Introduction
Temtamy syndrome (MIM no. 218340) is a rare disorder
which was first described in 1991 and then published a full formal
report in 1996, where three siblings affected appeared to have
inherited an autosomal recessive gene (1,2). It is
characterized with mental retardation and multiple congenital
anomaly, with symptoms including variable craniofacial dysmorphism,
ocular coloboma, seizures and abnormalities in the corpus callosum
and thalamus (1–3). Of note, the chromosome 12 open reading
frame 57 (C12orf57) encodes a 126-amino acid cytoplasmic
protein of unknown function was reported to be required for human
corpus callosum development (3,4). In
2013, pathogenic variants of C12orf57 were first reported to
cause Temtamy syndrome (3,4). At the present time, only seven
pathogenic variants have been illustrated (1–6). Those
of Middle Eastern descent are particularly susceptible to
C12orf57 pathogenic variants, with 54/56 (96.4%) of all the
reported patients from Middle Eastern countries, predominantly
Saudi Arabia (3–7).
The present study reported the first East Asian
patient with global developmental delay and epilepsy caused by a
novel homozygous C12orf57 pathogenic variant, and presented
a brief review of all the previously published cases.
Patients and methods
Patients
Ethics approval for this study was obtained through
the Institutional Review Board of Children's Hospital of Chongqing
Medical University. Written informed consent was obtained from the
parents of the patient for the publication of this case report and
accompanying images.
The present case is of a boy born to first-cousin
consanguineous Chinese parents of Hui nationality at 40 weeks and 5
days of gestation, gravida 2 para 2. The boy was born by normal
delivery with a birth weight of 3,510 g, while the birth length and
head circumference were unknown. His elder sister is 7 years old
and healthy as of October 2019.
The patient was admitted to the Neurology ward at
the Children's Hospital of Chongqing Medical University (May 2018)
at the age of 8 months and 28 days because of four tonic-clonic
seizures in 10 days, as well as pneumonia which had lasted for 1
week. All of the available clinical characteristics of the patient,
along with the aforementioned auxiliary examination results are
summarized in the present study.
Whole-exome sequencing and
bioinformatics analysis
Based on the hg19/GRCh37 reference, whole-exome
sequencing was performed on the proband and the parents at Beijing
Mygenostics Co., Ltd. Several online databases containing data from
different ethnic groups were used as follows: Genome Aggregation
Database (gnomAD, http://gnomad.broadinstitute.org/); 1000 genome
Project variants database (http://www.1000genomes.or/); Esp6500siv2_all
(http://evs.gs.washington.edu/EVS/),
Inhouse databases (http://192.168.0.69/db/inhouse/); Exome Variant Server
(version 0.0.30;http://evs.gs.washington.edu/EVS/); ExAC (http://exac.broadinstitute.org/); dbSNP (version
2.0; http://www.ncbi.nlm.nih.gov/projects/SNP/); and
Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/). The variants
were selected according to the following five-step process, to
select the potential pathogenic variants in the downstream
analysis: i) Mutation reads >10 and mutation ratio ≥30%; ii)
following removal of the mutation from the search, the frequency
showed more than 0.1% in the 1,000 Genomes, ESP6500, ExAC and
Inhouse databases; iii) if the mutations existed in the In Normal
database (MyGenostics), they were dropped; iv) removal of
synonymous mutations or mutations in intronic region, since they
were considered non-pathogenic; and v) after i), ii) and iii), if
the mutations were in introns or were synonymous and reported in
HGMD, they were included. All remaining variants were considered
pathogenic. Finally, seven homozygous mutations were identified.
