan, China) and performed in accordance with the
guidelines of the Laboratory Animal Science Association (IRB
approval no. FPHKA201512012).
Mouse OV treatment and AKI
induction
OV treatment was performed as previously described
(21). Briefly, mice were
administered with two intraperitoneal (i.p.) doses of OV 1 h apart.
At 1 h after the second OV injection, LPS was injected to induce
septic AKI as previously described (17). Animals were injected with LPS (10
mg/kg; cat. no. L2880; Sigma-Aldrich; Merck KGaA) i.p. The mice
were sacrificed 24 h post-injection, after which serum samples were
collected for renal function assessment by centrifugation at 10,000
× g for 10 min at 4°C and kidney samples were processed for
subsequent analysis. For ELISA, kidney tissue samples were
homogenized in RIPA buffer (Thermo Fisher Scientific, Inc.)
supplemented with protease inhibitor cocktail (Roche Diagnostics)
on ice, and then centrifuged at 10,000 × g for 10 min at 4°C to
pellet cell debris. The resulting supernatants were collected and
adjusted to a concentration of 1 mg/ml [concentrations were
determined using a NanoDrop™ One (Thermo Fisher Scientific, Inc.)],
and stored at −80°C until use. For histological analysis, kidney
samples were fixed with 4% paraformaldehyde for 16 h at room
temperature, embedded in paraffin and cut into 4 µm sections and
stored at room temperature until use.
ELISA
IL-6, TNF-α and IL-1β levels in cell culture
supernatants and murine kidney tissue samples were measured using
ELISA. For cell culture supernatants, cell debris was cleared by
centrifugation (400 × g, 5 min at 4°C). Tissue samples were
homogenized as described above. ELISA kits for IL-6 (cat. no.
5017218), TNF-α (cat. no. 5017331) and IL-1β (cat. no. 501129749)
were purchased from eBioscience; Thermo Fisher Scientific, Inc. and
used according to the manufacturer's protocols.
Co-immunoprecipitation (co-IP) and
western blot analysis
MPC5 cells or mouse kidney tissue samples were lysed
or homogenized in RIPA buffer (Thermo Fisher Scientific, Inc.)
supplemented with protease inhibitor cocktail (Roche Diagnostics)
and then centrifuged at 10,000 × g for 10 min at 4°C to remove cell
debris and tissue clumps. Cleared supernatants were either used for
co-IP or directly for western blot analysis. Co-IP was performed
using a Dynabeads™ co-immunoprecipitation kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. In
brief, rabbit anti-mouse MyD88 antibody (5 µg; cat. no. 4283; Cell
Signaling Technology, Inc.) or anti-TLR2 antibody (5 µg; cat. no.
AF1530; R&D systems, Inc.)-coupled Dynabeads were incubated
with cleared cell lysis supernatants overnight at 4°C. After washes
with wash buffer, proteins bound to the beads were eluted using
elution buffer. Vehicle treated cells were used as control.
Protein concentrations were determined using a
Bicinchoninic Acid assay (Thermo Fisher Scientific, Inc.). For
western blot analysis, lysed cell supernatants, homogenized tissue
supernatants or Co-IP samples were first resolved using 12%
SDS-PAGE at 20 µg per lane and then transferred to PVDF membranes
(Millipore; Merck KGaA). After blocking with 5% non-fat milk for 1
h at room temperature, the membranes were then sequentially
incubated with primary antibodies overnight at 4°C and with
HRP-conjugated secondary antibodies for 1 h at room temperature.
The membranes were then extensively washed with PBST and the
immune-bands were visualized using an ECL substrate (Millipore;
Merck KGaA) under a CCD camera (Bio-Rad Laboratories, Inc.).
Gray-scale analysis of the western blot bands was performed using
Image J (version 2; National Institutes of Health) (22). The following primary antibodies were
used in the current study (all at 1:1,000 dilution): Goat
anti-mouse TLR2 (cat. no. AF1530; R&D systems, Inc.), goat
anti-mouse GAPDH (cat. no. sc-48166; Santa Cruz Biotechnology,
Inc.), rabbit anti-mouse MyD88 (cat. no. 4283; Cell Signaling
Technology, Inc.), rabbit anti-mouse p65 (cat. no. 8242; Cell
Signaling Technology, Inc.) and rabbit anti-mouse phosphorylated
(p)-p65 (cat. no. 3033; Cell Signaling Technology, Inc.).
Horseradish peroxidase (HRP)-conjugated rabbit anti-goat IgG (H+L)
(cat. no. SA00001-4) and HRP-conjugated goat anti-rabbit IgG (H+L)
(cat. no. SA00001-2) secondary antibodies were purchased from
Proteintech Group, Inc.
Histology evaluation
Histological evaluation of kidney samples was
performed as previously described (17,18).
