Introduction
Infantile neuroaxonal dystrophy (INAD; Online
Mendelian Inheritance in Man no. 256600), is an extremely rare
autosomal recessive neurodegenerative disorder involving axons in
the central and peripheral nervous system. The diagnosis of INAD is
difficult due to the frequent occurrence of atypical cases and lack
of specific early signs (1). The
clinical manifestation includes progressive psychomotor regression
with onset between 6 months and 2 years of age, and usually leads
to death by the age of 10 years (2).
Most patients with INAD display a progressive disorder with motor
and mental deterioration, cerebellar ataxia, spastic tetraplegia,
hyperreflexia and early visual disturbances (3).
INAD is caused by loss of the ability of
calcium-independent group VI phospholipase A2 (PLA2G6) to catalyze
fatty acid release from phospholipids (4). The PLA2G6 gene is mapped to chromosome
22q and encodes a 85-kDa protein, which is also known as
calcium-independent phospholipase A2 β (5). At present, no effective treatments are
available for INAD. The carrier couple had a child affected by
INAD, which implies a 25% recurrence risk for future pregnancies.
Preimplantation genetic diagnosis (PGD) is a powerful tool for
preventing this neurodegenerative disorder without facing the
trauma of termination of pregnancy in the case of an affected
fetus.
PGD as an alternative to current prenatal diagnoses
for severe lethal inherited diseases has been in use for >2
decades since its introduction by Handyside et al (6) in 1990. The technique has been used not
only for single-gene disorders (SGD) to avoid the risk of having an
affected child (7), but also for
chromosomal abnormalities, human leukocyte antigen matching
(8), mitochondrial disease (9) and hereditary cancer syndrome (10).
Compared to prenatal diagnosis, PGD spares parents
from the physical and emotional trauma of the termination of
pregnancy in the case of an affected fetus. The sensitivity and
efficiency of PGD to detect SGDs is restricted by amplification
failure or allelic dropout due to the limited amount of template
DNA in a single embryonic cell (11). To solve these problems, blastocyst
biopsy for inputting more cells and whole genome amplification
(WGA) combined with linkage analysis (haplotyping analysis) have
been applied to improve the accuracy of PGD (12,13).
Linkage analysis is a method to deduce the
inheritance of mutation alleles by detecting short tandem repeats
(STRs) or single nucleotide polymorphisms (SNPs). The STR approach
is not only labor-intensive and time-consuming, but also often less
informative due to the limited number and uneven distribution of
polymorphic markers in certain pedigrees (14). The introduction of karyomapping
(15–17) hold promise to markedly change the way
molecular diagnostics are performed during PGD with higher
efficiency, accuracy and reliability. Karyomapping technology for
PGD makes use of the abundance of the ‘informative’ SNPs in
pedigrees, which allows for simultaneous detection of monogenic and
chromosomal disorders. However, karyomapping has its limitations.
For instance, for certain genes, which have low SNP coverage,
polymerase chain reaction (PCR) testing, including STR analysis or
direct mutation detection, is required to be performed in parallel
(18). Thus, it would be more
practical if specific SNPs were to be identified for rare inherited
diseases with higher SNP densities near the causal genes (14).
With the advent of next-generation sequencing (NGS)
techniques, an opportunity arises to perform cost-efficient genetic
testing by sequencing in different clinical scenarios, which may
contribute to definitive improvements in the genetic assessment of
embryos prior to transfer to the uterus (19). NGS-based methods, including Mutated
Allele Revealed by Sequencing with Aneuploidy and Linkage Analyses
and haplotyping analysis, have been successfully applied in SGDs
and chromosomal copy number assessment (7,18,20,21).
In the present study, NGS-based haplotyping analysis
combined with aneuploidy screening was applied in the PGD for INAD.
The patient got pregnant successfully in the second frozen-thawed
embryo transfer cycle. To the best of our knowledge, the present
study is the first to report on PGD for INAD.
Materials and methods
Participants
The couple (maternal age, 26 years and paternal age,
27 years) was referred to the Reproductive Medicine Center of The
First Affiliated Hospital of Anhui Medical University (Hefei,
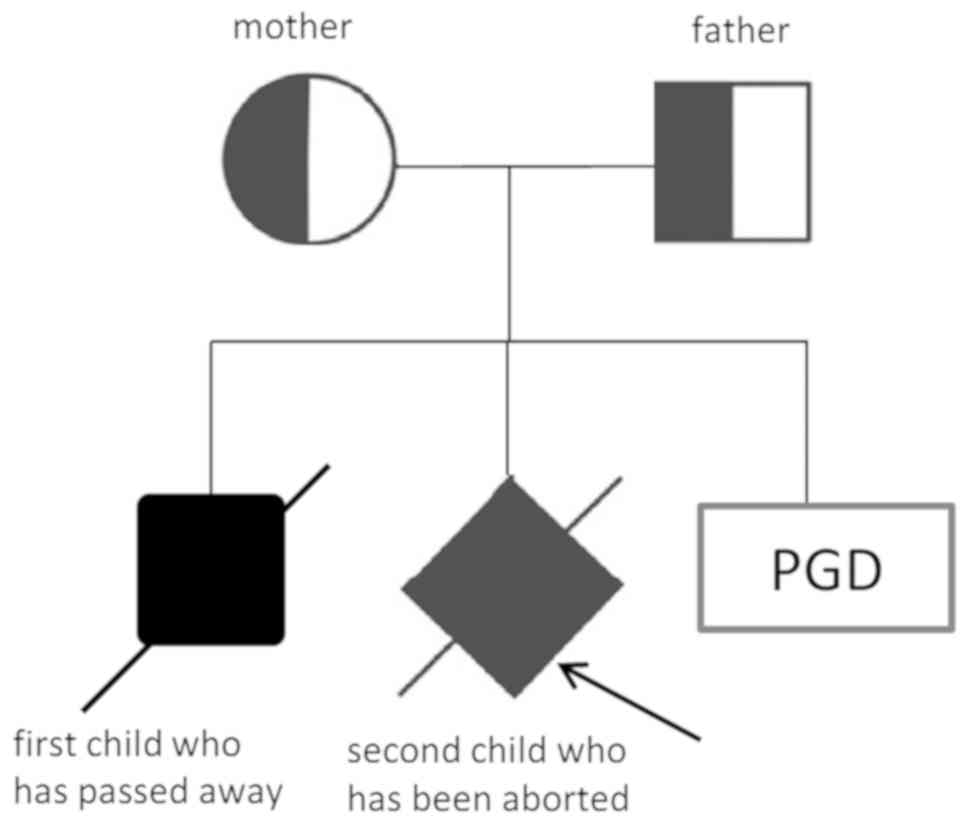
China) due to an adverse birth history in two cases (Fig. 1). Their first son suffered from
dystonia at 2 years of age, followed by the development of
retardation, and he died at 5 years of age with a diagnosis of
INAD. DNA testing of the son by targeted-next generation sequencing
of PLA2G6 revealed the compound heterozygous mutations c.692G>T
(p.G231V) in exon 5 and c.2213_2220delCAGACGGG (p.Asp739GlyfsX29)
in exon 16. The parents, with their son, had visited the Department
of Neurology in Beijing Children's Hospital (Beijing, China) in
February 2012 and the gene diagnosis of their first son had been
confirmed there. Segregation analysis revealed the c.692G>T
mutation in exon 5 in the mother and the c.2213_2220delCAGACGGG
mutation in exon 16 in the father in a heterozygous state. The
genetic diagnosis of amniocentesis for prenatal diagnosis during
the second pregnancy in May 2014 revealed the same compound
heterozygous mutations as those in their first son and labor
induction was therefore performed. The couple received counselling
regarding PGD in Reproductive Medicine Center of the First
Affiliated Hospital of Anhui Medical University in March 2016 in
order to have a child without INAD. This study underwent an EMRO
process
Fertilization, embryo culture, biopsy,
vitrification and WGA
A long protocol was used for ovarian stimulation.