Combined with the phenotype of the child, only the C12orf57
variant was confirmed as pathogenic. The C12orf57 pathogenic
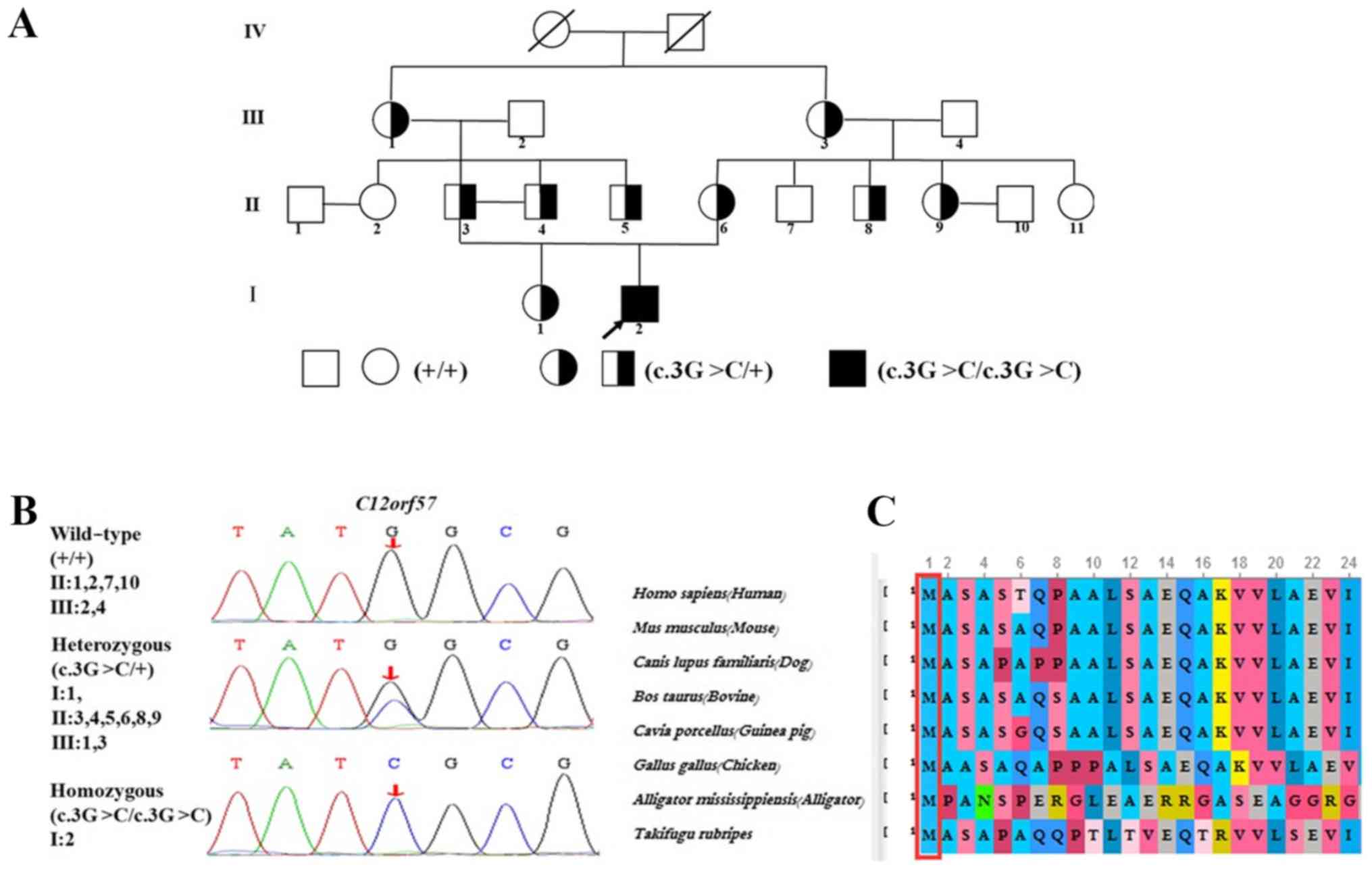
variant was screened using Sanger sequencing to amplify exon 1
(chr12-7053287) of C12orf57 (NM_138425). The segregation of
the identified pathogenic variant was investigated in all the
family members (Fig. 2A and B). The
prediction of mutations was assessed using software, including
Polyphen2_HVAR_score, Polyphen2_HVAR_pred, PolyPhen_2_Predict and
PolyPhen_2 (April 2010; http://genetics.bwh.harvard.edu/pph2/);
MutationTaster, Mutation Taster_Predict (April 2009; http://www.mutationtaster.org/ChrPos.html); SIFT,
SIFT_Predict (http://sift.jcvi.org/); LRT_score,
LRT_pred (November 2009; http://www.genetics.wustl.edu/jflab/lrt_query.html);
CADD_raw, CADD_phred (version 1.3; http://cadd.gs.washington.edu/).
Literature review
PubMed and Wanfang (http://www.wanfangdata.com.cn/index.html) databases
were searched to retrieve studies using the keywords
‘C12orf57’, from inception to March 2019. The publication
language was restricted to English and Chinese.
Results
Clinical manifestation
On admission, the patient was unable to hold his
head at 6 months, and was unable to sit up straight at 10 months.