Briefly, kidney samples were fixed with 4% paraformaldehyde for 16
h at room temperature, embedded in paraffin and cut into 4 µm
sections. Slides were then de-waxed, rehydrated in descending
alcohol series (100, 95, 70 and 50%) and stained with periodic acid
Schiff's reagent (PAS). For PAS staining, slides were sequentially
stained with periodic acid solution and Schiff's reagent for 5 and
15 min respectively, at room temperature. Stained slides were
visualized under a light microscope and images were captured at
×200 magnification using an NiU upright microscope coupled with a
DS-Fi3 camera (Nikon Corporation). A total of 10 kidney sections
per mouse were analyzed and two independent experienced
investigators randomly evaluated 20–30 glomeruli per kidney section
in a blinded manner. NIS-Element D imaging software (version
4.30.00; Nikon Corporation) was used for image capture. Kidney
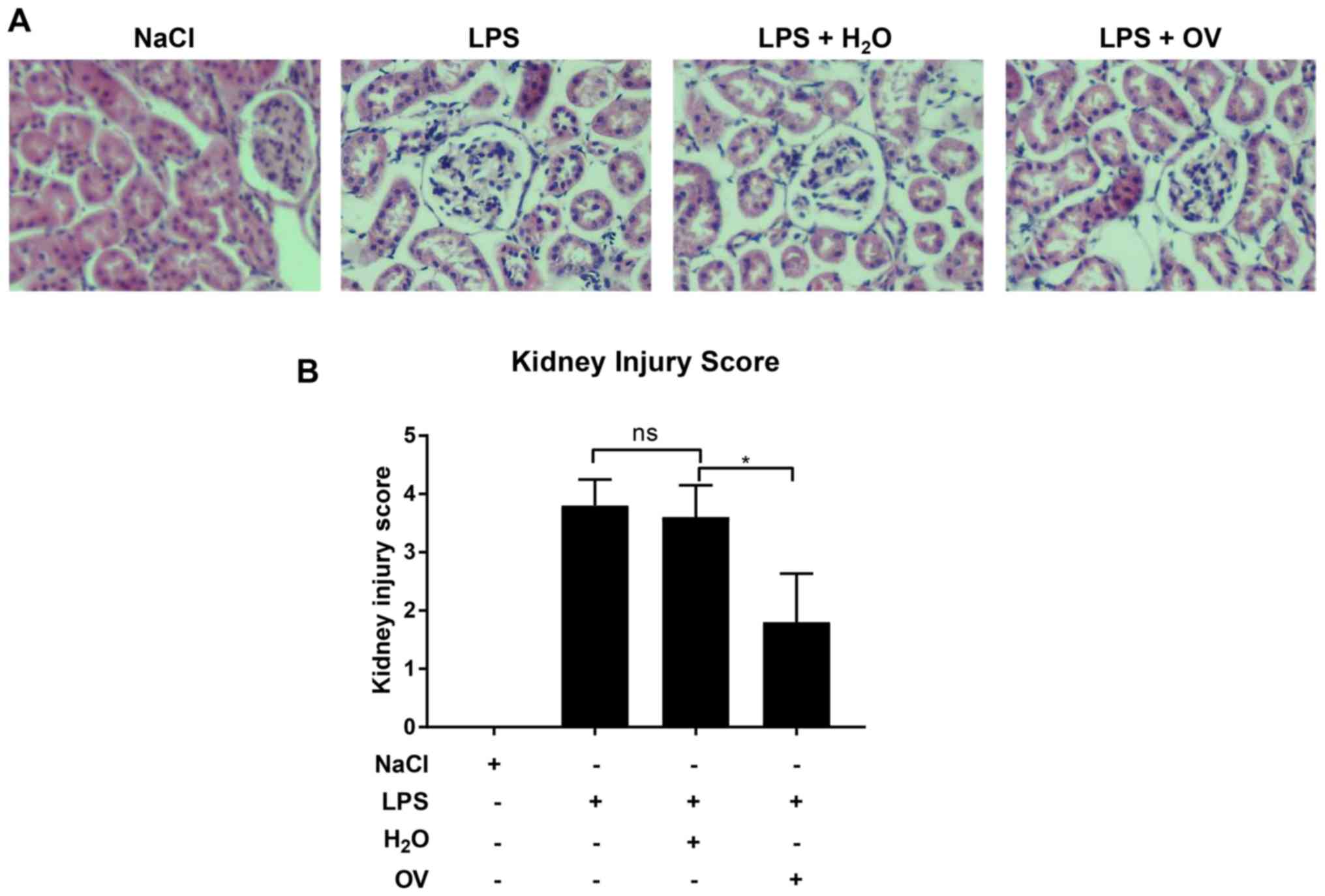
injury was assessed using a previously described criteria (23), which classifies the kidney injury
severity into a 0–4 scoring system: No injury, Score 0; <10%
injury, score 1; 10–25% injury, score 2; 25–75% injury, score 3;
and >75% injury, score 4.
Measurement of Blood Urea Nitrogen
(BUN) and Serum Creatinine (SCr)
At the time of mice sacrifice, blood samples were
collected and kept at room temperature undisturbed for 30 min to
enable blood clotting. Subsequently, blood samples were centrifuged
at 2,000 × g for 10 min at 4°C and sera (supernatants) were
subsequently collected. BUN and SCr concentrations in murine sera
were determined using a Hitachi 7060 automated Chemistry Analyzer
(Diamond Diagnostics, Inc.) according to the manufacturer's
protocols.
Statistical analysis
The data are expressed as mean ± standard deviation.
Student's t-test was applied for statistical comparisons between
two groups while a One-way ANOVA followed by Student-Newman-Keuls
post hoc test was used for comparisons between three or more
groups. P<0.05 was considered to indicate a statistically
significant difference. All analyses were performed using Prism
7.03 (GraphPad Software, Inc.).
Results
OV inhibits TLR2 signaling and
pro-inflammatory cytokine production in murine podocytes
Since TLR2 was previously found to be overexpressed
and activated in podocytes in septic AKI mouse (18), the potential anti-inflammation
effects of OV were first investigated on the murine podocyte cell
line MPC5. Cell cytotoxicity of OV was first measured by treating
MPC5 cells with increasing doses (0, 10, 50 and 100 µM) of OV for
24 h. No apparent cell cytotoxicity was observed within the dose
range tested (10–150 µM; Fig. 1A).
As OV at all tested doses showed similar results, a moderate dose
within the tested range was selected (final concentration of 50 µM)
for the subsequent experiments on cell lines.