Following ovarian stimulation, follicles were aspirated at 34–36 h
after human chorionic gonadotropin injection and fertilized by
intracytoplasmic sperm injection (ICSI). The following morning (day
1), each injected oocyte was checked for pronuclei to confirm
fertilization. All embryos were cultured to the blastocyst stage
(day 5 or 6) and scored according to Gardner's grading scale
(22). Hatched blastocysts were
biopsied using a 30-µm inner diameter biopsy pipette (Cook Medical,
Bloomington, IN, USA) with the ZILOS-tk laser (Hamilton Thorne,
Inc., Beverly, MA, USA). Biopsied trophectoderm cells were then
transferred to sterile 0.2-ml PCR tubes supplemented with 2.5 µl
PBS, which was later subjected to whole-genome amplification (WGA).
Biopsied blastocysts were then vitrified according to the protocol
recommended in the Kitazato vitrification kit using Kitazato
vitrification solution (Kitazato Biopharma Co. Ltd., Shizuoka,
Japan). Multiple displacement amplification (MDA) was performed
using a REPLI-g Single Cell kit (Qiagen, Hilden, Germany) for
whole-genome amplification.
Linkage analysis by NGS-based SNP
haplotyping and aneuploidy screening
Regarding the PLA2G6 gene (GenBank ID, NM_003560.2;
chr22:38507502-38577857; reverse transcription product length, 70
Kb) as a target region, a panel of 100 high-frequency SNP markers
located 2 Mb upstream and downstream of the PLA2G6 gene in the
genomes of Han Chinese in Beijing and Southern Han Chinese from the
1,000 Genomes Project and the maternal mutation site were selected
for NGS-based SNP haplotyping. Primers were designed using AmpliSeq
Designer (https://www.ampliseq.com). Target
regions were amplified by multiplex PCR (23). The WGA products and designed primers
were used for library preparation with Ion AmpliSeq Library Kits
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). The template
preparation was performed using an Ion One Touch 2 system and an
Ion One Touch ES following instructions of the latest version of
the manuals (Ion Onetouch Template kit; Thermo Fisher Scientific,
Inc.). The template positive Ion Sphere Particles were sequenced on
an Ion Torrent Personal Genome Machine (PGM) (Life Technologies
Ltd.) according to the instructions of the Ion Sequencing kit v2.0
(Thermo Fisher Scientific, Inc.). The remaining WGA products were
simultaneously subjected to aneuploidy screening by NGS according
to a standard protocol using the Ion Torrent System (Thermo Fisher
Scientific, Inc.) as previously described (24). The data from the PGM sequencing were
analyzed by Peking Jabrehoo Med Tech., Ltd. (Beijing, China).
Sanger sequencing for the paternal mutation sites was performed in
order to verify the NGS results.
Blastocysts thawing and transfer
At three months after oocyte retrieval, the patient
was treated with oral Estradiol Valerate from day 3 to prepare the
endometrium for frozen embryo transfer. Luteal support by
administration of intramuscular progesterone was applied when a
satisfactory endometrial development (thickness, ≥8 mm) was
confirmed on ultrasound. The embryo unaffected by INAD, which had
normal chromosomes or mosaicism, was thawed and transferred.
Clinical pregnancy was confirmed when an intrauterine gestational
sac with heartbeat was observed by ultrasound examination at 35 and
65 days after embryo transfer. Amniocentesis was performed at 18
weeks of gestation. Linkage analysis and aneuploidy screen were
performed using NGS. Karyotype analysis was also performed.
Results
ICSI, biopsy and MDA
A total of 40 cumulus-oocyte complexes were
retrieved, and 40 metaphase-II oocytes were subjected to ICSI for
fertilization. A total of 15 embryos reached the hatched blastocyst
stage of development on day 5 or 6 post-ICSI. Biopsies (5–10
trophoblastic ectoderm cells) from all blastocysts were
successfully amplified by MDA.
Linkage analysis by NGS-based SNP
haplotyping and NGS-based aneuploidy screen
Based on the SNPs in the proband, heterozygote SNPs
in the father and heterozygote SNPs in the mother were selected at
the same loci to construct the haplotypes of the blastocysts. The
haplotypes were constructed by including 77 informative SNPs from
100 selected high-frequency SNP markers and the maternal mutation
site (Fig. 2). The remaining MDA
products were simultaneously subjected to aneuploidy screening
(Fig. 3; Table I).
