Introduction
Colorectal cancer is ranked as the second most
commonly diagnosed cancer type in males and third most commonly
diagnosed cancer type in females (1). Chemotherapy using agents such as
cisplatin and oxaliplatin is the conventional treatment approach
for colorectal cancer patients (2,3); however
development of chemoresistance frequently occurs leading to a
mortality rate of >33% in developed countries (4). Due to an increased understanding of
colorectal cancer molecular pathogenesis, multiple target therapy
agents have been used for colorectal cancer patients (5,6);
however, the clinical beneficial rate is often unsatisfactory
(7). Therefore, there is an urgent
need for research into chemoresistance in order to provide novel
targets for colorectal cancer therapy.
The fibroblast growth factor (FGF) signaling pathway
is involved in regulation of homeostasis, angiogenesis and
organogenesis (8,9). Aberrant activation of FGF signaling is
observed in cancer cells and considered a critical step during
carcinogenesis (10). During
activation of FGF signaling, FGFs bind to high affinity tyrosine
kinase FGF receptors (FGFRs) on the surface of cells (11). FGF9 is highly conserved and
ubiquitously expressed in embryos (12,13).
Overexpression of FGF9 is observed in several types of cancer and
its expression is associated with prognosis (14,15).
Recent research has determined that FGF9 exerts oncogenic activity
in cancer cells via regulating expression of several key genes such
as T-box 3 and vascular endothelial growth factor A (16,17). In
colorectal cancer cells, FGF9 protein expression is maintained at a
high level via translational activation (18). To date, whether FGF9 mediates
cisplatin resistance in colorectal cancer and the underlying
mechanisms remain not fully understood.
Overactivation of the Wnt signaling pathway is an
important step during cancer initiation and development (19). Following Wnt binding to receptors,
the signal is transduced to the nucleus leading to stabilization of
transcription co-activator β-catenin and activation of Wnt target
gene expression (20). Activity of
β-catenin is tightly controlled in cells. Negative regulators of
the Wnt/β-catenin pathway, such as tumor suppressor adenomatous
polyposis coli (APC), determine the stability and cellular location
of β-catenin (21). In particular,
the Wnt/β-catenin pathway is a well-known mediator of cancer
stemness and promotes chemotherapy resistance (22). In aldehyde dehydrogenase-positive
colorectal cancer, activation of the Wnt/β-catenin pathway
facilitates development of cisplatin resistance (23).
The present study determined that FGF9 was elevated
in colorectal tumors compared with matched normal tissue. In
colorectal cancer cells, FGF9 overexpression decreased
cisplatin-induced cell apoptosis whilst FGF9 silencing increased
cisplatin-induced cytotoxicity. Mechanistically, FGF9 repressed APC
expression and activated the Wnt/β-catenin signaling pathway.
Notably, FGF9 and β-catenin protein expression increased whilst APC
protein expression decreased in the LoVo cisplatin resistant cell
line (LoVo/cisplatin). FGF9 knockdown reversed cisplatin resistance
of LoVo/cisplatin cells. In conclusion, the results demonstrated
that FGF9 activated the Wnt signaling pathway and was a mediator of
cisplatin resistance in colorectal cancer.
Materials and methods
Tissue samples from patients
Tumor tissue and matched normal tissue (5 cm away
from the tumor) was collected from 20 patients with colorectal
cancer (age, 47–64 years old; mean age, 54.3±7.2 years old; 14 male
and 6 female) at the Shanghai Eighth People's Hospital between
March 2015 and October 2016. Patients who received any prior
radiotherapy or chemotherapy treatment were excluded from
enrollment. Written consent was provided by all enrolled patients.
All experiments were approved by the Ethics Committee of the
Shanghai Eighth People's Hospital. Samples were immediately frozen
at −80°C following collection. The stage of colon cancer was
defined according to the TNM system classification of the American
Joint Committee on Cancer (AJCC, 7th edition) (24).
