Introduction
Lung cancer, including small cell lung cancer (SCLC)
and non-SCLC (NSCLC) (1,2), may be regarded as the most common tumor
type and major contributor to the high tumor-associated mortality
rate worldwide (3). Recent
epidemiological data have revealed that it affected 1.8 million
patients and resulted in 1.6 million deaths in 2012 (4). As one of the most frequent histological
subtypes of lung cancer, lung adenocarcinoma (LACA) is a major
cause of tumor-associated death (5,6). Despite
recent advances in surgical techniques, radiotherapeutic
interventions and combined chemotherapy strategies, the long-term
survival rate of patients diagnosed with primary LACA has not
significantly improved (7,8). Due to tumor heterogeneity factors and
different molecular subtypes of LACA, its treatment faces large
challenges. In this light, it is significant to identify specific
details regarding characteristic molecules in LACA tissues to
delineate the heterogeneity of LACA and develop strategies for
personalized therapy.
In recent years, epigenetics, which has a critical
role in carcinogenesis, has gained attention (9). Aberrant DNA methylation, as the core
element of epigenetic modification, influences certain tumor
suppressor genes and regulates gene functions (10,11).
Increasing studies also demonstrated that DNA methylation is
associated with genome stability, gene imprinting and cell
differentiation (12,13). Thus, the methylation level is deemed
a molecular biomarker for the diagnosis and prognostication of
patients with tumors. However, the current expertise on the
association between the epigenetic modifications and the clinically
predicted outcomes of LACA is limited. Thus, in the present study,
distinctive DNA methylation data for LACA vs. control tissues were
acquired to evaluate the prognostic significance of distinctive DNA
methylation patterns and provide insight regarding survival
prediction for patients with LACA.
Materials and methods
Data processing
Original, publicly available and open-access genetic
representation data of LACA samples and relevant clinical
information of the patients obtained from The Cancer Genome Atlas
(TCGA) database (http://cancergenome.nih.gov/) were included in the
present study. The clinical information included the following
attributes: Age, sex, ethnicity, stage and histological type of
LACA. The exclusion criteria were as follows: i) Samples without
clinical information, ii) samples without survival data, and iii)
samples from patients that survived for <1 month. Ultimately,
447 LACA samples with DNA methylation data and clinical information
were screened for further testing. The data were provided by TCGA
and the study was performed in compliance with the TCGA publication
guidelines (14).
Selection of differential DNA
methylation sites
In the present study, aggregation and collection of
DNA methylation information was performed by using R. First, the
data were normalized by log2 transformation. The Limma package was
employed for analyzing the differential DNA methylation sites
between LACA tumor tissues and normal tissues. The fold changes
(FCs) of DNA methylation were also calculated and significant
aberrations in gene methylation were defined as those having a
log2|FC|>2.0, P-value <0.01 and beta value
>0.1. The least absolute shrinkage and selection operator
(LASSO) method, which is suitable for regression of
high-dimensional data (15), was
used to select the most useful predictive features from the primary
data set. The potential association of the CpG-based signature with
LACA status was first assessed in the primary cohort and then
validated in the validation cohort using a Mann-Whitney U-test.
With this CpG-based signature, patients in each dataset were
classified into a high-risk group and a low-risk group by using the
median risk score. Kaplan-Meier curves and log-rank analysis were
then performed to calculate the association between the CpG-based
signature and patient's OS in the two groups with high-risk and
low-risk CpG-based signatures. The CpG-based signature, which was
significantly associated with OS (P<0.001), was then subjected
to receiver operating characteristic (ROC) curve analysis to
evaluate the predictive accuracy and sensitivity of the prognostic
model. The area under the ROC curve (AUC) was also calculated. In
the Kaplan-Meier curve, log-rank test and ROC analysis, the
significance was defined as P<0.05.
Functional and pathway enrichment
analysis
To further elucidate the biological functions of the
mapped genes and the molecular mechanisms, a functional enrichment
analysis was performed using the Database for Annotation,
Visualization and Integrated Discovery (DAVID) (16). Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses
with P<0.05 were identified and biological process terms were
further clustered using the Enrichment Map Plugin of Cytoscape
(17).
Statistical analyses
Fundamental characteristics of the sample in the
study were summarized by using descriptive statistics. Data for
continuous variables, which were expressed as the mean ± standard
deviation, were compared using the Student's t-test, Mann-Whitney
U-test, Kruskal-Wallis H-test or one-way ANOVA with post hoc
Student-Newman-Keuls tests, depending on the normality of data
distribution as tested by Kolmogorov-Smirnov tests; data for
categorical variables, which were presented as percentages, were
compared using the Chi-square test. Statistical analyses were
performed using SPSS 17.0 (SPSS Inc.) and R version 3.5.1 software
(http://www.r-project.org/) (18). P<0.05 was considered to indicate a
statistically significant difference.