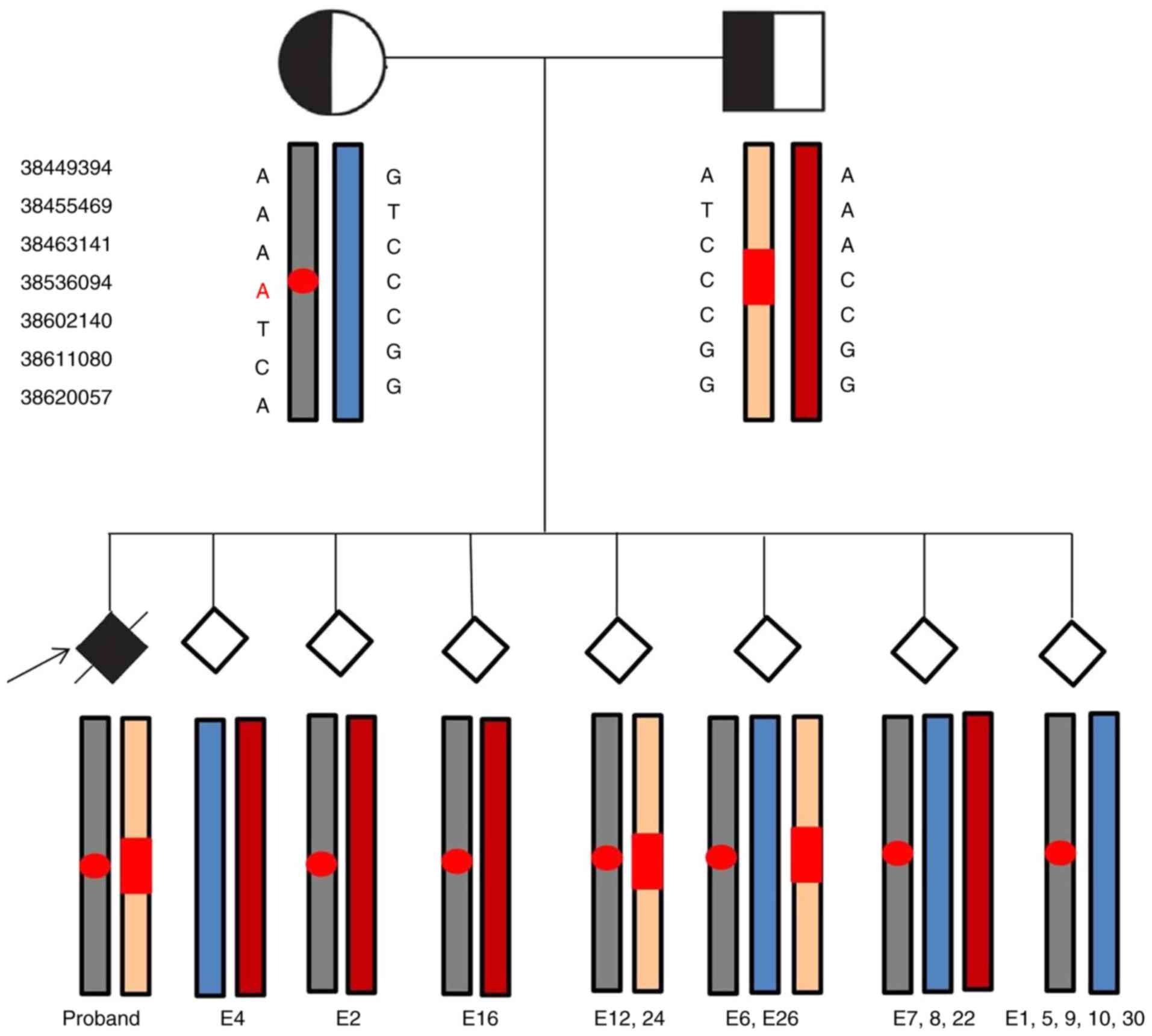 | Figure 2.Next-generation sequencing-based
single-nucleotide polymorphism haplotyping for infantile
neuroaxonal dystrophy diagnosis. Gray and blue represent pathogenic
and normal haplotypes of the mother, respectively, while beige and
red represent the pathogenic and normal haplotypes of the father,
respectively. Gene mutation sites were marked with red circles
(maternal) and red rectangles (paternal). E4 was genotypically
normal, embryos 2 and 16 exhibited a carrier pattern, and E12 and
−24 were affected. E6, −26, −7, −8 and −22 were diagnosed as having
trisomies. Uniparental disomy occurred in E1, −5, −9, −10 and −30.
E, embryo. |
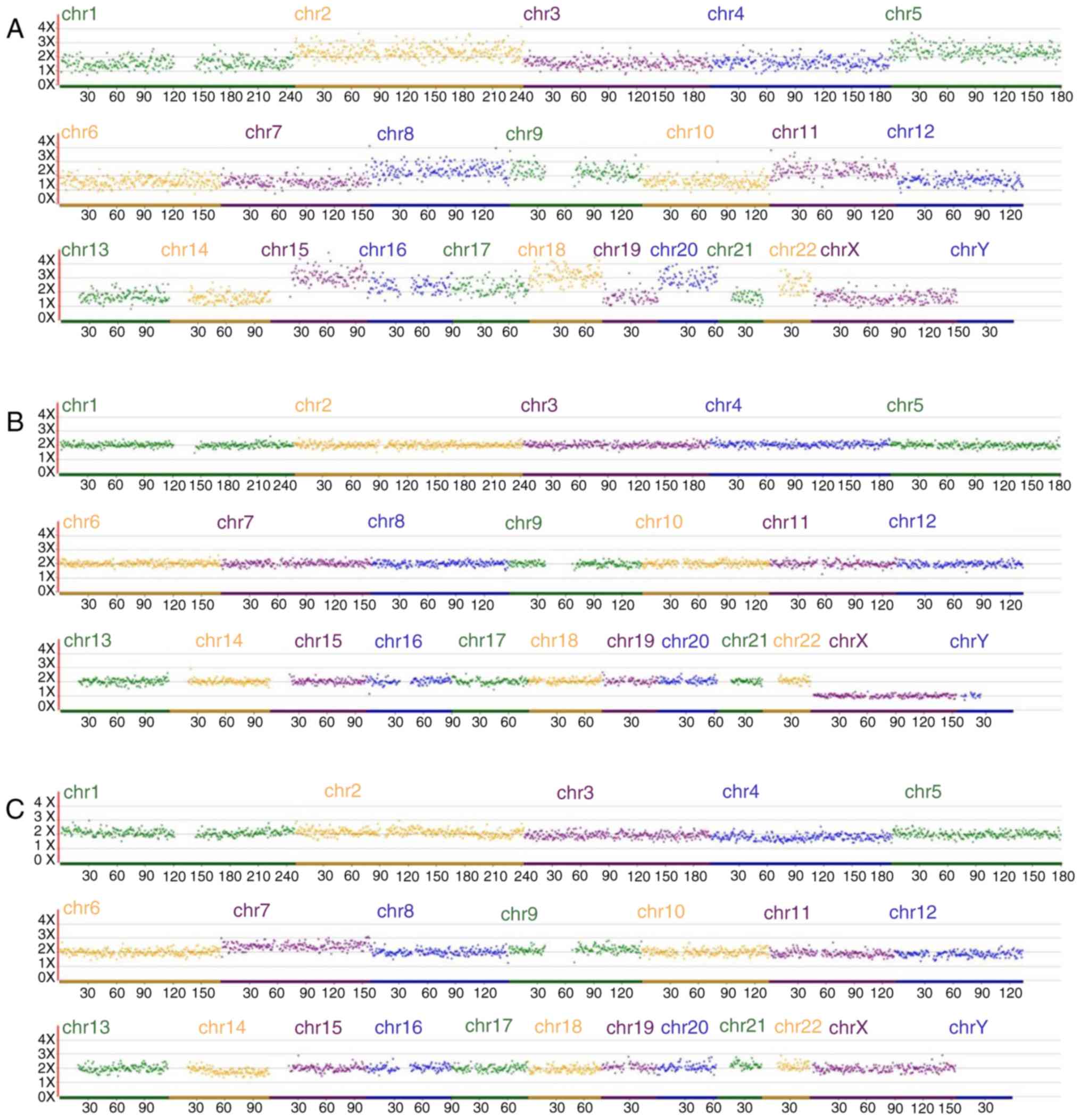 | Figure 3.NGS-based aneuploidy screen for
embryos. (A) NGS-based aneuploidy screen for embryo 1: Segmental
imbalances for chr 2, 5, 8, 9, 11, 15, 16, 18, 20 and 22. (B)
NGS-based aneuploidy screen for embryo 4: Normal. (C) NGS-based
aneuploidy screen for embryo 2: Mosaicism for trisomy 7. NGS,
next-generation sequencing; chr, chromosome. |
 | Table I.Summary of pre-implantation genetic
diagnosis results regarding infantile neuroaxonal dystrophy. |
Table I.
Summary of pre-implantation genetic
diagnosis results regarding infantile neuroaxonal dystrophy.
| Embryo no. | NGS-based linkage
analysis | NGS-based
aneuploidy screening |
|---|
| 1 | UPD | +2(p25.3->p25.1)
(9.82 Mb) |
|
|
|
+5(p15.33->q13.3) (71.48 Mb) |
|
|
| +8(q22.3->q24.3)
(33.71 Mb) |
|
|
| +9(p24.3->p21.1)
(29.72 Mb) |
|
|
|
+11(p15.1->p14.1) (8.71 Mb) |
|
|
|
+15(q11.1->q26.3) (78.30 Mb) |
|
|
|
+16(p13.3->p11.2) (23.34 Mb) |
|
|
| +18(p11.32->q23)
(73.91 Mb) |
|
|
| +20(p13->q13.33)
(59.93 Mb) |
|
|
|
+22(q11.1->q13.33) (33.13 Mb) |