Cell culture
The colorectal cancer cell line LoVo (parental) was
purchased from American Type Culture Collection (Manassas, VA,
USA). Cells were cultured in Ham's F-12K medium (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
under standard conditions (37°C; 5% CO2).
Cisplatin was purchased from Selleck Chemicals
(Houston, TX, USA). To determine cisplatin sensitivity, cells were
treated for 48 h with various concentrations of cisplatin (1, 2, 4,
8 and 16 µM).
To establish the cisplatin resistant LoVo subline
(LoVo/cisplatin), LoVo cells were divided into two groups and
treated with gradually increasing concentrations of cisplatin (100,
200, 400 nm, 1, 2, 5 µM, for 1 month) or dimethyl sulfoxide (DMSO)
for 6 months. The cell viability (data not shown) of LoVo cells
which was determined by Cell Counting Kit-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) was initially significantly
suppressed by cisplatin; however, following 6 months of cisplatin
treatment, cells no longer responded to 5 µM cisplatin and were
considered to be a cisplatin resistant subline (LoVo/cisplatin).
The LoVo cells treated with DMSO were considered as the parental
cell line (LoVo/parental).
Construction of plasmid and
overexpression of FGF9
Full length FGF9 cDNA was amplified from LoVo cells
by polymerase chain reaction (PCR) using a Taq DNA polymerase
SuperMix kit (Invitrogen; Thermo Fisher Scientific, Inc.) under the
following thermocycling conditions: 94°C for 2 min followed by 35
cycles of 94°C for 2 sec, 60°C for 60 sec and 72°C for 1 min and
ligated into pcDNA3.1 vector (Addgene, Inc., Cambridge, MA, USA)
using the restriction sites for HindIII and XhoI (New England
Biolabs, Inc.). The primer sequences for FGF9 were as listed:
Forward, 5′-AAGCTTATGGCTCCCTTAGGTGAAGT-3′, and reverse,
5′-CTGCAGTCAACTTTGGCTTAGAATAT-3′. The amplified sequence was
verified by sequencing. For overexpression of FGF9, 2 µg
pcDNA3.1-FGF9 vector was mixed with Lipofectamine 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) in serum-free Ham's F-12K medium
for 15 min, then added to cells cultured in 6-well plates
(1×106 cells/well). In the control group, 2 µg pcDNA3.1
was mixed with Lipofectamine 3000 in serum-free Ham's F-12K medium
for 15 min, then added to cells cultured in 6-well plates
(1×106 cells/well). The cells were maintained for 48 h,
and transfection efficiency was confirmed by western blotting
before experimentation.
Silencing of FGF9
Control small interfering RNA (siRNA,
5′-CAGUACUUUUGUGUAGUACAA-3′) and FGF9 siRNA
(5′-GCGAUACUAUGUUGCAUUATT-3′, 3′-UAAUGCAACAUAGUAUCGCCT-5′) were
purchased from GenePharma Co. Ltd. (Suzhou, China). For knockdown
of FGF9, FGF9 siRNA was mixed with Lipofectamine RNAiMax
(Invitrogen) in serum-free Ham's F-12K medium for 5 mins then the
mixture was added into culture medium of 6-well plates
(1×106 cells/well). Following incubation for 48 h, cells
were collected and transfection efficiency was confirmed by western
blot before the subsequent experiments.