Results
Patient characteristics
The data of all 447 samples with clinical
information and methylation data available were obtained from the
TCGA database. The samples included 440 LACA tissues and 7 normal
tissues. Table I lists the detailed
clinical characteristics of patients in the initial stage and
specific groups (primary and validation cohort), including age at
diagnosis, sex, ethnicity, disease stage and survival status. The
results revealed that there were no major distinctions between the
two groups in terms of these five clinical characteristics.
 | Table I.Baseline clinical characteristics of
the patients in the primary and validation cohorts. |
Table I.
Baseline clinical characteristics of
the patients in the primary and validation cohorts.
| Characteristic | Primary cohort
(n=220) | Validation cohort
(n=440) | P-value |
|---|
| Age at diagnosis
(years) | 66.6±9.9 | 65.4±10.1 | 0.86 |
| Sex |
|
| 1.00 |
|
Female | 120 (54.5) | 240 (54.5) |
|
| Male | 100 (45.5) | 200 (45.5) |
|
| Ethnicity |
|
| 0.08 |
|
Caucasian | 176 (80.0) | 340 (77.3) |
|
| Of
African descent | 22 (10.0) | 50 (11.4) |
|
|
Othersa | 22 (10.0) | 50 (11.4) |
|
| Stage |
|
| 0.45 |
|
I/II | 175 (77.3) | 336 (76.4) |
|
|
III/IV | 40 (18.2) | 97 (22.0) |
|
| NA | 5 (2.3) | 7 (1.6) |
|
| Survival
status |
|
| 0.86 |
|
Alive | 139 (63.2) | 275 (62.5) |
|
|
Dead | 81 (36.8) | 165 (37.5) |
|
Differential methylation sites in
LACA
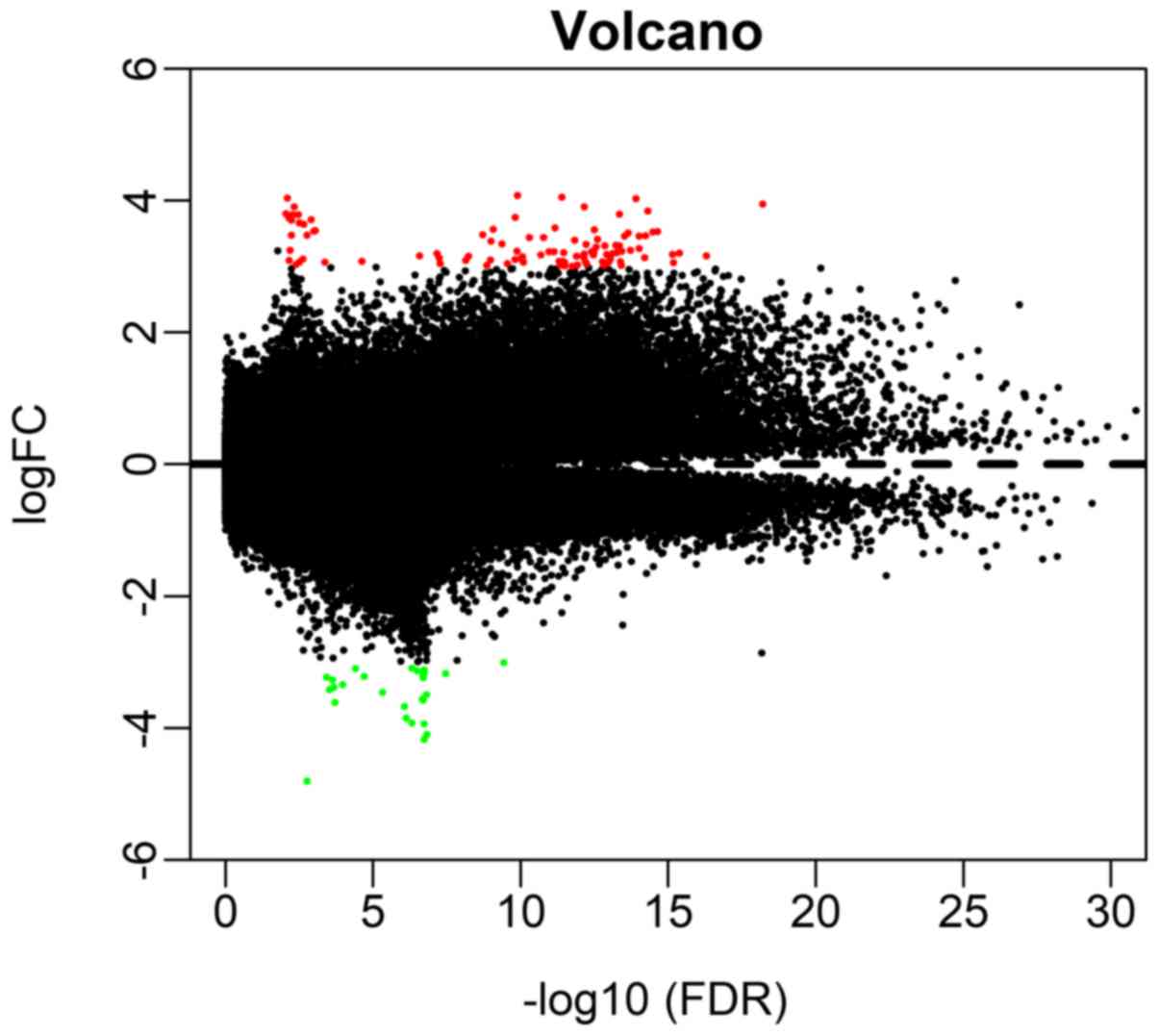
A total of 209 differential methylation sites were
recognized between the LACA and regular tissue samples in the
present study, including 133 hypermethylation and 76
hypomethylation sites. The distribution of hypermethylated and
hypomethylated sites were visualized in Fig. 1. Furthermore, the methylation data
were analyzed using the Limma incremental model. The five most
hypermethylated sites (cg16306898, cg00648301, cg01869632,
cg18837178 and cg22449330) and hypomethylated sites (cg05100666,
cg15998127, cg07764932, cg12581354 and cg27649653) between LACA and
normal tissue samples are listed in Table II.