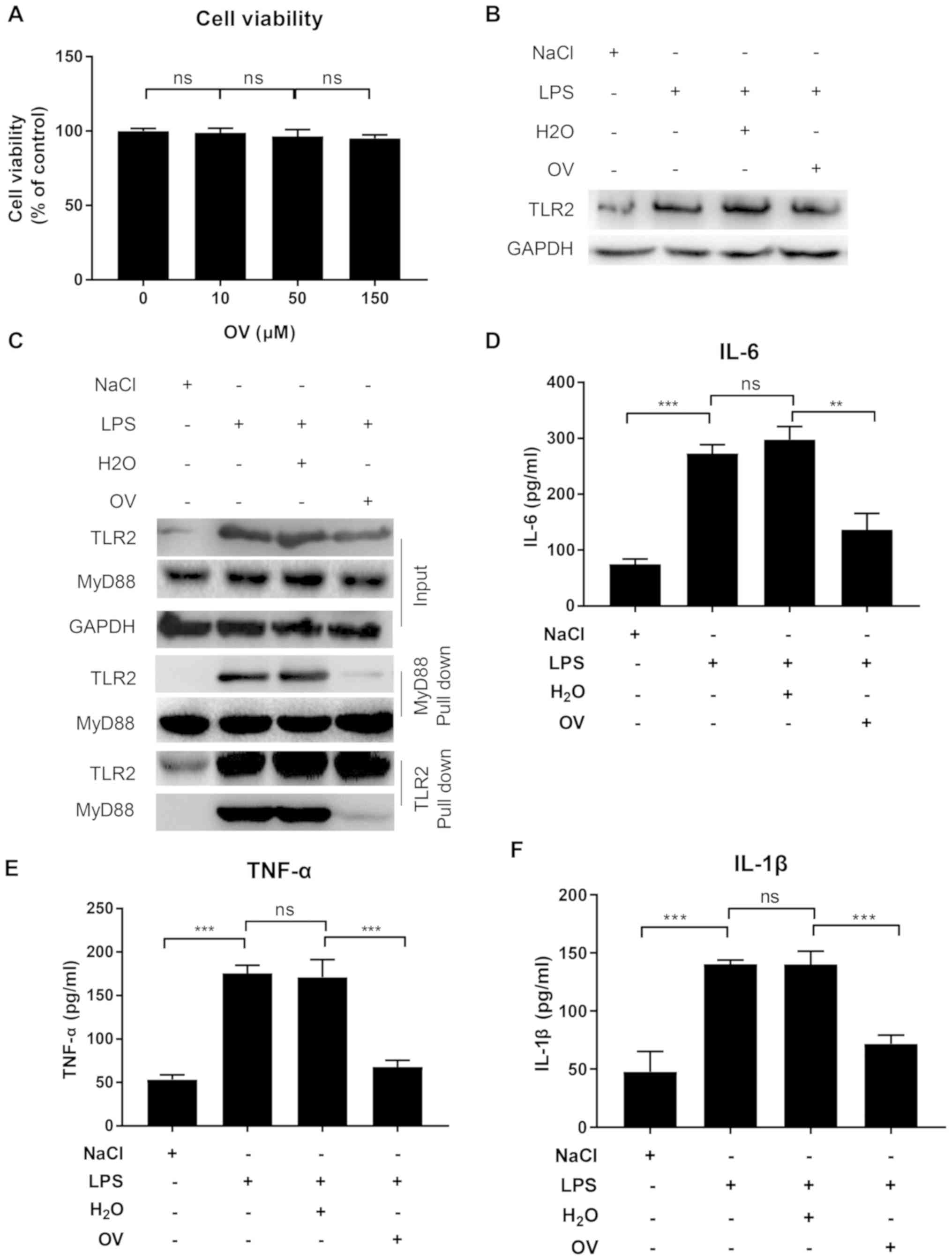 | Figure 1.OV inhibits TLR2 signaling in the
murine podocyte cell line MPC5. (A) OV cytotoxicity assay. MPC5
cells were treated with increasing doses of OV for 24 h, following
which an MTT assay was performed to assess the cytotoxicity of OV.
Data are presented as the mean ± SD from three independent
experiments. MPC5 cells were pretreated with or without OV, and
then treated with vehicle control or stimulated with LPS for 24 h.
After stimulation, (B) TLR2 expression was determined using western
blotting. Data shown are representative of three independent
experiments. (C) TLR2-MyD88 interaction was assessed using
co-immunoprecipitation assay. Data shown are one representative of
three independent experiments. (D) IL-6, (E) TNF-α and (F) IL-1β
expression in cell culture supernatant as measured using ELISA.
Data are presented as the mean ± SD from three independent
experiments. ns, not statistically significant; **P<0.01 and
***P<0.001. OV, ortho-vanillin; LPS, lipopolysaccharide; TLR2,
toll-like receptor 2; MyD88, myeloid differentiation primary
response 88; IL, interleukin; TNF-α, tumour necrosis factor-α; SD,
standard deviation. |
The impact of OV treatment on TLR2 expression was
subsequently determined. MPC5 cells pretreated with or without OV
were stimulated with LPS for 24 h, and then TLR2 expression was
determined using western blot analysis. In accordance with previous
findings, LPS treatment markedly increased TLR2 expression, but
treatment with OV did not result in any changes to LPS-induced TLR2
expression (Fig. 1B) (18).
TLR2 is activated upon recognition of PAMPs, which
bind to the intracellular adaptor protein MyD88 to activate a
number of signaling pathways that lead to the expression of
pro-inflammatory cytokines (24). To
investigate if OV could inhibit TLR2 signaling by interfering with
TLR2-MyD88 interaction, a Co-IP assay was performed with lysates
from MPC5 cells with or without OV treatment and LPS stimulation
using an anti-MyD88 antibody. TLR2 was not associated with MyD88 in
the absence of LPS stimulation, but TLR2-My88 complexes were
detected upon LPS stimulation (Fig.
1C). Of note, when cells were pretreated with OV, the
TLR2-MyD88 association was considerably reduced, suggesting an
inhibition of TLR2-MyD88 interaction following OV treatment
(Fig. 1C). To further confirm the
findings, an additional Co-IP with anti-TLR2 antibody was
performed, and the results were consistent with the anti-MyD88
Co-IP. Namely, LPS stimulation induced TLR2-MyD88 interaction,
while such interaction was inhibited by OV treatment (Fig. 1C).
To determine whether the interrupted TLR2-MyD88
interaction induced by OV treatment resulted in reduced
inflammation downstream, the supernatants of MPC5 cells with or
without OV treatment and LPS stimulation were tested for IL-6,
TNF-α and IL-1β expression, which are pro-inflammatory cytokines
that have been previously reported to serve important roles in AKI
pathogenesis (25–27). The ELISA results revealed that when
compared with vehicle control, LPS stimulation significantly
increased the release of IL-6, TNF-α and IL-1β, which was inhibited
by OV treatment (Fig. 1D-F).
In conclusion, these results demonstrated that OV
exhibited no cell toxicity and treatment of MPC5 cells with OV
reduced inflammatory cytokine production by inhibiting the
TLR2-MyD88 interaction in podocytes.