Hospitalization had occurred three times because of severe
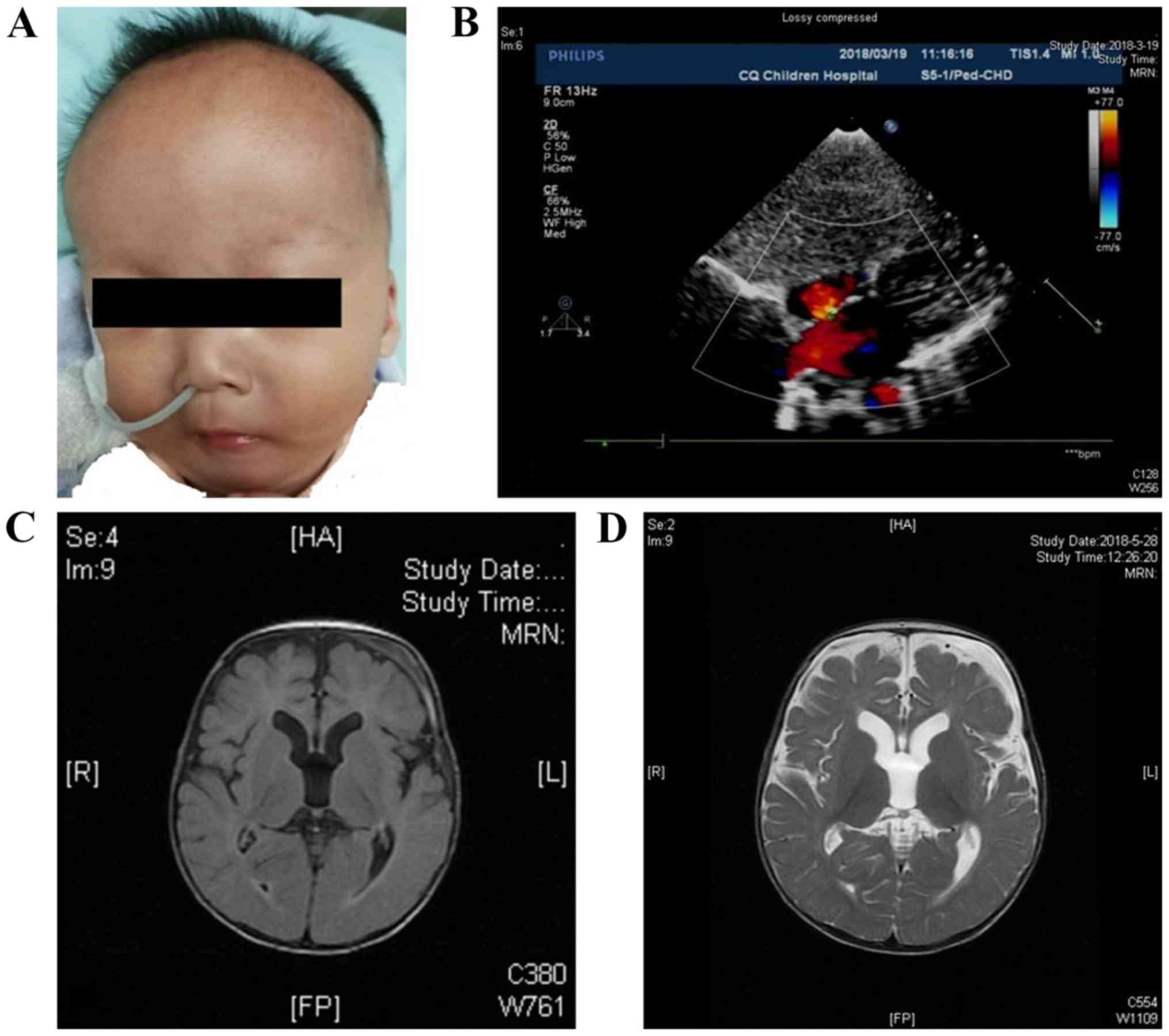
pneumonia. Also, abnormal dysmorphic features (Figs. 1A and Video S1), such as frontal bossing, low-set
ears, depressed nasal bridge, ocular hypertelorism, micrognathia
and single transverse palmar crease were observed. Referring to the
World Health Organization Anthro guidelines, the patient was 7 kg
(Z-score=−2.17 SD) in weight and 65 cm (Z-score=−2.75 SD) in
length; the head circumference was 45 cm (Z-score=0.02 SD).
An atrial septum defect (4.5 mm) was found by color
Doppler ultrasonography (Fig. 1B).