| Table II.Top 5 hyper- and hypomethylated sites
of differential methylation. |
Table II.
Top 5 hyper- and hypomethylated sites
of differential methylation.
| A, Hypermethylated
sites |
|---|
|
|---|
| Composite | Log FC | adj.P-val | Gene |
|---|
| cg16306898 | 4.07515 |
1.28×10−10 | TMEM240 |
| cg00648301 | 4.04696 |
3.99×10−12 | INSM1 |
| cg01869632 | 4.034999 |
8.17×10−3 | DUSP26 |
| cg18837178 | 4.034999 |
8.17×10−3 | LINC01194 |
| cg22449330 | 4.034999 |
8.17×10−3 | WDPCP |
|
| B,
Hypomethylated sites |
|
|
Composite | Log FC |
adj.P-val | Gene |
|
| cg05100666 | −4.80454 |
1.76×10−3 | BRD9 |
| cg15998127 | −4.80454 |
1.76×10−3 | HDAC4 |
| cg07764932 | −4.17089 |
1.87×10−7 | ARHGAP6 |
| cg12581354 | −4.17089 |
1.87×10−7 | RP11-175P13.2 |
| cg27649653 | −4.17089 |
1.87×10−7 | AC010642.1 |
Methylated loci signature
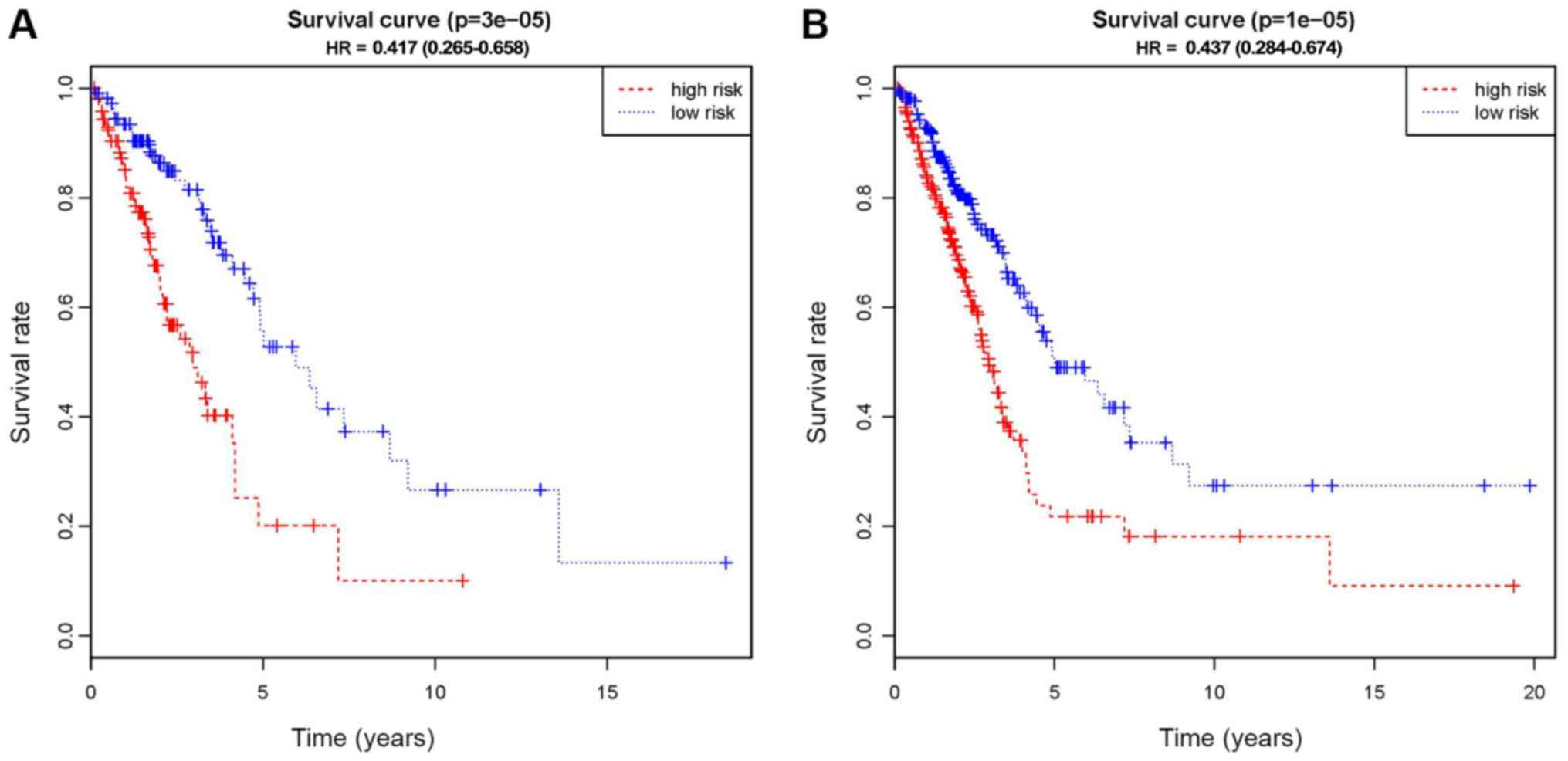
Based on the LASSO regression, 13 potential
predictors were showed in the primary cohort (Fig. S1). As indicated in Fig. 2, Kaplan-Meier curves were drawn and