OV treatment reduces LPS-induced AKI
in mice
The renal protective effects of OV were determined
using an LPS-induced AKI mouse model. BALB/c mice were first
pretreated with OV before being challenged with LPS, and renal
function was subsequently assessed using staining. A period of 24 h
after LPS challenge, severe renal pathological lesions, including
glomeruli abnormality in morphology, loss of brush border with
notable inflammatory cell infiltration were observed in renal
tissues (Fig. 2A and B). In
contrast, significantly attenuated injury score was identified in
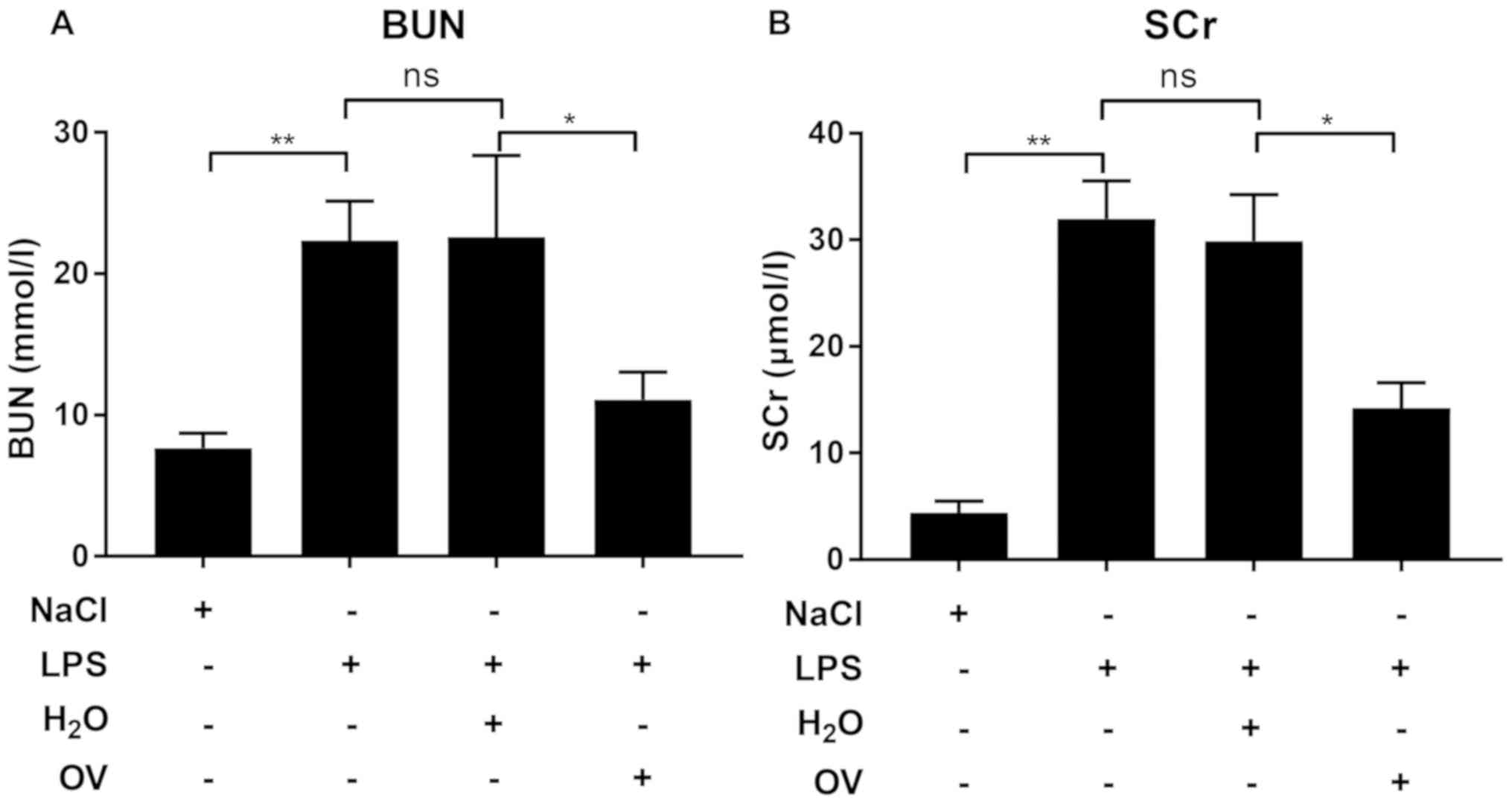
kidney tissues isolated from mice pre-treated with OV. Measurements
of BUN and SCr, which are two biomarkers for septic AKI, were also
consistent with the histological evaluation, in that LPS challenge
increased BUN and SCr levels in the blood when compared with
vehicle control, an effect that was significantly reduced by OV
treatment (Fig. 3A and B). Taken
together, these data demonstrated that OV conferred protective
properties against LPS-induced AKI.
OV treatment reduces proinflammatory
cytokine production in LPS-induced AKI mice
The hypothesis that OV treatment also inhibited
inflammatory responses that are induced by LPS challenge was
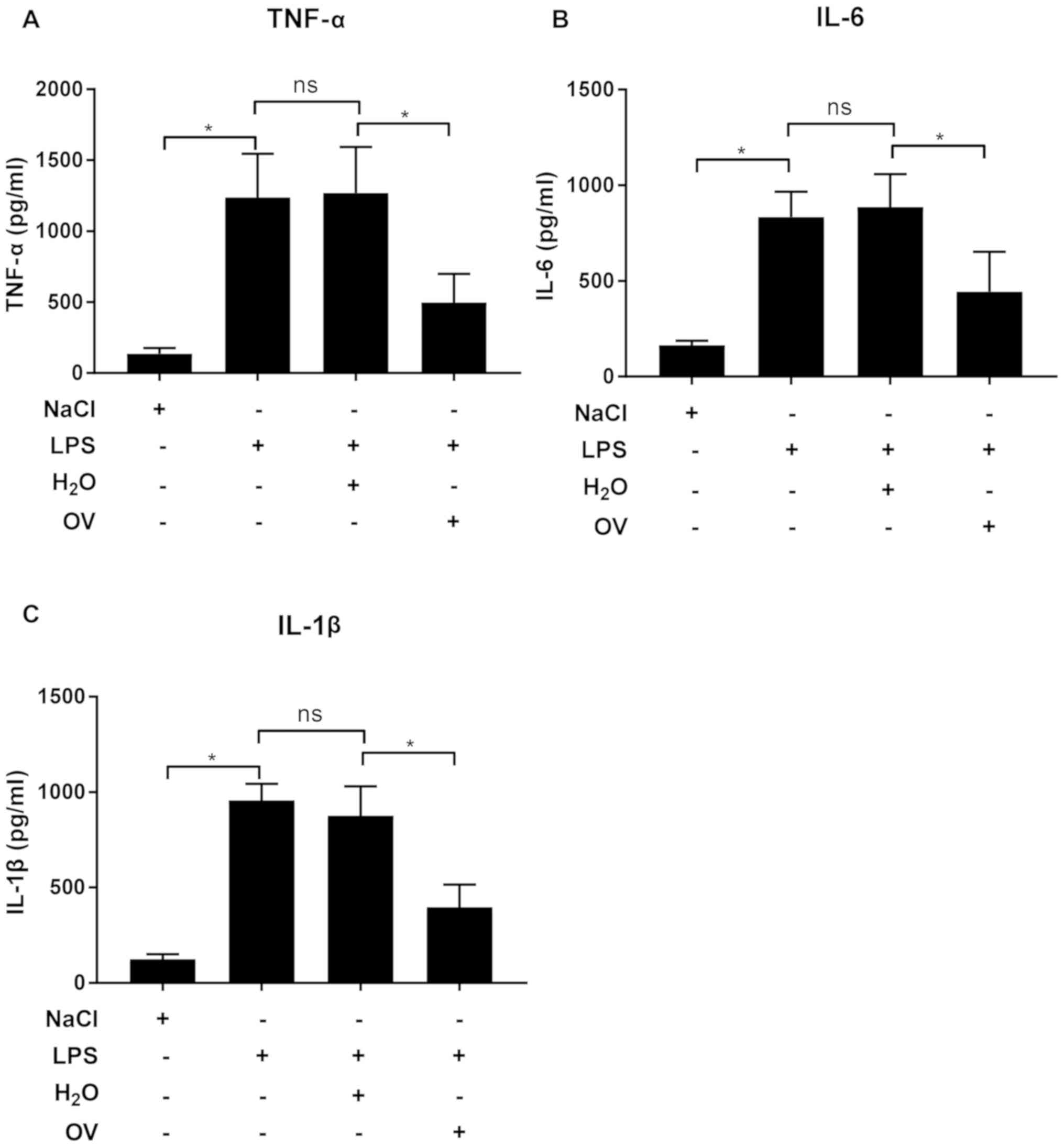
subsequently investigated using ELISA. The levels of
pro-inflammatory cytokines TNF-α, IL-6 and IL-1β in kidney tissue
extracts were significantly increased following LPS challenge,
which was prevented by OV treatment (Fig. 4A-C). These data indicated that OV
treatment could alleviate renal damage by inhibiting the expression
of pro-inflammatory cytokines.
OV treatment inhibits the NF-kB
signaling pathway in LPS-induced AKI mice
Previous studies have revealed that TLR2 induces
inflammation by activating the NF-κB signaling pathway in
LPS-treated AKI mice (17,18). Since OV is a small molecule inhibitor
against TLR2 and treatment with this inhibitor has been previously
demonstrated to reduce TLR2 activation and downstream inflammatory
cytokine production, it was hypothesized that the NF-κB signaling
pathway may also be inhibited by OV. To test this, TLR2 expression
was measured in kidney tissues. In similarity with the data
obtained from MPC5 cells, LPS challenge significantly increased
TLR2 expression, compared with that in vehicle control, but this
was not alleviated by treatment with OV (Fig. 5A and C). The expression of p65 and
p-p65 were then measured in kidney tissues. In comparison with
vehicle control, LPS challenge significantly upregulated the
expression of p65 and p-p65 (Fig. 5B and
D). OV treatment did not induce the expression of p65 but