Cell viability assay
LoVo cells were seeded in 96-well plates at a
density of 1×103 cells/well. The cell viability of each
well was determined by Cell Counting Kit-8 (Dojindo Molecular
Technologies) according to manufacturer's protocol. Cell viability
was evaluated by detection of the absorbance of each well at 450 nm
using a plate reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from tissues and cells were extracted
using TRIzol reagent (Invitrogen) according to the manufacturer's
instructions. RNA concentration was detected using Nanodrop 1000
(Thermo Fisher). RNA was reverse transcribed into cDNA with
PrimeScript RT reagent Kit (Takara Bio, Inc., Otsu, Japan). RT-qPCR
was performed with a SYBR Premix Ex Taq Kit (Takara) on a CFX-96
Real-Time PCR Detection System (Bio-Rad). The following
thermocycling conditions were used: Denaturing, 95°C for 30 sec;
annealing, 95°C for 5 sec; elongation, 60°C for 30 sec, for 40
cycles. The primer sequences were as follows: FGF9, forward
5′-ATGGCTCCCTTAGGTGAAGTT-3′ and reverse
5′-CCCAGGTGGTCACTTAACAAAAC-3′; APC, forward
5′-GGAGACAGAATGGAGGTGCT-3′ and reverse 5′-TCTTCAGTGCCTCAACTTGC-3′;
and GAPDH, forward 5′-GGACTCATGACCACAGTCCATGCC-3′ and reverse
5′-TCAGGGATGACCTTGCCCACAG-3′. Quantification was carried out by the
2−ΔΔCq method (25).
Western blot analysis
Lysates were prepared using radioimmunoprecipitation
lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China)
following the manufacturer's instruction. Protein concentration was
determined by the BCA method. For western blot, proteins in lysates
(20 µg/lane) were separated by 8% SDS-PAGE, transferred to a
polyvinylidene difluoride membrane, then blocked with 5% non-fat
milk. Membranes were incubated with primary antibody against APC
(cat. no. 2504; 1:1,000; Cell Signaling Technology, Danvers, MA,
USA), β-catenin (cat. no. 848; 1:1,000; Cell Signaling Technology),
GAPDH (cat. no. sc-47724; 1:1,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) and FGF9 (cat no. sc-8413; 1:1,000; Santa Cruz
Biotechnology, Inc.) at 4°C overnight. Membranes were then
incubated with horseradish peroxidase-labeled secondary antibodies
against mouse (cat no. ab6289; 1:10,000; Abcam, Cambridge, UK) and
rabbit (cat no. ab6721; 1:10,000; Abcam) at room temperature for 2
h. Protein bands were visualized using enhanced chemiluminescence
western blot substrate (Pierce; Thermo Fisher Scientific, Inc.).
The intensity of bands was calculated using Image J software
version 1.8.0 (National Institutes of Health, Bethesda, MD) and
normalized to GAPDH.
Immunohistochemical staining
The normal tissues and tumor tissues were fixed with
4% paraformaldehyde (PFA) solution at 4°C overnight, embedded in
paraffin and sliced into 4-µm sections. Immunohistochemistry (IHC)
was performed as follows: The sections were first deparaffinized
and rehydrated in 0.03% H2O2 in 95% methanol
for 20 min at room temperature to block the endogenous peroxidase
activity; antigen retrieval was carried out by water bath
(Immunosaver; Nisshin EM, Tokyo, Japan) for 45 min at 98°C;
sections were blocked with normal horse serum (Thermo Fisher
Scientific, Inc.) for 20 min at room temperature to eliminate
non-specific staining prior to incubation with an anti-FGF9
antibody (cat no. ab71395; 1:1,000; Abcam) overnight at 4°C; the
next day, sections were incubated with horseradish peroxidase (HRP)
conjugated goat anti-rabbit IgG (cat no. ab205718; 1:2,000; Abcam)
for 1 h at room temperature; color was developed by 3,
3′-diaminobenzidine (ZsBio, Beijing, China), sections were
counterstained with hematoxylin for 30 sec at room temperature.
Images from 6 individual fields of view were captured using a light
microscope and the attached camera (×40, Nikon, Tokyo, Japan).
Cell apoptosis assay
The Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) Apoptosis Detection kit (Becton,
Dickinson and Company, Franklin Lakes, NJ, USA) was used to
investigate apoptosis, according to manufacturer's protocol.