the log-rank test was performed to evaluate the association between
the CpG-based signals and the survival rates of patients with LUAD.
The 13 methylated CpG signature, including cg00002719, cg02769743,
cg05239163, cg05507908, cg07918170, cg08213398, cg08516516,
cg08623223, cg12748948, cg14904034, cg16007456, ch.6.2958553R and
cg19868631, was a significant predictor of OS in the primary cohort
(P<0.01; Fig. 2A). The risk-score
formula is as follows: (0.93× cg00002719) + (1.15× cg02769743) +
(0.24× cg05239163) + (−1.20× cg05507908) + (1.31× cg07918170) +
(0.83× cg08213398) + (0.54× cg08516516) + (0.06× cg08623223) +
(0.12× cg12748948) + (0.03× cg14904034) + (−0.36× cg16007456) +
(−0.36× ch.6.2958553R) + (−0.21× cg19868631). The mapped genes of
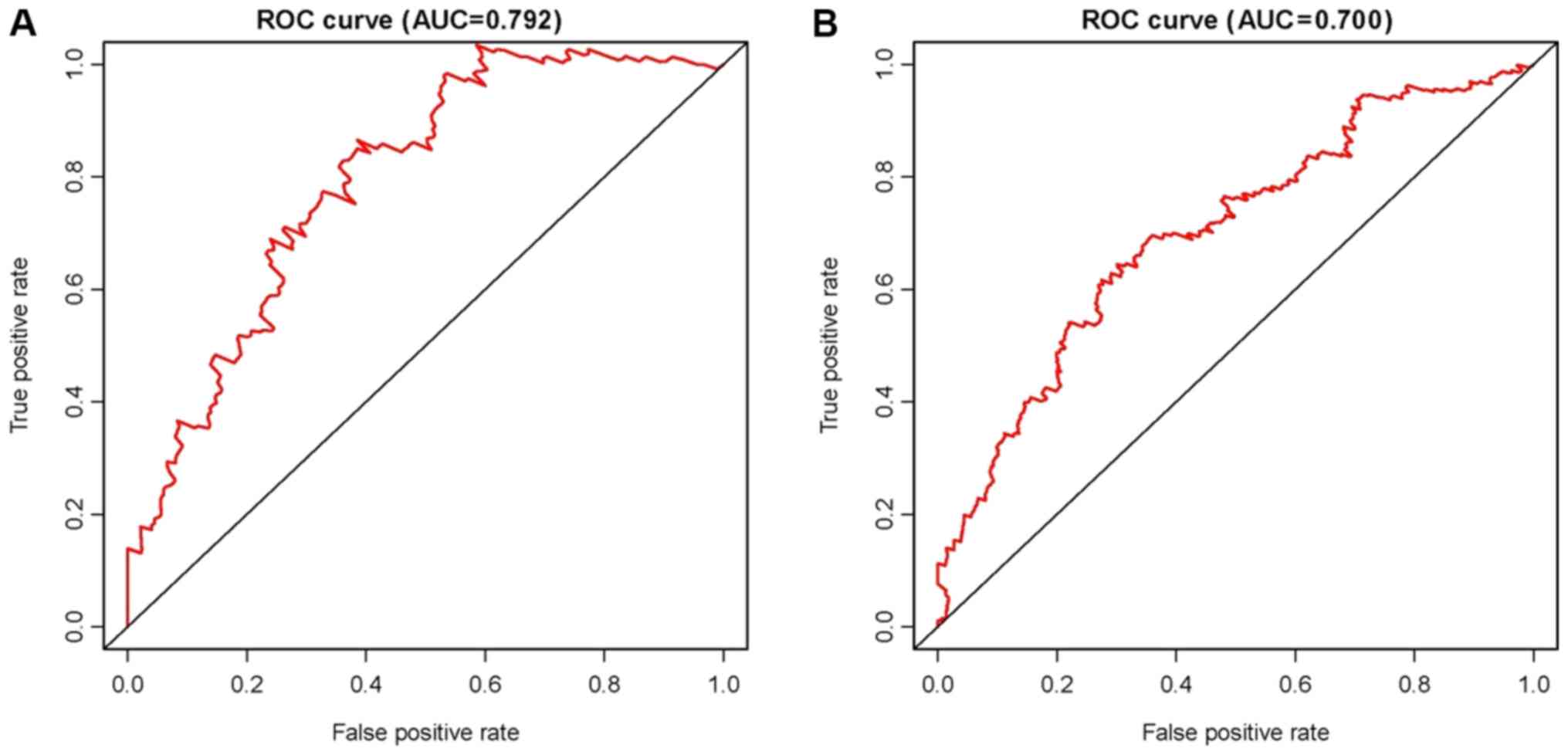
the methylation sites are also provided in Table III. In addition, for identifying
LACA, the approach of ROC curve analysis was pursued (Fig. 3). The AUC was 0.79, and the optimal
cut-off value was 0.11 (Fig.