significantly inhibited the phosphorylation of this protein,
suggesting that OV inhibited the activation of the NF-κB signaling
pathway (Fig. 5B and D). Total p65
levels were unchanged by treatment with OV. Under some
circumstances, the upregulation of p65 expression together with
phosphorylation have been previously observed (28,29).
These findings support the results of the current study, which
determines the expression of p65 was upregulated in the kidney upon
LPS stimulation.
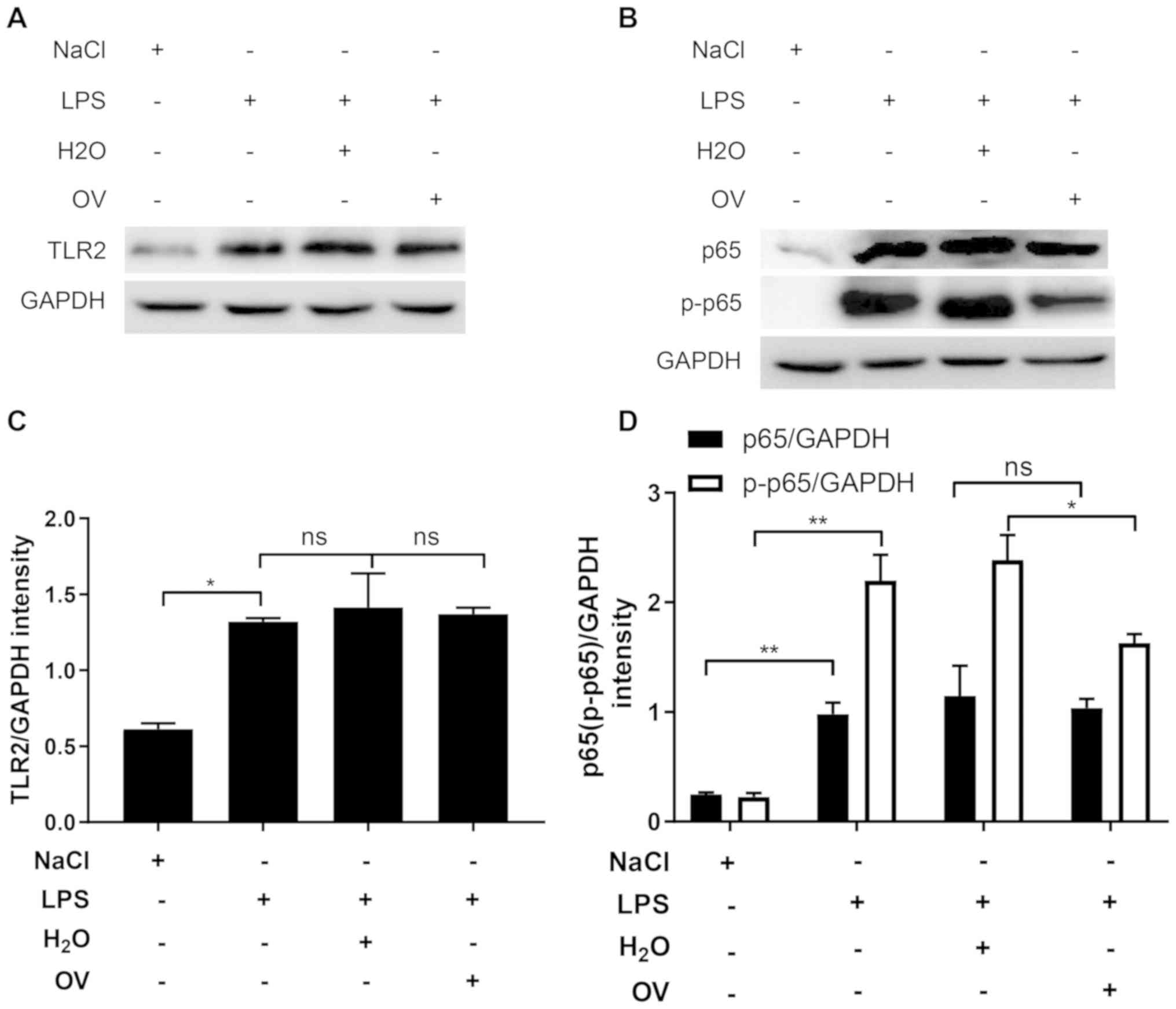 | Figure 5.OV treatment inhibits NF-κB signaling
activation in LPS-induced AKI mice. Mice were pretreated with OV
and then challenged with LPS and were sacrificed 24 h later where
kidney samples were harvested and homogenized. (A) TLR2, (B) p65
and p-p65 in the kidney tissue lysates were quantified using
western blot analysis. One representative image from three is
shown. (C) Quantified densitometric values of TLR2, (D) p65 and
p-p65 performed using ImageJ. Data are presented as the mean ±
standard deviation (n=5 mice/group) of three independent
experiments. *P<0.05 and **P<0.01. ns, not statistically
significant; AKI, acute kidney injury; OV, ortho-vanillin; LPS,
lipopolysaccharide; IL, interleukin; TNF-α, tumour necrosis
factor-α; TLR2, toll-like receptor 2; p65, NF-κΒ p65 subunit;
p-p65, phosphorylated p65. |
Taken together, the results of the present study
revealed that OV inhibited the activation of the TLR2 signaling
pathway and subsequent inflammatory cytokine production both in
vitro and in vivo. Furthermore, treatment with OV can
effectively protect against LPS-induced AKI via inhibiting
TLR2/NF-κB signaling pathway, representing a novel therapeutic
approach for the treatment and possible prevention of AKI.
Discussion
As one of the most severe complications in
hospitalized patients, AKI is a multi-factorial disease that is
associated with a rapid loss of renal function. AKIs are
categorized into 3 groups: Pre-renal (reduced blood supply into the
kidney), renal (kidney tissue damage, including sepsis and
accident) and post-renal (urine retention in kidney). Among these
different AKIs, ~50% of the cases are septic AKI (30). Excessive inflammation is generally
considered to be a key factor in triggering and aggravating AKI,
whilst AKI can in turn aggravate inflammation further, resulting in
exacerbated renal dysfunction and tissue damage (31). Given the roles of inflammation in AKI
initiation and progression, preventing inflammation represents a
promising therapeutic strategy for use in AKI (32).
As a general mechanism to combat harmful pathogens