| 2 | Carrier | Mosaicism for
trisomy 7 (q33->q36.3) (21.50 Mb) |
| 4 | Normal | Normal |
| 5 | UPD | Normal |
| 6 | Trisomy | +4(p15.1->q34.3)
(136.59 Mb), |
|
|
| +6(p25.3->p21.1)
(42.11 Mb), |
|
|
|
+8(p23.3->q21.11) (72.54 Mb), |
|
|
|
+12(q24.11->q24.31) (14.08 Mb) |
| 7 | Trisomy | Normal |
| 8 | Trisomy | +2(q37.1->q37.3)
(7.14 Mb), |
|
|
| +3(p26.3->p22.2)
(36.54 Mb), |
|
|
| +3(p13->q29)
(120.18 Mb), |
|
|
| -X(q11.1->q28)
(88.10 Mb) |
| 9 | UPD | Normal |
| 10 | UPD | Normal |
| 12 | Pathogenic | Normal |
| 16 | Carrier |
+1(p36.13->p35.2) (11.94 Mb), |
|
|
| +2(p23.1->p22.3)
(4.21 Mb), |
|
|
| −4(q33->q34.3)
(7.25 Mb), |
|
|
| +5(p13.3->q14.3)
(56.95 Mb), |
|
|
|
+6(p22.3->p21.31) (12.90 Mb) |
| 22 | Trisomy | Abnormal |
| 24 | Pathogenic | Normal |
| 26 | Trisomy |
+1(p36.21->p32.1) (45.95 Mb), |
|
|
| −2(p25.1->p24.2)
(6.64 Mb), |
|
|
| +3(p26.3->p25.2)
(11.43 Mb), |
|
|
| −4(q12->q13.1)
(5.49 Mb) |
| 30 | UPD |
−1(p36.21->p36.12)(5.66Mb), |
|
|
|
+1(q25.1->q25.2)(5.58Mb), |
|
|
|
+2(p24.1->p23.2)(6.26Mb), |
|
|
|
−3(p26.3->p25.1)(15.52Mb), |
|
|
|
+4(q21.1->q22.1)(13.23Mb) |
Embryo 4 was diagnosed as being unaffected by INAS
and exhibiting euploidy. Embryos 2 and 16 were diagnosed as
carriers of INAS, with embryo 2 inheriting the maternal affected
haplotype and the normal paternal haplotype, whereas embryo 16
inheriting the paternal affected haplotype and the normal maternal
haplotype. Embryos 12 and 24 had pathogenic haplotypes, inheriting
both affected parental haplotypes similar to the previous affected
offspring. Embryos 6, 7, 8, 22 and 26 were diagnosed as having
trisomy. Embryos 1, 5, 9, 10 and 30 were identified to have
uniparental disomy (UPD), inheriting both maternal haplotypes.