3A).
| Table III.Methylation loci significantly
associated with survival. |
Table III.
Methylation loci significantly
associated with survival.
| Composite | Chromosome | Start | End | Gene |
|---|
| cg00002719 | 1 | 169427468 | 169427469 | CCDC181 |
| cg02769743 | 1 | 9608344 | 9608345 | TMEM201 |
| cg05239163 | 1 | 154218790 | 154218791 | C1orf43 |
| cg05507908 | 5 | 75237454 | 75237455 | ANKRD31 |
| cg07918170 | 17 | 82932272 | 82932273 | TBCD |
| cg08213398 | 11 | 9722579 | 9722580 | SWAP70 |
| cg08516516 | 5 | 115816795 | 115816796 | CDO1 |
| cg08623223 | 8 | 11283906 | 11283907 | AF131216.1 |
| cg12748948 | 1 | 21779802 | 21779803 | USP48 |
| cg14904034 | 2 | 10389150 | 10389151 | HPCAL1 |
| cg16007456 | 1 | 100539082 | 100539083 | GPR88 |
| ch.6.2958553R | 6 | 152198831 | 152198831 | SYNE1 |
| cg19868631 | 7 | 54542083 | 54542084 | VSTM2A |
Diagnostic and prognostic validation
of the signature
To assess the utility of the 13 CpG-based signature
in the diagnosis and prognosis of LUAD, the above-mentioned
analyses were performed using the validation cohort. There was a
marked distinction between the high-risk and low-risk groups in the
primary cohort (P<0.01), which successfully provided
confirmation in this process of validation (Fig. 2B). Subsequently, the signatures were
tested using an ROC analysis of the validation cohort and the
results revealed the AUC was 0.70, indicating that the signature
was an effective predictor for LACA, although the AUC was lower
than that in the primary cohort (Fig.
3B).
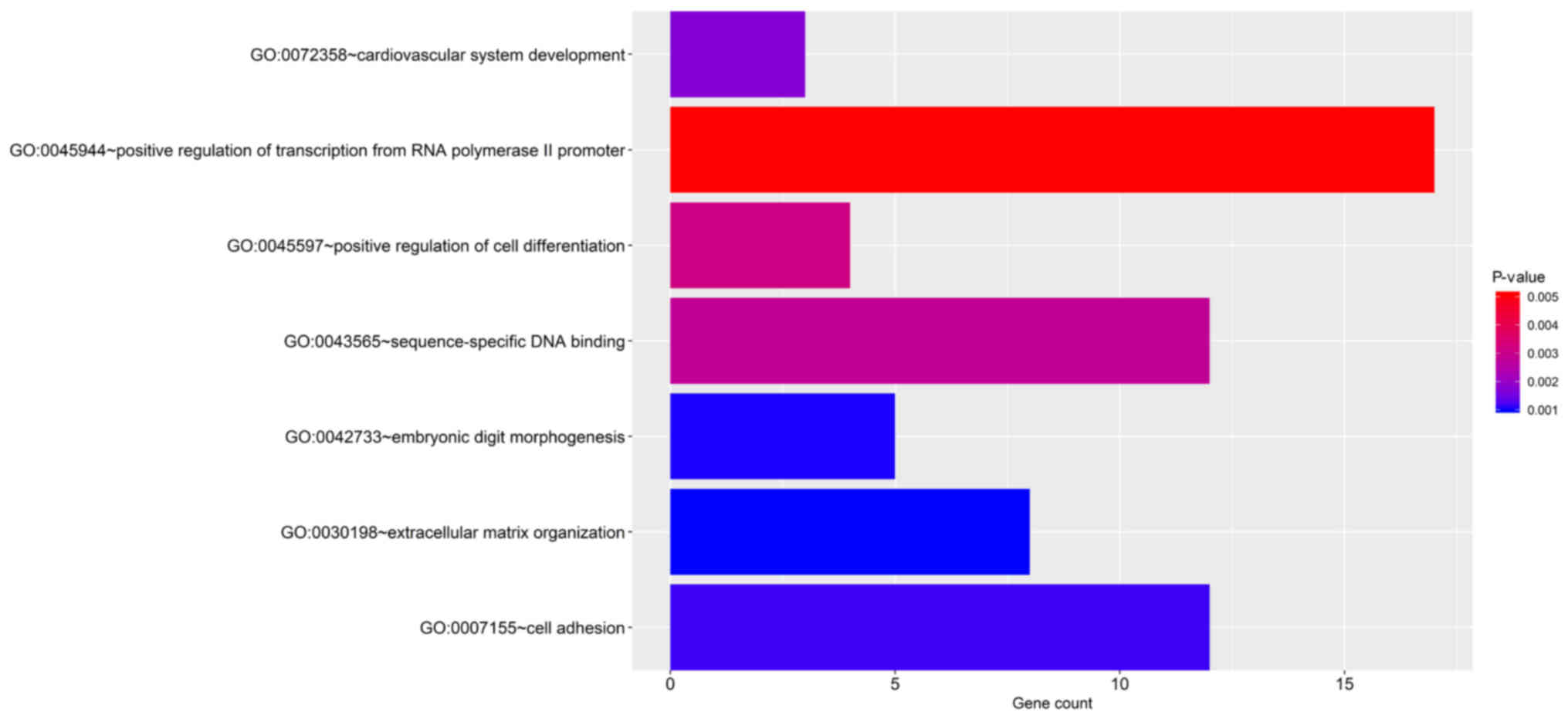
Functional enrichment analysis
The significant terms of the GO enrichment analysis
performed by DAVID and the KEGG pathways are provided in Fig. 4 and Fig.
S2. The genes were significantly enriched KEGG pathways
including extracellular matrix (ECM)-receptor interaction, cell
adhesion molecules, PI3K-Akt signaling pathway, dilated
cardiomyopathy and cysteine and methionine metabolism. In addition,
the GO biological process terms were mainly enriched in ECM
organization, embryonic digit morphogenesis, cell adhesion,
cardiovascular system development, sequence-specific DNA binding,
positive regulation of transcription from RNA polymerase II