and remove damaged cells, inflammation can be triggered by a number
of factors, and distinctive pathways can lead to the expression of
effectors such as pro-inflammatory cytokines (33). The initiation of inflammation is
usually mediated by immune cells that reside in the tissue, which
can recognize PAMPs and damage-associated molecular patterns
(DAMPs) through their surface pattern recognition receptors (PPRs).
Upon recognizing PAMPs or DAMPs, PPRs are activated and release
inflammatory mediators through highly regulated signaling pathways.
Since inflammation is a multi-factorial and highly regulated
cascade response, anti-inflammation strategies can be developed to
target each of these factors and/or steps, including the
elimination of harmful stimuli, inhibition of pro-inflammation
pathways, neutralization of pro-inflammatory mediators and
enhancement of anti-inflammatory mediators. LPS is a substance that
can cause septic AKI, where the infusion of alkaline phosphatase
dephosphorylates LPS to the nontoxic, monophosphorylated form of
LPS has prevented renal damage in patients with AKI (34,35).
Elevated levels of pro-inflammatory cytokines, including TNF-α and
IL-1β, are potent mediators of immunopathological responses during
LPS-induced septic AKI, such that the neutralization of TNF-α has
been previously demonstrated to reduce both mortality and renal
failure in animal studies (36–38). In
another study, injection of an immunosuppressive cytokine IL-35 has
been demonstrated to reduce renal damage in a LPS-induced AKI mouse
model (11). Previous studies
performed in the Laboratory of the Intensive Care Unit, The First
People's Hospital of Kunshan, Jiangsu, China have demonstrated that
TLR2 is excessively activated in LPS-induced AKI, and triggers the
production of pro-inflammatory cytokines, subsequently leading to
aggravated renal damage. This finding gave rise to the hypothesis
that targeting TLR2 may be a promising approach for septic AKI
prevention or treatment (17,18).
Therefore, in the present study, the potential protective effects
of a TLR2 small molecule inhibitor OV was investigated in a
LPS-induced AKI mouse model. OV treatment significantly alleviated
renal damage in LPS-induced AKI mice, and this protective effect of
OV appeared to be the result of an inhibition of TLR2/NF-κB
signaling that was induced by OV. Of note, despite the apparent
success achieved by the various anti-inflammation strategies, a
single approach does not appear to be sufficient to fully prevent
or treat AKI (5,7). Although it is beyond the scope of the
present study, it would potentially be useful to assess the
protective efficacies of combined therapies in AKI.