The magnetic resonance imaging (MRI) scans showed slightly expanded
lateral ventricles and increased extra-axial spaces (Fig. 1C and D). The interictal
electroencephalography showed sporadic spike-slow wave during
waking. Any abnormalities of the eyes were not detected during the
ophthalmological consultation. Furthermore, this patient was
administered oxcarbazepine (8–15 mg/kg/day) and anti-infection
therapy (ceftazidime 100 mg/kg/day (intravenous infusion) for 5
days, followed by cefixime 5 mg/kg/day (oral administration) for 5
days. After 2 weeks, the patient was discharged and transferred to
a local hospital for the recovery phase management.
During the 1-year follow-up, the patient was
seizure-free for 1 month after the oxcarbazepine (26 mg/kg/day) was
administered, and could only sit up straight for <30 sec at the
age of 14 months. While there were still another six
hospitalizations for pneumonia, only two had occurred in the
previous 6 months and the patient had recovered easily. The atrial
septum defect was not observed by color Doppler ultrasonography at
the age of 19 months.
Genetic analysis
Exome sequencing revealed a homozygous pathogenic
variant, c.3G>C (p.Met1IIe), that was confirmed by Sanger
sequencing. Furthermore, the pathogenic variant was segregated
according to a strictly recessive model with full penetrance. The
parents were tested as possible heterozygous carriers. A total of
four heterozygous carriers of the C12orf57 pathogenic
variant were detected; the patient's parents and their second
degree relatives inherited the pathogenic variants from their
grandparents (Fig. 2A and B).
C12orf57 encodes a protein that is
evolutionarily conserved across representative species (Fig. 2C), according to HomoloGene
(https://www.ncbi.nlm.nih.gov/homologene/). The actual
in silico results are as follows: SIFT_score (0); SIFT_pred
(D); Polyphen2_HDIV_score (0.072); Polyphen2_HDIV_pred (B);
Polyphen2_HVAR_score (0.008); Polyphen2_HVAR_pred (B); LRT_score
(0); LRT_pred (D); CADD_raw (3.109); CADD_phred (22.5); Pathogenic
variant Taster_score (1); Pathogenic
variant Taster_pred (D). As a result, most of the in silico
results predicted this biallelic missense pathogenic variant to be
deleterious, wherein the pathogenic variant is localized at the
start codon, abolishing the translation of C12orf57.
Literature review
Temtamy syndrome is not easy to distinguish
clinically from other syndromes with similar phenotypes. As such,
the present study only summarized cases with C12orf57
mutations. A total of 56 cases with C12orf57 pathogenic
variants (Fig. S1) have been
reported (3–7). All the cases were early onset, and the
clinical features are summarized in Table I.
 | Table I.Chromosome 12 open reading frame 57
pathogenic variants and phenotypes of all the affected cases from
the literature and the current report. |
Table I.
Chromosome 12 open reading frame 57
pathogenic variants and phenotypes of all the affected cases from
the literature and the current report.
| Characteristic | The present case | Literature
(n=56) | Total, % |
|---|
| Basic
characteristics |
|
|
|
| Early
onset | 1 | 56 | 100 |
|
Consanguineous | 1 | 49 | 87.7 |
| Ethnic origin |
|
|
|
| Saudi
Arabia |
| 25 | 43.9 |
|
Kuwait |
| 9 | 15.8 |
| United
Arab Emirates |
| 9 | 15.8 |
|
Libya |
| 4 | 7.0 |
|
Palestine |
| 4 | 7.0 |
| Oman |
| 3 | 5.3 |
|
Germany |
| 2 | 3.5 |
|
China | 1 |
| 1.8 |
| Genetics |
|
|
|
|
c.1A>G, p.(Met1?) |
| 45 | 78.9 |
|
c.3G>C, p.(Met1I) | 1 |
| 1.8 |
|
c.53-2A>G |
| 3 | 5.3 |
|
c.184C>T, p.(Gln62*) |
| 2 | 3.5 |
|
c.-3_2delinsG |
| 2 | 3.5 |
|
c.43C>T, p.(Gln15*) |
| 2 | 3.5 |
|
c.229+2T>C |
| 1 | 1.8 |
|
c.152T>A, p.(Leu51Gln) |
| 1 | 1.8 |
| Phenotype |
|
|
|
|
Neurological findings |
|
|
|
|
Developmental
delay | 1 | 56/56 | 100 |
|
Seizures | 1 | 41/56 | 73.7 |
|
Absent speech | N/A | 41/55 | 74.5 |
|
Generalized
hypotonia | 1 | 40/56 | 71.9 |
|
Delayed
speech | 1 | 20/55 | 37.5 |
|
Autistic
behavior | N/A | 40/55 | 72.7 |
|
Spasticity | 0 | 20/56 | 35.1 |
|
Dysmorphic features |
|
|
|
|
Dysmorphic