promoter and positive regulation of cell differentiation.
Discussion
Due to high invasion and poor prognosis, the outcome
for patients with LACA remains unsatisfactory, with a 5-year OS
rate of 4–17%, depending on the stage and regional differences
(19,20). Most lung cancer patients are in the
advanced stages at the time of diagnosis. It is achievable to
enhance the efficiency of diagnosing and prognosticating LACA
patients once the indication of the tumor's presence is able to be
detected and explored at an early stage. Thus, in-depth studies on
the aetiological elements and progressive mechanisms, early
detection of the prognostic markers and identification of specific
methylation CpG sites are urgently required.
For the purpose of clear classification of different
secondary types of cancer, making use of CpG methylation locations
is probably more efficient than it would be to collect detailed
information on genetic representation based on covalent chemical
alterations and steady hysterogenic markers of conjugated
duplication (21). Hence, in the
present study, a signature of 13 CpG sites with differential
methylation was established, which were as follows: cg00002719,
cg02769743, cg05239163, cg05507908, cg07918170, cg08213398,
cg08516516, cg08623223, cg12748948, cg14904034, cg16007456,
ch.6.2958553R and cg19868631. The 13 CpG signature, which was
significantly associated with the OS of patients with LACA, was
also recognized as an independent element for diagnosing LACA and
predicting the prognosis of the patients. Furthermore, compared
with that of the high-risk patients, low-risk patients had better
OS, and the 5-year survival rate in low-risk patients was also
higher than that in high-risk patients. The functional enrichment
analysis for the mapped genes of the CpG methylation sites were all
presented by means of approaches of the fields of biology and
information technology. The outcomes indicate that the methylation
sites included in the 13-CpG-based prognostic signature may have a
role in the molecular pathogenetic mechanisms and clinical
progression in LACA patients, and this provides novel information
for survival prediction and personalized treatment of patients with
LACA.