Following treatment with siRNA and/or cisplatin, cells were
harvested and suspended in 1X binding buffer. Annexin V-FITC and PI
solution were then added into cell suspension and incubated for 15
min. Finally, the cells were analyzed using a FACSCalibur system
(Becton, Dickinson and Company). The % apoptotic cells was
calculated with FlowJo software version 10.2 (FlowJo LLC, Ashland,
OR).
Statistical analysis
All data were analyzed using Graphpad Prism 6.0
(GraphPad Software, Inc., La Jolla, CA, USA) and expressed as mean
± standard deviation. The two-tailed Student's t-test was used to
compare two groups and determine the P-value. Three or more groups
were compared with one-way analysis of variance followed by Newman
Keul's test. P<0.05 was considered to indicate statistical
significance.
Results
Overexpression of FGF9 in colorectal
tumor tissues and normal tissues
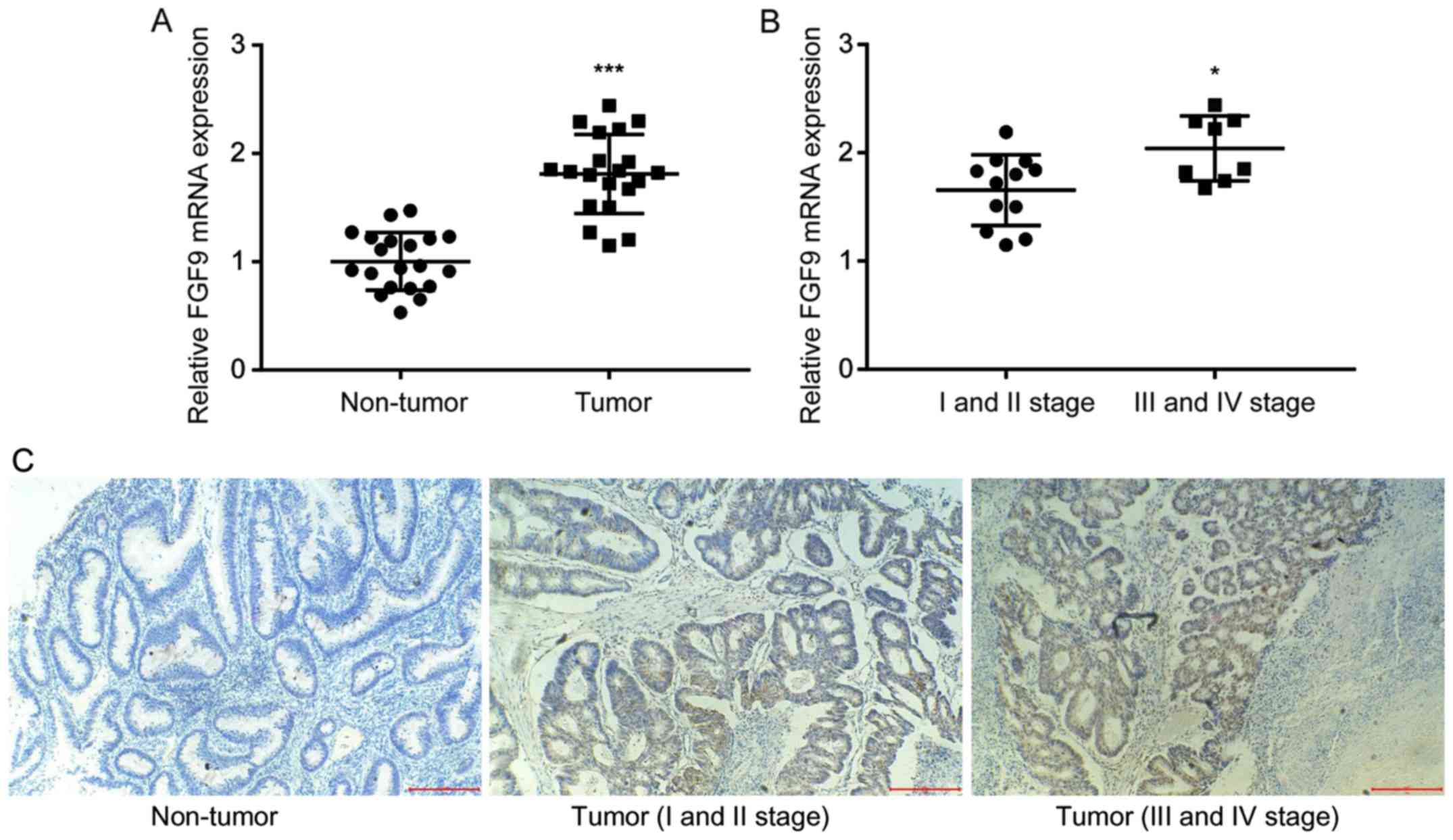
To investigate the role of FGF9 in colorectal
cancer, RT-qPCR was used to detect FGF9 mRNA expression in tumor
tissues and matched normal tissues from 20 colorectal cancer
patients. There was a significant increase of FGF9 mRNA expression
levels in colorectal tumor tissue compared with normal tissue
(Fig. 1A), which was consistent with
previous literature (18). In
addition, clinicopathological analysis revealed that higher FGF9
expression was associated with late stage colorectal cancer (III
and IV stage) rather than early stage colorectal cancer (I and II
stage) (Fig. 1B). IHC staining
demonstrated that FGF9 expression was markedly higher in tumor
tissues (I and II stage; III and IV stage) compared with the