faces | 1 | 36/55 | 66.1 |
|
Microphthalmia | 1 | 9/55 | 14.3 |
|
Ophthalmology |
|
|
|
|
Ocular
anomalies | 0 | 26/55 | 46.4 |
|
Coloboma | 0 | 8/55 | 14.5 |
|
Congenital heart disease |
|
|
|
|
Atria septal
defect | 1 | 16/55 | 30.4 |
|
Ventricular septal
defect | 0 | 2/19 | 10.0 |
|
Pulmonic
stenosis | 0 | 3/26 | 11.1 |
| Brain
imaging |
|
|
|
|
Abnormal corpus
callosum | 0 | 34/54 | 61.8 |
|
Abnormal thalamic
size | 0 | 20/51 | 38.5 |
|
Abnormal septum
pellucidum | 0 | 19/50 | 37.3 |
|
Abnormal white
matter | 0 | 19/50 | 37.3 |
|
Abnormal anterior
commissure | 0 | 11/51 | 21.2 |
|
Ventriculomegaly | 1 | 17/50 | 35.3 |
|
Others |
|
|
|
|
Recurrent
pneumonia | 1 | N/A |
|
Discussion
Temtamy syndrome is an extremely rare disorder, and
only a limited number of studies have been conducted (1–7). The
present study reports the first East Asian patient with a novel
C12orf57 homozygous pathogenic variant; all other
heterozygous carriers in the family were clinically healthy. The
inheritance pattern of this pedigree was in accordance with
autosomal recessive inheritance with complete penetrance.
Pathogenic variants in C12orf57 were first
reported to cause Temtamy syndrome in 2013 (3–5), and the
affected individuals were recently described to have variable
phenotypes beyond this syndrome (5).
The c.1A>G pathogenic variant in the C12orf57 start codon
has been reported as the most frequent pathogenic variant (45/56,
80.3%). Interestingly, the pathogenic variant in this start codon
can severely reduce the protein levels, suggesting a loss of
function as the mechanism of action behind the disease. Moreover,
the pathogenic variant (c.3G>C) in the current case was located
at the same start codon of C12orf57, but at different bases.
This suggests that the c.3G>C pathogenic variant may affect the
protein expression level and may lead to the occurrence of disease.
Bioinformatics analysis indicated that the aforementioned
pathogenic variant is the most likely mechanism of action behind
the pathogenesis. Since the parents of the patient are first-cousin
consanguineous relatives, whether the patient had other additional
homozygous pathogenic variants was investigated, but no positive
results were obtained.
All the patients with C12orf57 pathogenic
variants identified in the literature review exhibited global
developmental delay in concurrence with hypotonia. Compared to the
previously reported cases of epilepsy (73.2%), relatively
refractory (37.5%) and low frequency of seizure-free (15.6%), as
found in the literature review, the seizures in the current case
were controlled. No recurrence of seizures occurred after the
patient was medicated with oxcarbazepine. Although the dysmorphic
features are not distinctly recognized among patients with
C12orf57 pathogenic variants, the current case had many
similar dysmorphic facial features (65.5%), including frontal
bossing, low set and posteriorly rotated ears, depressed nasal
bridge, hypertelorism, micrognathia, up-slanted palpebral fissures
and microphthalmia, but without epicanthal folds. Previously
reported abnormalities of the eyes (such as chorioretinal
coloboma), iris or optic nerve were not found in this present case.
C12orf57 was once reported to cause colobomatous
microphthalmia, which seems to be invariably associated with
profound global developmental delay, seizures and defects of the
corpus callosum, but these cases were later summarized to be
Temtamy syndrome by the same research group (4,5). This
present study identified colobomatous microphthalmia to be a clear
sign of this syndrome.
Abnormalities of the corpus callosum were included
in the classical characteristics of this disease and may be
observed in approximately two-thirds of patients with
C12orf57 pathogenic variants. However, no similar corpus
callosum changes were demonstrated and MRI scanning of the brain
revealed only slight cerebral dysplasia in the present case.
Reportedly, atrial septum defects are the most frequent congenital
heart defect, which was also observed in the present case, at 4.5
mm.