As an important epigenetic mechanism in tumors, DNA
methylation has a critical role in carcinogenesis (22). It regulates the extent of gene
expression to control the function of the biomolecules encoded by
those genes (23,24). Numerous studies have revealed that
DNA methylation has a significant role in the initiation,
progression and metastasis of cancer by controlling different
aspects, including DNA repair, cell cycle regulation, angiogenesis
and apoptosis (25). In the present
study, 13 mapped genes were identified to be aberrantly methylated
in LACA, which were as follows: Coiled-coil domain containing 181
(CCDC181), transmembrane protein 201 (TMEM201), chromosome 1 open
reading frame 43 (C1orf43), ankyrin repeat domain 31 (ANKRD31),
tubulin folding cofactor D (TBCD), switching B cell complex subunit
SWAP70 (SWAP70), cysteine dioxygenase type 1 (CDO1), AF131216.1,
ubiquitin specific peptidase 48 (USP48), hippocalcin like 1
(HPCAL1), G protein-coupled receptor 88 (GPR88), spectrin repeat
containing nuclear envelope protein 1 (SYNE1) and V-set and
transmembrane domain containing 2A (VSTM2A). Previous studies have
directly shown that CCDC181, CDO1 and SYNE1 are associated with
LACA. Gao et al (26) stated
that CCDC181 could be a provisional prognostic biomarker of LACA.
Moreover, Diaz-Lagares et al (27) indicated that there is a possibility
for the cancer-specific methylation of CDO1 to enhance the
diagnosis approach at the early stage and also achievements for
patients. The methylation status of SYNE1 has also been valuable in
estimating the sporadic lung cancer prognosis (28). However, no previous studies have
reported on the association of TMEM201, C1orf43, ANKRD31, TBCD,
SWAP70, AF131216.1, USP48, HPCAL1, GPR88 or VSTM2A and LACA. Hence,
it is necessary to perform further studies on the methylation of
these 10 genes and LACA.
In addition, GO annotations and KEGG pathways were
established to provide detailed information regarding the molecular
functions of the genes. The differentially methylated genes were
mainly enriched in ECM, embryonic morphogenesis, cell adhesion and
vascular system development; these are why high-risk tumors are
more biologically aggressive. Generated through direct diffusion,
lymphatic and vascular metastases are the common metastases in
LACA. Once metastasis occurs, the prognosis of patients with LACA
is poor. Increasing evidence has indicated that the ECM has a
significant role in tumor occurrence and progression (29). Lim et al (30) developed a 29-gene ECM-associated
signature to predict the prognosis of the patients at the early
stage of NSCLC. Furthermore, it has been well established that the
ECM is involved in regulating metastasis and invasion of lung
cancer (31,32). Thus, it is necessary to perform
in-depth research on these molecules to confirm these predictions,
and simultaneously develop novel therapeutic interventions for
LACA.
The present study does have certain limitations.
First, in view of the LACA cohort exhibiting a reasonably high
censored rate, this probably had an effect on the credibility of
the Kaplan-Meier evaluation. Furthermore, as all of the samples
analyzed in the present study were acquired from TCGA only,
in-depth verification should be performed using independent
datasets. In addition, the mechanistic role of each components of
the prognostic signature remains to be investigated. Therefore,
experimental research on cancer cell lines may provide significant
information to further the understanding of their functional
roles.
In conclusion, a 13 CpG-based prognostic signature
for OS prediction in patients with LACA was obtained through
comprehensively analyzing DNA methylation. The present results
suggest that further research is required to validate the
diagnostic ability of the novel diagnostic model in LACA.
Retrospective and prospective studies may be performed to verify
the prognostic utility of the CpG-based signature model.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RZ, HX, WM, ZD and MH participated in the study
design, analysis and interpretation of data and drafting of the
manuscript. MW performed the research and revised the paper. XG
contributed to the acquisition of data, collection of relevant
literature and drafting of the article together. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung carcinoma
|
|
LACA
|
lung adenocarcinoma
|
|
TCGA
|
The Cancer Genome Atlas
|
|
FC
|
fold change
|
|
OS
|
overall survival
|
|
ROC
|
receiver operating characteristic
|
|
LASSO
|
least absolute shrinkage and selection
operator
|
|
DAVID
|
Database for Annotation, Visualization
and Integrated Discovery
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
AUC
|
area under the ROC curve
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Locher C, Debieuvre D, Coëtmeur D, Goupil
F, Molinier O, Collon T, Dayen C, Le Treut J, Asselain B, Martin F,
et al: Major changes in lung cancer over the last ten years in
France: The KBP-CPHG studies. Lung Cancer. 81:32–38. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
B'chir F, Laouani A, Ksibi S, Arnaud MJ
and Saguem S: Cigarette filter and the incidence of lung
adenocarcinoma among Tunisian population. Lung Cancer. 57:26–33.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Woods LM, Coleman MP, Lawrence G, Rashbass
J, Berrino F and Rachet B: Evidence against the proposition that
‘UK cancer survival statistics are misleading’: Simulation study
with National Cancer Registry data. BMJ. 342:33992011. View Article : Google Scholar
|
|
4
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. Biochim
Biophys Acta. 1856:189–210. 2015.PubMed/NCBI
|
|
6
|
Travis WD: Pathology of lung cancer. Clin
Chest Med. 32:669–692. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ganti AK and Mulshine JL: Lung cancer
screening: Panacea or Pipe dream? Ann Oncol. 16:215–219. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Field JK and Raji OY: The potential for
using risk models in future lung cancer screening trials. F1000 Med
Rep. 24:382010.
|
|
9
|
Momparler RL: Cancer epigenetics.