non-tumor tissues. In addition, FGF9 expression was obviously
increased in tumor tissues at III and IV stage compared with
tissues at I and II stage (Fig. 1C).
These results suggested that FGF9 overexpression might promote
colorectal cancer progression.
FGF9 regulates cisplatin sensitivity
in colorectal cancer cells
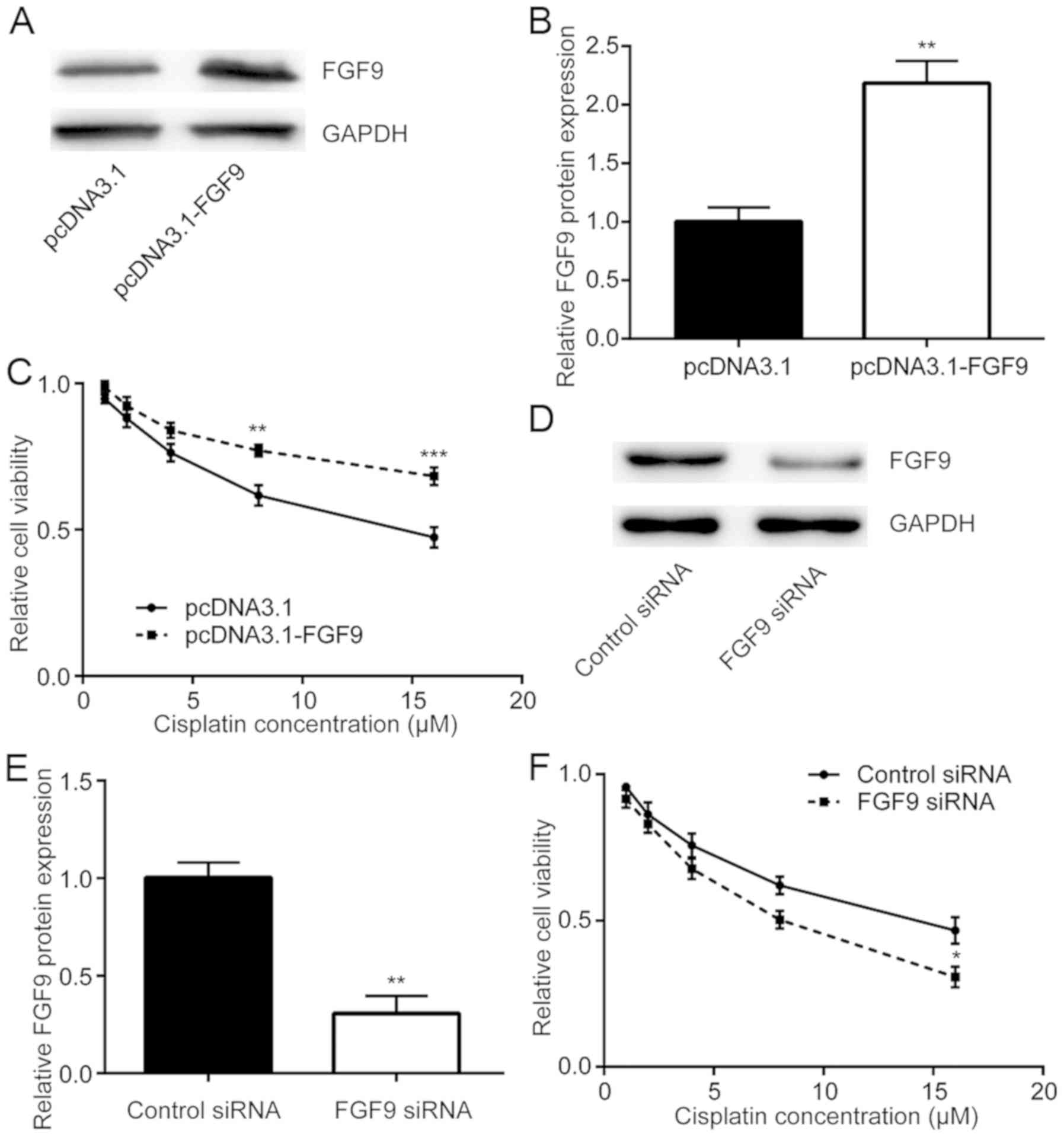
To study whether FGF9 expression was related to
cisplatin resistance, FGF9 was overexpressed in LoVo cells then
sensitivity to cisplatin was detected. Transfection of recombinant
FGF9 increased FGF9 expression in LoVo cells (Fig. 2A and B). Cisplatin treatment
decreased cell viability in a dose dependent manner whilst
overexpression of FGF9 decreased this cytotoxic effect of cisplatin
(Fig. 2C). By contrast, silencing of