NGS-based aneuploidy screening revealed that embryos 4, 5, 7, 9,
10, 12 and 24 were normal and embryo 2 displayed mosaicism for
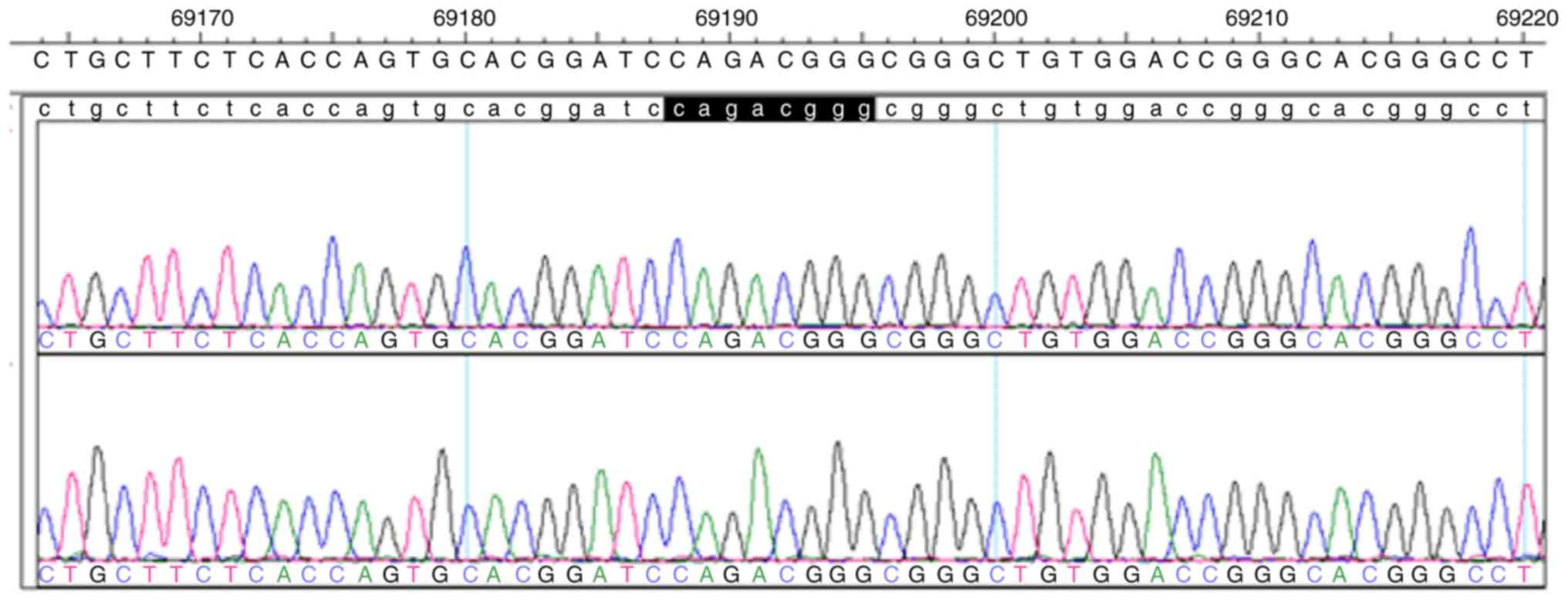
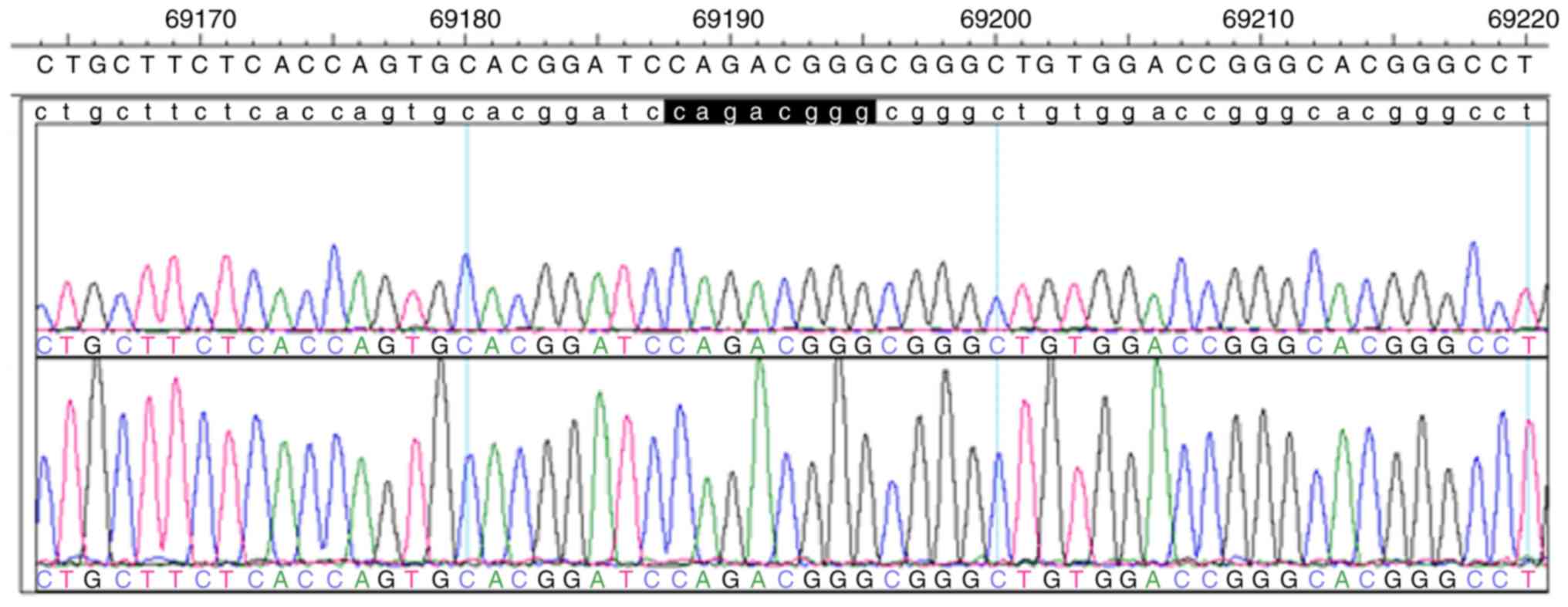
trisomy 7 (Fig. 3). Sanger
sequencing analysis of the paternal mutation site indicated that
embryos 2 and 4 were void of this mutation site (Figs. 4 and 5). On the basis of the linkage analysis and
aneuploidy results, only embryos 2 and 4 were considered for
transfer.
Clinical outcome
The unaffected embryo 4 was successfully thawed
three months after it was generated. However, no clinical pregnancy
was achieved. In the subsequent frozen-thawed cycle, the couple
asked for transfer of the carrier embryo 2, although it was mosaic.