In the present study, OV was used as the TLR2
inhibitor to block TLR2-mediated inflammation in LPS-induced AKI
mice for a number of reasons. OV is an organic compound present in
many plants and has been demonstrated to exhibit no cytotoxicity in
numerous studies so far, making this inhibitor a suitable candidate
with great potential for drug development (21,39). In
particular, OV has been proven to specifically inhibit mouse and
human TLR2 signaling, which can facilitate the translation of
research from animal to a more clinical setting (21). The mechanism in which OV inhibits
TLR2 activity has been elucidated. TLR2 signaling is initiated by
dimerization of intracellular Toll/IL-1 receptor resistance (TIR)
domains, where the resultant downstream signaling relies on the
binding of MyD88 to TIR domains. OV can bind in the BB loop pocket
of TIR domains, which inhibits TLR2 trimerization and MyD88 binding
(21). However, other measures to
inhibit TLR2 activity may still need further investigation to
optimize strategies for septic AKI prevention and/or treatment. A
number of studies have been previously performed that investigate a
number of different TLR2 inhibition strategies for the treatment of
diseases that may also be applicable to septic AKI. In the
treatment of acute gut inflammation, Shmuel-Galia et al
(40) indicated that inhibiting TLR2
dimerization by a TLR2 transmembrane peptide significantly reduced
monocyte activation and pro-inflammatory cytokine production. In
human cytomegalovirus infection, the virus-derived
microRNA-UL112-3p could effectively inhibit the activation of the
TLR2/NF-κB signaling pathway by targeting TLR2 (41). One structural study has also revealed
that staphylococcal superantigen-like protein 3, which is secreted
by staphylococcus aureus, can antagonize TLR2 by inhibiting
its ligand binding and subsequent receptor dimerization (42). Although each disease has its own
characteristics and requires unique treatment formulations, it
would still be warranted to investigate if the aforementioned TLR2
inhibition strategies could also apply to the prevention and/or
treatment of septic AKI.
In terms of the dose used, a previous study has
determined that OV administrated at a dose of 1.314 mM/g twice i.p.
exhibited good TLR2 inhibition in mice (21). As a proof-of-concept study, this
administration route and dose for OV was adopted in septic AKI
prevention in the present study. However, to determine the
protective effects of OV, a systematic evaluation of different
administration routes and doses is required. In addition, as with
many other studies in septic AKI treatment, only the preventative
effects of OV and not effects on AKI post-injury were examined.
Although this is beyond the scope of the present study, it would be
interesting to investigate whether OV could also have exhibit
effects following AKI injury.
In summary, the present study revealed that
targeting TLR2 signaling by treatment with OV could effectively
alleviate LPS-induced AKI in vitro and in vivo, by
inhibiting the TLR2/NF-κB signaling pathway. Although further
characterization is required, OV, and the general targeting of
TLR2, represents a promising target for the prevention of septic
AKI.
Acknowledgements
Not applicable.
Funding
This work was supported by the Clinical Development
Project of Jiangsu University (grant no. JLY20160060), Kunshan
Science and Technology Program for Social Development (grant no.
0012018ZX03), the Fundamental and Clinical Research Team for Brain
Disease Study in Kunshan Hospital Affiliated to Jiangsu University
(grant no. KYC004) and the Kunshan Science and Technology Planning
Project (grant no. KS18060).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YP and SL designed the study. YP, LL, YW, JY, FJ,
TT, HY performed the experiments. YP, LL, HY and LS analyzed the
data. YP and SL wrote the manuscript. All the authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All protocols involving animals in the present study
were reviewed and approved by the Bioethics Committee of the First
People's Hospital of Kunshan (Kunshan, China) and performed in
accordance with the guidelines of the Laboratory Animal Science
Association (IRB approval no. FPHKA201512012).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mehta RL, Cerda J, Burdmann EA, Tonelli M,
García-García G, Jha V, Susantitaphong P, Rocco M, Vanholder R,
Sever MS, et al: International society of nephrology's 0 by 25
initiative for acute kidney injury (zero preventable deaths by
2025): A human rights case for nephrology. Lancet. 385:2616–2643.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rewa O and Bagshaw SM: Acute kidney
injury-epidemiology, outcomes and economics. Nat Rev Nephrol.
10:193–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang L, Xing G, Wang L, Wu Y, Li S, Xu G,
He Q, Chen J, Chen M, Liu X, et al: Acute kidney injury in China: A
cross-sectional survey. Lancet. 386:1465–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jha V and Parameswaran S:
Community-acquired acute kidney injury in tropical countries. Nat
Rev Nephrol. 9:278–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cerda J, Bagga A, Kher V and Chakravarthi
RM: The contrasting characteristics of acute kidney injury in
developed and developing countries. Nat Clin Pract Nephrol.
4:138–153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chawla LS, Amdur RL, Amodeo S, Kimmel PL
and Palant CE: The severity of acute kidney injury predicts
progression to chronic kidney disease. Kidney Int. 79:1361–1369.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zuk A and Bonventre JV: Acute kidney
injury. Annu Rev Med. 67:293–307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Makris K and Spanou L: Acute kidney
injury: Definition, pathophysiology and clinical phenotypes. Clin
Biochem Rev. 37:85–98. 2016.PubMed/NCBI
|
|
9
|
Friedewald JJ and Rabb H: Inflammatory
cells in ischemic acute renal failure. Kidney Int. 66:486–491.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bonventre JV and Zuk A: Ischemic acute
renal failure: An inflammatory disease? Kidney Int. 66:480–485.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu L, Chen C, Zhang J, Wu K, Zhang X, Liu
H and Hou J: IL-35 pretreatment alleviates
lipopolysaccharide-induced acute kidney injury in mice by
inhibiting NF-κB activation. Inflammation. 40:1393–1400. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zuk A, Gershenovich M, Ivanova Y,
MacFarland RT, Fricker SP and Ledbetter S: CXCR4
antagonism as a therapeutic approach to prevent acute kidney
injury. Am J Physiol Renal Physiol. 307:F783–F797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gigliotti JC, Huang L, Ye H, Bajwa A,
Chattrabhuti K, Lee S, Klibanov AL, Kalantari K, Rosin DL and Okusa
MD: Ultrasound prevents renal ischemia-reperfusion injury by
stimulating the splenic cholinergic anti-inflammatory pathway. J Am
Soc Nephrol. 24:1451–1460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Castoldi A, Braga TT, Correa-Costa M,
Aguiar CF, Bassi ÊJ, Correa-Silva R, Elias RM, Salvador F,
Moraes-Vieira PM, Cenedeze MA, et al: TLR2, TLR4 and the MYD88
signaling pathway are crucial for neutrophil migration in acute
kidney injury induced by sepsis. PLoS One. 7:e375842012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gluba A, Banach M, Hannam S, Mikhailidis
DP, Sakowicz A and Rysz J: The role of Toll-like receptors in renal
diseases. Nat Rev Nephrol. 6:224–235. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alves-Filho JC, Freitas A, Souto FO,
Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM,
Ferreira SH and Cunha FQ: Regulation of chemokine receptor by
Toll-like receptor 2 is critical to neutrophil migration and
resistance to polymicrobial sepsis. Proc Natl Acad Sci USA.