FGF9 expression by transfection of FGF9 siRNA enhanced the
cytotoxic effect of cisplatin on LoVo cells (Fig. 2D-F). Therefore, FGF9 expression was
associated with cisplatin sensitivity of colorectal cancer
cells.
FGF9 inhibits cisplatin-induced cell
apoptosis in colorectal cancer cells
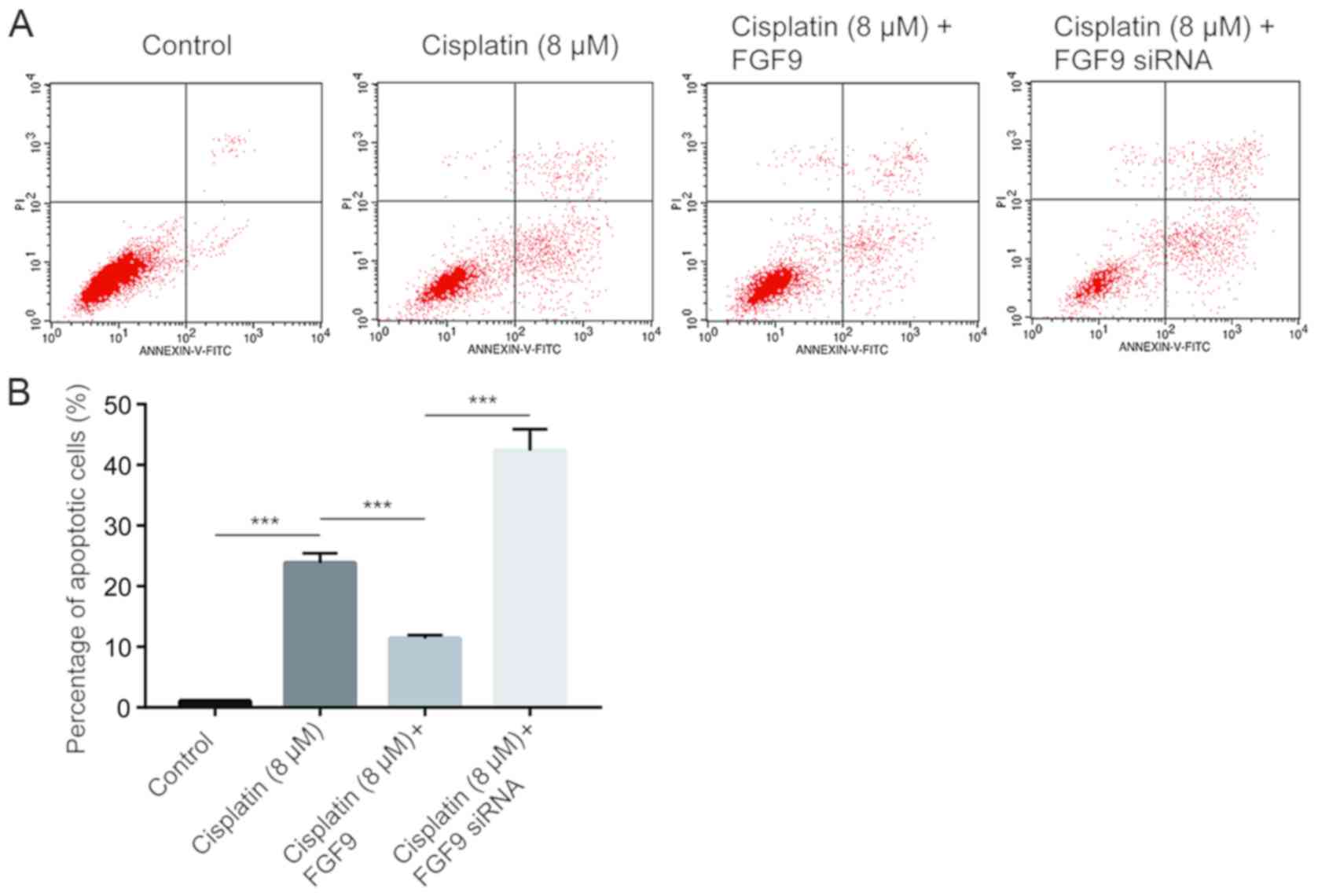
Cisplatin inhibits tumor cell growth via induction
of cell apoptosis (26). Using flow
cytometry analysis, it was determined that cisplatin (8 µM)
significantly increased the % of apoptotic LoVo cells compared with
the control (Fig. 3). Notably,
overexpression of FGF9 decreased cisplatin-induced cell apoptosis.
By contrast, silencing of FGF9 increased cell apoptosis (Fig. 3), which suggested that FGF9
sensitized colorectal cancer cells towards cisplatin and induced
cell apoptosis.