The couple informed of relevant risks prior to transfer and a
consent form was signed. Embryo 2 was then transferred and resulted
in a successful pregnancy. Transvaginal ultrasonography examination
on day 35 and 65 revealed a single intrauterine gestational sac
with a normal fetal heartbeat. Prenatal diagnosis was performed
using amniotic fluid cells, whose results were consistent with
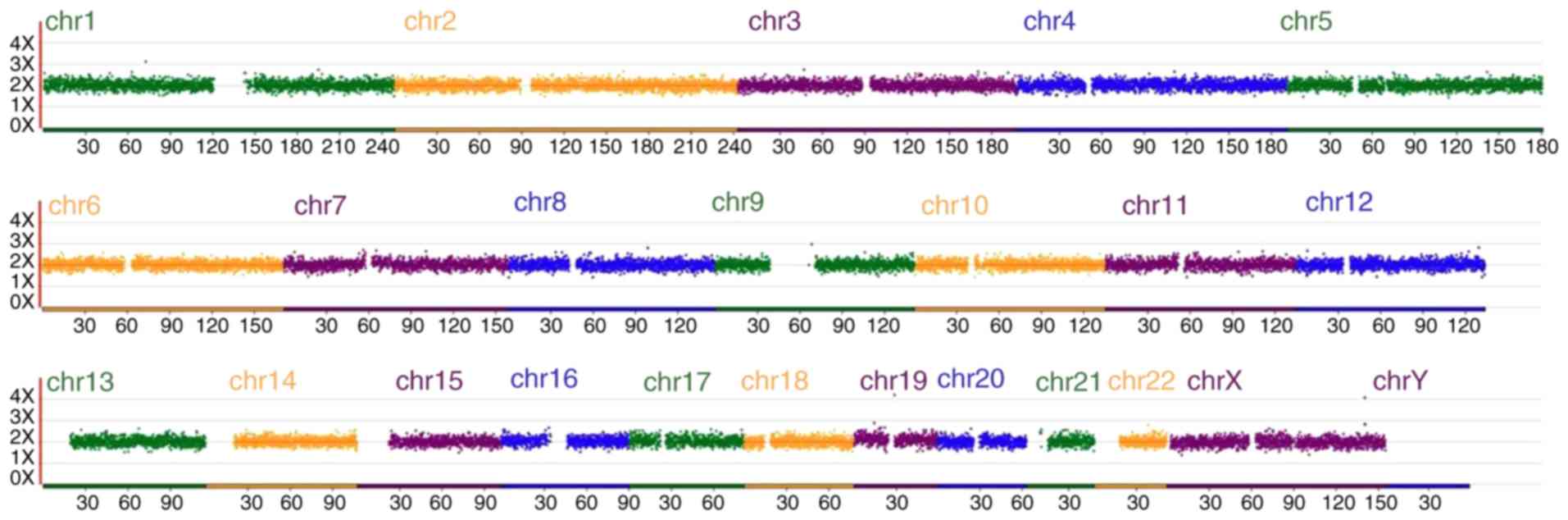
those of the PGD. The aneuploidy screen (Fig. 6) and karyotype analysis indicated
that the chromosomes of the fetus were normal and not mosaic. The
full-term female baby was born by normal Apgar score, with a birth
weight of 3600 g, and a head circumference was 50 cm.
Discussion
To the best of our knowledge, the present study was
the first to report on PGD in parallel with an aneuploidy screen
for INAD.
INAD is an extremely rare, lethal, autosomal
recessive neurodegenerative disorder. Disease progression is rapid,
and numerous affected children never learn to walk or lose the
ability shortly after attaining it. Severe spasticity, progressive
cognitive decline and visual impairment typically result in death
during the first decade of life and no effective treatments are
available, thus emphasizing the importance of preventing this
lethal disease (25). In the present
study, during PGD, detection of SNPs in the vicinity of the
mutation by NGS offers the advantage of a one platform technology
with less processing of samples and lower labor costs. Compared
with previous methods of linkage analysis by short tandem repeats
(STR), NGS-based SNP haplotyping provides more informative genetic
markers in high throughput (7) and
the inclusion of more multiple linked SNPs close to the targeted
mutation in NGS allows for more accurate measurement than STR. On
the basis of NGS-based linkage analysis, trisomies and monosomies
may be identified by the presence of both haplotypes from one
parent or absence of either chromosome haplotype from the parent of
origin (26). In the present study,
NGS-based linkage analysis revealed trisomies in five embryos (nos.
6, 7, 8, 22 and 26), with the extra haplotype being the maternal
one.
Furthermore, when compared to other techniques,
including array-based comparative genomic hybridization (CGH) and
quantitative fluorescent PCR, this method has the advantage that
general features of the proband's haplotype may be detected,
including UPD and recombination. For embryos 1, 5, 9, 10 and 30,
examination of the NGS revealed that no paternal chromosomes were
present and both haplotypes were from the maternal pair, which is
called heterodisomy. UPD for a complete chromosome may appear due
to post-fertilization error, gamete complementation,
trisomic/monosomic rescue, mitotic error, isochromosome formation,
deletion and duplication (27).
Segmental UPD may arise from a postzygotic somatic recombination
between the maternal and paternal homologue, or in connection with
numerical and/or structural chromosomal aberrations (27). It is also a novel mechanism for the
occurrence of INAD (28). In the
present study, 5 embryos were diagnosed with UPD by NGS-based SNP
haplotyping, exemplifying the high efficiency of this
technique.
Given the high prevalence of embryonic aneuploidy,
particularly in mothers of advanced reproductive age, unaffected
embryos remain at high risk of implantation failure or pregnancy
loss due to aneuploidy (29).
Therefore, single-gene PGD in conjunction with aneuploidy screening
is recommended, as a normal genotyping result does not necessarily
guarantee that the embryo is also euploid. The importance of
screening for aneuploidy and SGDs were also demonstrated in the
present study. Embryos 1, 6, 8, 16, 22, 26 and 30 were aneuploidy.
As sequencing costs decrease further, allowing for a greater read
depth per sample for the same or a reduced price, NGS approaches
provide simultaneous evaluation of single-gene disorders and
translocations with comprehensive aneuploidy screening from the
same biopsy without the requirement for multiple technological
platforms (7,18,30,31).