Additionally, the present patient was particularly
susceptible to infection, developing severe pneumonia every 2–3
months leading to hospitalization for several weeks. No similar
recurrent pneumonia has been mentioned in the previous studies.
During the follow-up, the recurrent infections were less frequent
in the latter 6 months; therefore, it is considered that the
recurrent infections may be partly associated with the atrial
spectrum defects, which was not detected at previous visits.
Therefore, additional cases could substantiate the findings if
recurrent infection is a feature of patients with C12orf57
pathogenic variants. Currently, the patient is 20 months old and no
more new outcomes have been observed. Follow-up observations are
needed to confirm the presence of further phenotypes.
The present study has some limitations. Detailed
information on reported cases of pathogenic C12orf57
pathogenic variants is limited (for example, familial aggregation,
life expectancy and type of seizures). Furthermore, the
phenotype-genotype correlation cannot be confirmed since the
majority (45/57) of the reported cases had the same pathogenic
variant, c.1A>G, p. (Met1?), and the other different genetic
variants were only observed in one or two cases.
In conclusion, a novel homozygous C12orf57
pathogenic variant (c.3G>C) was identified in a patient with
developmental delay, epilepsy and dysmorphic facial appearance. The
C12orf57 pathogenic variant site has not been published
previously, and this was the first reported East Asian case with
Temtamy syndrome to the best of our knowledge. The present results
expanded the spectrum of C12orf57 pathogenic variants, as
well as the ethnic backgrounds of the affected cases.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was partially supported by a grant
obtained from the Children's Hospital of Chongqing Medical
University (grant no. HJYN2013-4).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YW, YL and ML carried out the diagnosis and
treatment of the case, collected the data, and performed literature
review. MZ designed and supervised the study. XZ, SL, LJ and XL
were involved in the management of the patient and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The ethics approval for this study was obtained
through the Institutional Review Board of Children's Hospital of
Chongqing Medical University (Chongqing, China; no. 2018-64).
Patient consent for publication
The parents provided consent for the case report and
for images of the patient's face to be published for educational
purposes.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Temtamy SA, Salam MA, Aboul-Ezz EH,
Hussein HA, Helmy SA and Shalash BA: New autosomal recessive
multiple congenital abnormalities/mental retardation syndrome with
craniofacial dysmorphism absent corpus callosum, iris colobomas and
connective tissue dysplasia. Clin Dysmorphol. 5:231–240. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Temtamy SA, Abdel Salam M, Aboul-Ezz EH,
Hussein HA, Helmy SMH and Shalash BA: A new autosomal recessive
MCA/MR syndrome with craniofacial dysmorphism, absent corpus
callosum, iris colobomas and connective tissue dysplasia.
(Abstract) Am J Hum Genet. 49 (Suppl):S1671991.
|
|
3
|
Akizu N, Shembesh NM, Ben-Omran T, Bastaki
L, Al-Tawari A, Zaki MS, Koul R, Spencer E, Rosti RO, Scott E, et
al: Whole-exome sequencing identifies mutated c12orf57 in recessive
corpus callosum hypoplasia. Am J Hum Genet. 92:392–400. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zahrani F, Aldahmesh MA, Alshammari MJ,
Al-Hazzaa SA and Alkuraya FS: Mutations in c12orf57 cause a
syndromic form of colobomatous microphthalmia. Am J Hum Genet.
92:387–391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alrakaf L, Al-Owain MA, Busehail M,
Alotaibi MA, Monies D, Aldhalaan HM, Alhashem A, Al-Hassnan ZN,
Rahbeeni ZA, Murshedi FA, et al: Further delineation of temtamy
syndrome of corpus callosum and ocular abnormalities. Am J Med
Genet A. 176:715–721. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Platzer K, Huning I, Obieglo C,
Schwarzmayr T, Gabriel R, Strom TM, Gillessen-Kaesbach G and Kaiser
FJ: Exome sequencing identifies compound heterozygous mutations in
C12orf57 in two siblings with severe intellectual disability,
hypoplasia of the corpus callosum, chorioretinal coloboma, and
intractable seizures. Am J Med Genet A 164A. 1976–1980. 2014.
View Article : Google Scholar
|
|
7
|
Salih MA, Tzschach A, Oystreck DT, Hassan
HH, AlDrees A, Elmalik SA, El Khashab HY, Wienker TF, Abu-Amero KK
and Bosley TM: A newly recognized autosomal recessive syndrome
affecting neurologic function and vision. Am J Med Genet A 161A.
1207–1213. 2013. View Article : Google Scholar
|