106:4018–4023. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peng Y, Zhang X, Wang Y, Li S, Wang J and
Liu L: Overexpression of toll-like receptor 2 in glomerular
endothelial cells and podocytes in septic acute kidney injury mouse
model. Ren Fail. 37:694–698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng Y, Zhang X, Wang Y, Yuan H, Zhang S
and Liu L: Toll like receptor 2 induces kidney inflammation via
MyD88/NF-κB signaling pathway. Int J Clin Exp Med. 11:3494–3503.
2018.
|
|
19
|
Chen Y, Lin L, Tao X, Song Y, Cui J and
Wan J: The role of podocyte damage in the etiology of
ischemia-reperfusion acute kidney injury and post-injury fibrosis.
BMC Nephrol. 20:1062019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perkin WH and Robinson R: CCXXII. - Some
derivatives of ortho-vanillin. J Chem Soc Transactions.
105:2376–2392. 1914. View Article : Google Scholar
|
|
21
|
Mistry P, Laird MH, Schwarz RS, Greene S,
Dyson T, Snyder GA, Xiao TS, Chauhan J, Fletcher S, Toshchakov VY,
et al: Inhibition of TLR2 signaling by small molecule inhibitors
targeting a pocket within the TLR2 TIR domain. Proc Natl Acad Sci
USA. 112:5455–5460. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rueden CT, Schindelin J, Hiner MC, DeZonia
BE, Walter AE, Arena ET and Eliceiri KW: ImageJ2: ImageJ for the
next generation of scientific image data. BMC Bioinformatics.
18:5292017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu L, Chen C, Zhang J, Wu K, Zhang X, Liu
H and Hou J: IL-35 Pre-treatment alleviates
lipopolysaccharide-induced acute kidney injury in mice by
inhibiting NF-κB activation. Inflammation. 40:1393–1400. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y, Yin H, Zhao M and Lu Q: TLR2 and
TLR4 in autoimmune diseases: A comprehensive review. Clin Rev
Allergy Immunol. 47:136–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chawla LS, Seneff MG, Nelson DR, Williams
M, Levy H, Kimmel PL and Macias WL: Elevated plasma concentrations
of IL-6 and elevated APACHE II score predict acute kidney injury in
patients with severe sepsis. Clin J Am Soc Nephrol. 2:22–30. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paladino JD, Hotchkiss JR and Rabb H:
Acute kidney injury and lung dysfunction: A paradigm for remote
organ effects of kidney disease? Microvasc Res. 77:8–12. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu C, Chang A, Hack BK, Eadon MT, Alper SL
and Cunningham PN: TNF-mediated damage to glomerular endothelium is
an important determinant of acute kidney injury in sepsis. Kidney
Int. 85:72–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ajuwon KM and Spurlock ME: Adiponectin
inhibits LPS-induced NF-kappaB activation and IL-6 production and
increases PPARgamma2 expression in adipocytes. Am J Physiol Regul
Integr Comp Physiol. 288:R1220–F1225. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cascinu S, Scartozzi M, Carbonari G,
Pierantoni C, Verdecchia L, Mariani C, Squadroni M, Antognoli S,
Silva RR, Giampieri R and Berardi R: COX-2 and NF-κB overexpression
is common in pancreatic cancer but does not predict for COX-2
inhibitors activity in combination with gemcitabine and
oxaliplatin. Am J Clin Oncol. 30:526–530. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zarjou A and Agarwal A: Sepsis and acute
kidney injury. J Am Soc Nephrol. 22:999–1006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grigoryev DN, Liu M, Hassoun HT, Cheadle
C, Barnes KC and Rabb H: The local and systemic inflammatory
transcriptome after acute kidney injury. J Am Soc Nephrol.
19:547–558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ye HY, Jin J, Jin LW, Chen Y, Zhou ZH and
Li ZY: Chlorogenic acid attenuates lipopolysaccharide-induced acute
kidney injury by inhibiting TLR4/NF-κB signal pathway.
Inflammation. 40:523–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dorak MT: Basic immunology: Functions and
disorders of the immune system. Am J Epidemiol. 155:185–186. 2012.
View Article : Google Scholar
|
|
34
|
Gustafson GL and Rhodes MJ: A rationale
for the prophylactic use of monophosphoryl lipid A in sepsis and
septic shock. Biochem Biophys Res Commun. 182:269–275. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Poelstra K, Bakker WW, Klok PA, Hardonk MJ
and Meijer DK: A physiologic function for alkaline phosphatase:
Endotoxin detoxification. Lab Invest. 76:319–327. 1997.PubMed/NCBI
|
|
36
|
Cavaillon JM, Adib-Conquy M, Fitting C,
Adrie C and Payen D: Cytokine cascade in sepsis. Scand J Infect
Dis. 35:535–544. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cunningham PN, Ying W, Guo R, Gang H and
Quigg RJ: Role of toll-like receptor 4 in endotoxin-induced acute
renal failure. J Immunol. 172:2629–2635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fiedler VB, Loof I, Sander E, Voehringer
V, Galanos C and Fournel MA: Monoclonal antibody to tumor necrosis
factor-alpha prevents lethal endotoxin sepsis in adult rhesus
monkeys. J Lab Clin Med. 120:574–588. 1992.PubMed/NCBI
|
|
39
|
Pasquet V, Perwuelz A, Behary N and Isaad
J: Vanillin, a potential carrier for low temperature dyeing of
polyester fabrics. J Cleaner Product. 43:20–26. 2013. View Article : Google Scholar
|
|
40
|
Shmuel-Galia L, Aychek T, Fink A, Porat Z,
Zarmi B, Bernshtein B, Brenner O, Jung S and Shai Y: Neutralization
of pro-inflammatory monocytes by targeting TLR2 dimerization
ameliorates colitis. EMBO J. 35:685–698. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Landais I, Pelton C, Streblow D,
DeFilippis V, McWeeney S and Nelson JA: Human Cytomegalovirus
miR-UL112-3p targets TLR2 and modulates the TLR2/IRAK1/NFκB
signaling pathway. PLoS Pathog. 11:e10048812015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koymans KJ, Feitsma LJ, Brondijk TH, Aerts
PC, Lukkien E, Lössl P, van Kessel KP, de Haas CJ, van Strijp JA
and Huizinga EG: Structural basis for inhibition of TLR2 by
staphylococcal superantigen-like protein 3 (SSL3). Proc Natl Acad
Sci USA. 112:11018–11023. 2015. View Article : Google Scholar : PubMed/NCBI
|