Compared to array CGH or SNP array, the NGS approach not only
detects mosaic blastocysts, but may also be effective in
characterizing small abnormal chromosomal fragments (24). In the present study, NGS-based
24-aneuploidy screening revealed that embryo 2 was mosaic; the
method allows for accurate detection of segmental imbalances as
small as ~4 Mb in size due to the high resolution. During the
second frozen-thawed embryo transfer cycle, although embryo 2 was
mosaic for trisomy 7, it had the potential to achieve a full-term
pregnancy (32–34). The couple had a strong demand for
transferring this mosaic embryo and were informed of relevant
risks. Fortunately, the result of the prenatal diagnosis revealed
that the chromosomes were normal, indicating the value of this
mosaic embryo.
Nowadays, karyomapping has the potential for
providing a simultaneous identification of aneuploidy and SGD, but
since karyomapping has not yet been fully validated for this
purpose, it was decided that array CGH is used in parallel in the
present study, in order to provide information regarding the
chromosomal status (15).
Karyomapping is not validated for microdeletions (26), and cannot detect sequence-identical
chromosome duplication that may result from malsegregation of
chromosomes during the early cleavage divisions of the embryo
(1). Furthermore, the dependence of
DNA samples from family members may limit its application.
NGS-based linkage analysis may still correctly diagnose the embryos
by using the affected embryo as the proband under the circumstance
of the absence of suitable affected family members (18). Chromosomal copy number assessment
based on NGS may offer two major advantages: i) Enhanced detection
of partial or segmental aneuploidies as a result of the potential
increase in chromosomal analysis resolution to a few Mb, and ii)
the potential automation of the sequencing library preparation to
minimize human errors, reduce hands-on time, and achieve higher
throughput and consistency (35,36).
In summary, the present study was the first to
report on successful PGD for INAD. The feasibility of an NGS-based
linkage analysis method for the selection of embryos for couples
carrying PLA2G6 mutations was demonstrated. With reduced cost, it
is expected that NGS-based analysis may enable PGD in parallel with
aneuploidy screen for all types of monogenetic disorders with a
known pathogenic gene mutation, thus preventing the occurrence of
severe genetic diseases, which will ultimately bring benefits for
the whole population.
Acknowledgements
Not applicable.
Funding
The current study is financially supported through
grants from Anhui provincial science and technology research
project (grant no. 1604a0802077), College Natural Science Project
of Anhui Province (grant no. KJ2019A0287) and the Central Guiding
Science and Technology Development of the Local Anhui Province
(grant no. 2018080802D0081).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YH contributed to writing the manuscript,
preparation of the manuscript and whole-genome amplification. DC,
GZ, XL and LX contributed to NGS data analysis. ZZ contributed to
embryo biopsy and embryo culture. PZ, ZW, XX and YC collected the
data of the patient, performed ovarian stimulation and revised the
manuscript. XH, ML, DJ, BC performed NGS. WZ, HW and YL contributed
to the literature review and vitrified embryos. All authors read
and approved the final manuscript.
Ethical approval and consent to
participate
The study was approved by the Ethics Committees of
the Anhui Medical University (approval no. 2017002). This study
underwent an EMRO process. The patients provided written informed
consent for undertaking PGD. Furthermore, the parents provided
written informed consent regarding the use of any embryos that are
not viable for implantation for scientific research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Iodice A, Spagnoli C, Salerno GG, Frattini
D, Bertani G, Bergonzini P, Pisani F and Fusco C: Infantile
neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: An
update for the diagnosis. Brain Dev. 39:93–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y,
Xiong H, Chang X, Bao X, Zhang Y, Xiao J and Wu X: Clinical study
and PLA2G6 mutation screening analysis in Chinese patients with
infantile neuroaxonal dystrophy. Eur J Neurol. 16:240–245. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khateeb S, Flusser H, Ofir R, Shelef I,
Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K and Birk
OS: PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J
Hum Genet. 79:942–948. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Engel LA, Jing Z, O'Brien DE, Sun M and
Kotzbauer PT: Catalytic function of PLA2G6 is impaired by mutations
associated with infantile neuroaxonal dystrophy but not
dystonia-parkinsonism. PLoS One. 5:e128972010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morgan NV, Westaway SK, Morton JE, Gregory
A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, et
al: PLA2G6, encoding a phospholipase A2, is mutated in
neurodegenerative disorders with high brain iron. Nat Genet.
38:752–754. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Handyside AH, Kontogianni EH, Hardy K and
Winston RM: Pregnancies from biopsied human preimplantation embryos
sexed by Y-specific DNA amplification. Nature. 344:768–770. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Treff NR, Fedick A, Tao X, Devkota B,
Taylor D and Scott RT Jr: Evaluation of targeted next-generation
sequencing-based preimplantation genetic diagnosis of monogenic
disease. Fertil Steril. 99:1377–1384.e6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kurekci E, Küpesiz A, Anak S, Öztürk G,
Gürsel O, Aksoylar S, Ileri T, Kuşkonmaz B, Eker İ, Cetin M, et al:
Hematopoietic stem cell transplantation using preimplantation
genetic diagnosis and human leukocyte antigen typing for human
leukocyte antigen-matched sibling donor: A Turkish Multicenter
Study. Biol Blood Marrow Transplant. 23:790–794. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sallevelt SC, Dreesen JC, Drüsedau M,
Hellebrekers DM, Paulussen AD, Coonen E, van Golde RJ, Geraedts JP,
Gianaroli L, Magli MC, et al: PGD for the m.14487 T>C mitoch
ondrial DNA mutation resulted in the birth of a healthy boy. Hum
Reprod. 32:698–703. 2017.PubMed/NCBI
|
|
10
|
Lee VC, Chow JF, Lau EY, Kwong A, Leung
SY, Yeung WS, Ho PC and Ng EH: Preimplantation genetic diagnosis
for hereditary cancer syndrome: Local experience. Hong Kong Med J.