Oncogene. 22:6479–6483. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng H, Cao Y, Qin J, Song X, Zhang Q, Shi
Y and Cao L: DNA methylation, its mediators and genome integrity.
Int J Biol Sci. 11:604–617. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klutstein M, Nejman D, Greenfield R and
Cedar H: DNA methylation in cancer and aging. Cancer Res.
76:3446–3450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berdasco M and Esteller M: Aberrant
epigenetic landscape in cancer: How cellular identity goes awry.
Dev Cell. 19:698–711. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Senner CE: The role of DNA methylation in
mammalian development. Reprod Biomed Online. 22:529–535. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Cancer Institute, . The Cancer
Genome Atlas Program. http://cancergenome.nih.gov/publications/publicationguidelinesDecember
21–2015
|
|
15
|
Sauerbrei W, Royston P and Binder H:
Selection of important variables and determination of functional
form for continuous predictors in multivariable model building.
Stat Med. 26:5512–5528. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang DW, Sherman BT, Tan Q, Kir J, Liu D,
Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki RA:
DAVID Bioinformatics Resources: Expanded annotation database and
novel algorithms to better extract biology from large gene lists.
Nucleic Acids Res. 35:169–175. 2007. View Article : Google Scholar
|
|
17
|
Merico D, Isserlin R, Stueker O, Emili A
and Bader GD: Enrichment map: A network-based method for gene-set
enrichment visualization and interpretation. PLoS One. 5:139842010.
View Article : Google Scholar
|
|
18
|
R Core Team, . R: A language and
environment for statistical computingR Foundation for Statistical
Computing; Vienna: 2014
|
|
19
|
American Cancer Society, . Cancer facts
& figures 2015American Cancer Society; Atlanta, GA: 2015
|
|
20
|
Grinberg-Rashi H, Ofek E, Perelman M,
Skarda J, Yaron P, Hajdúch M, Jacob-Hirsch J, Amariglio N, Krupsky
M, Simansky DA, et al: The expression of three genes in primary
non-small cell lung cancer is associated with metastatic spread to
the brain. Clin Cancer Res. 15:1755–1761. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moore LD, Le T and Fan G: DNA methylation
and its basic function. Neuropsychopharmacology. 38:23–38. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lorincz MC and Schübeler D: Evidence for
converging DNA methylation pathways in placenta and cancer. Dev
Cell. 43:257–258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding YX and Cui H: Integrated analysis of
genome-wide DNA methylation and gene expression data provide a
regulatory network in intrauterine growth restriction. Life Sci.
179:60–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lim JH, Kim SY, Han JY, Kim MY, Park SY
and Ryu HM: Comprehensive investigation of DNA methylation and gene
expression in trisomy 21 placenta. Placenta. 42:17–24. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Feng H, Li D, Liu S, Amizuka N
and Li M: Identification of differentially expressed genes induced
by aberrant methylation in oral squamous cell carcinomas using
integrated bioinformatic analysis. Int J Mol Sci. 19:16982018.
View Article : Google Scholar
|
|
26
|
Gao C, Zhuang J, Li H, Liu C, Zhou C, Liu
L and Sun C: Exploration of methylation-driven genes for monitoring
and prognosis of patients with lung adenocarcinoma. Cancer Cell
Int. 18:1942018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Diaz-Lagares A, Mendez-Gonzalez J, Hervas
D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga
LM, Zulueta J, et al: A novel epigenetic signature for early
diagnosis in lung cancer. Clin Cancer Res. 22:3361–3371. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tessema M and Belinsky SA: Mining the
epigenome for methylated genes in lung cancer. Proc Am Thorac Soc.
5:806–810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Multhaupt HA, Leitinger B, Gullberg D and
Couchman JR: Extracellular matrix component signaling in cancer.
Adv Drug Deliv Rev. 97:28–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lim SB, Tan SJ, Lim WT and Lim CT: An
extracellular matrix-related prognostic and predictive indicator
for early-stage non-small cell lung cancer. Nat Commun. 8:17342017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng DH, Ungewiss C, Tong P, Byers LA,
Wang J, Canales JR, Villalobos PA, Uraoka N, Mino B, Behrens C, et
al: ZEB1 induces LOXL2-mediated collagen stabilization and
deposition in the extracellular matrix to drive lung cancer
invasion and metastasis. Oncogene. 36:1925–1938. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stevens LE, Cheung WKC, Adua SJ,
Arnal-Estapé A, Zhao M, Liu Z, Brewer K, Herbst RS and Nguyen DX:
Extracellular matrix receptor expression in subtypes of lung
adenocarcinoma potentiates outgrowth of micrometastases. Cancer
Res. 77:1905–1917. 2017. View Article : Google Scholar : PubMed/NCBI
|