22:289–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dreesen J, Destouni A, Kourlaba G, Degn B,
Mette WC, Carvalho F, Moutou C, Sengupta S, Dhanjal S, Renwick P,
et al: Evaluation of PCR-based preimplantation genetic diagnosis
applied to monogenic diseases: A collaborative ESHRE PGD consortium
study. Eur J Hum Genet. 22:1012–1018. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen J, Cram DS, Wu W, Cai L, Yang X, Sun
X, Cui Y and Liu J: Successful PGD for late infantile neuronal
ceroid lipofuscinosis achieved by combined chromosome and TPP1 gene
analysis. Reprod Biomed Online. 27:176–183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Trachoo O, Satirapod C, Panthan B,
Sukprasert M, Charoenyingwattana A, Chantratita W, Choktanasiri W
and Hongeng S: First successful trial of preimplantation genetic
diagnosis for pantothenate kinase-associated neurodegeneration. J
Assist Reprod Genet. 34:109–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu X, Xu Y, Sun J, Zhang Z, Wang J, Ding
C, Zheng SL, Xu J and Zhou C: Preimplantation genetic haplotyping
for six Chinese pedigrees with thalassemia using a single
nucleotide polymorphism microarray. Prenat Diagn. 37:460–468. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giménez C, Sarasa J, Arjona C, Vilamajó E,
Martínez-Pasarell O, Wheeler K, Valls G, Garcia-Guixé E and Wells
D: Karyomapping allows preimplantation genetic diagnosis of a
de-novo deletion undetectable using conventional PGD technology.
Reprod Biomed Online. 31:770–775. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Handyside AH, Harton GL, Mariani B,
Thornhill AR, Affara N, Shaw MA and Griffin DK: Karyomapping: A
universal method for genome wide analysis of genetic disease based
on mapping crossovers between parental haplotypes. J Med Genet.
47:651–658. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Natesan SA, Bladon AJ, Coskun S, Qubbaj W,
Prates R, Munne S, Coonen E, Dreesen JC, Stevens SJ, Paulussen AD,
et al: Genome-wide karyomapping accurately identifies the
inheritance of single-gene defects in human pre-implantation
embryos in vitro. Genet Med. 16:838–845. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren Y, Zhi X, Zhu X, Huang J, Lian Y, Li
R, Jin H, Zhang Y, Zhang W, Nie Y, et al: Clinical applications of
MARSALA for preimplantation genetic diagnosis of spinal muscular
atrophy. J Genet Genomics. 43:541–547. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Martín J, Cervero A, Mir P,
Martinez-Conejero JA, Pellicer A and Simón C: The impact of
next-generation sequencing technology on preimplantation genetic
diagnosis and screening. Fertil Steril. 99:1054–1061.e3. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan L, Huang L, Xu L, Huang J, Ma F, Zhu
X, Tang Y, Liu M, Lian Y, Liu P, et al: Live births after
simultaneous avoidance of monogenic diseases and chromosome
abnormality by next-generation sequencing with linkage analyses.
Proc Natl Acad Sci USA. 112:15964–15969. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen L, Diao Z, Xu Z, Zhou J, Wang W, Li
J, Yan G and Sun H: The clinical application of preimplantation
genetic diagnosis for the patient affected by congenital
contractural arachnodactyly and spinal and bulbar muscular atrophy.
J Assist Reprod Genet. 33:1459–1466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gardner DK, Lane M, Stevens J, Schlenker T
and Schoolcraft WB: Blastocyst score affects implantation and
pregnancy outcome: Towards a single blastocyst transfer. Fertil
Steril. 73:1155–1158. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dreesen JC, Jacobs LJ, Bras M, Herbergs J,
Dumoulin JC, Geraedts JP, Evers JL and Smeets HJ: Multiplex PCR of
polymorphic markers flanking the CFTR gene; a general approach for
preimplantation genetic diagnosis of cystic fibrosis. Mol Hum
Reprod. 6:391–396. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ou J, Wang W, Feng T, Liao L, Meng Q, Zou
Q, Ding J, Zheng A, Duan C, Li P, et al: Identification of small
segmental translocations in patients with repeat implantation
failure and recurrent miscarriage using next generation sequencing
after in vitro fertilization/intracytoplasmic sperm injection. Mol
Cytogenet. 8:1052015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goyal M, Bijarnia-Mahay S, Kingsmore S,
Farrow E, Saunders C, Saxena R and Verma IC: Molecular diagnosis of
infantile neuro axonal dystrophy by next generation sequencing.
Indian J Pediatr. 82:474–477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thornhill AR, Handyside AH, Ottolini C,
Natesan SA, Taylor J, Sage K, Harton G, Cliffe K, Affara N,
Konstantinidis M, et al: Karyomapping-a comprehensive means of
simultaneous monogenic and cytogenetic PGD: Comparison with
standard approaches in real time for Marfan syndrome. J Assist
Reprod Genet. 32:347–356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liehr T: Cytogenetic contribution to
uniparental disomy (UPD). Mol Cytogenet. 3:82010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Solomons J, Ridgway O, Hardy C, Kurian MA,
Jayawant S, Hughes S, Pretorius P and Németh AH: Infantile
neuroaxonal dystrophy caused by uniparental disomy. Dev Med Child
Neurol. 56:386–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goldman KN, Nazem T, Berkeley A, Palter S
and Grifo JA: Preimplantation genetic diagnosis (PGD) for monogenic
disorders: The value of concurrent aneuploidy screening. J Genet
Couns. 25:1327–1337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tan Y, Yin X, Zhang S, Jiang H, Tan K, Li
J, Xiong B, Gong F, Zhang C, Pan X, et al: Clinical outcome of
preimplantation genetic diagnosis and screening using next
generation sequencing. Gigascience. 3:302014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wells D, Kaur K, Grifo J, Anderson S,
Taylor J, Fragouli E and Munne S: A novel embryo screening
technique provides new insights into embryo biology and yields the
first pregnancies following genome sequencing. Hum Reprod.
28:i262013.
|
|
32
|
Lledó B, Morales R, Ortiz JA, Blanca H,
Ten J, Llácer J and Bernabeu R: Implantation potential of mosaic
embryos. Syst Biol Reprod Med. 63:206–208. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Greco E, Minasi MG and Fiorentino F:
Healthy babies after intrauterine transfer of mosaic aneuploid
blastocysts. N Engl J Med. 373:2089–2090. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Besser AG and Mounts EL: Counselling
considerations for chromosomal mosaicism detected by
preimplantation genetic screening. Reprod Biomed Online.
34:369–374. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fiorentino F, Bono S, Biricik A,
Nuccitelli A, Cotroneo E, Cottone G, Kokocinski F, Michel CE,
Minasi MG and Greco E: Application of next-generation sequencing
technology for comprehensive aneuploidy screening of blastocysts in
clinical preimplantation genetic screening cycles. Hum Reprod.
29:2802–2813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Handyside AH: 24-chromosome copy number
analysis: A comparison of available echnologies. Fertil Steril.
100:595–602. 2013. View Article : Google Scholar : PubMed/NCBI
|