FGF9 activates the Wnt/β-catenin
pathway via repression of APC
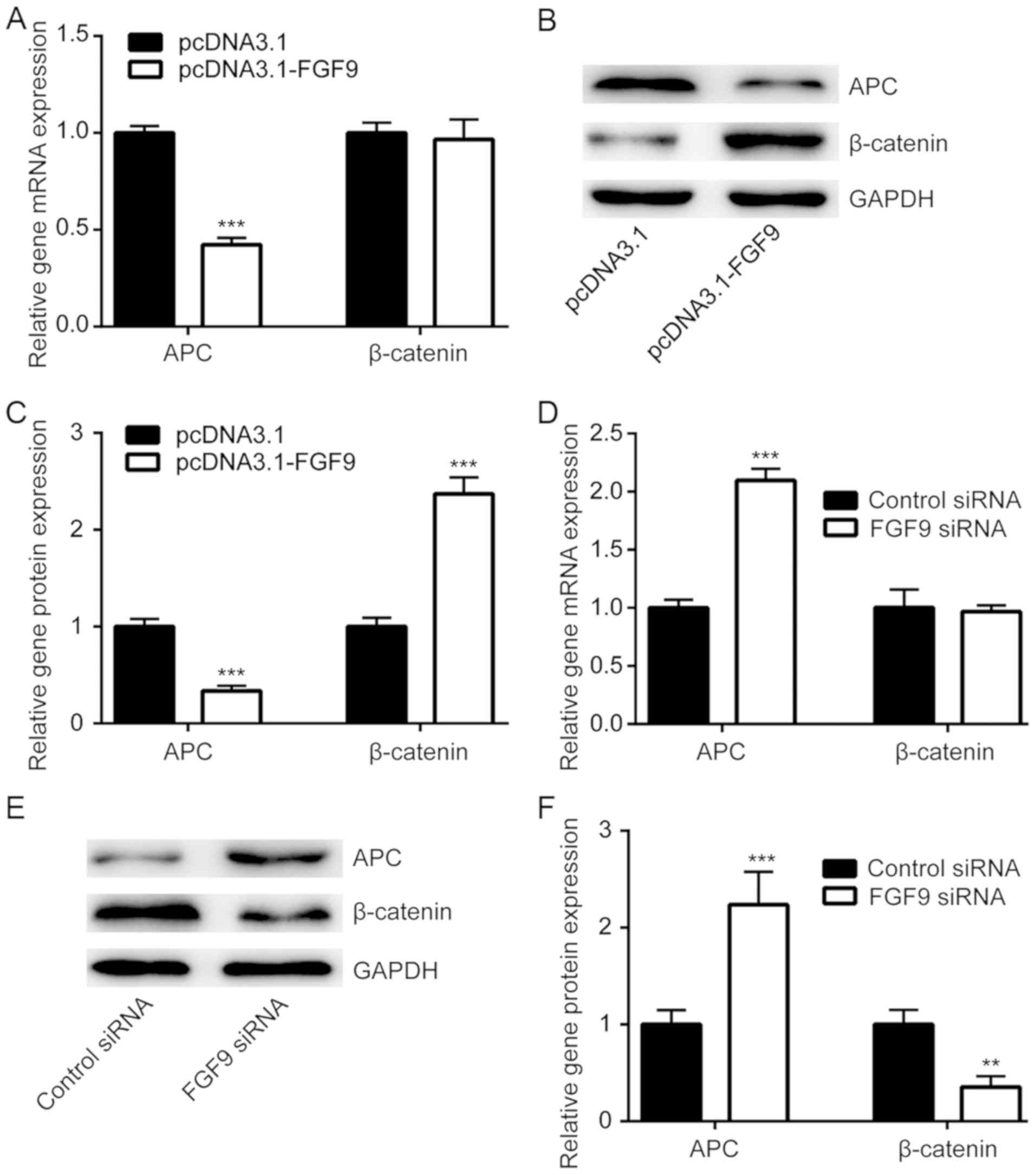
The Wnt/β-catenin signaling pathway is responsible
for chemoresistance in cancer cells (27). APC decreases β-catenin expression and
is a negative regulator of Wnt/β-catenin signaling (28). Using RT-qPCR, it was demonstrated
that FGF9 overexpression significantly decreased APC mRNA
expression, but not β-catenin mRNA expression in LoVo cells
(Fig. 4A). Western blot analysis
demonstrated that APC protein expression was downregulated and
β-catenin protein expression was elevated upon transfection of
pcDNA3.1-FGF9 (Fig. 4B and C). By
contrast, silencing of FGF9 significantly increased APC mRNA
expression and protein levels, whilst β-catenin protein levels
decreased (Fig. 4D and F). These
results suggested that FGF9 might regulate cisplatin sensitivity of
colorectal cancer cells via activation of the Wnt/β-catenin
pathway.
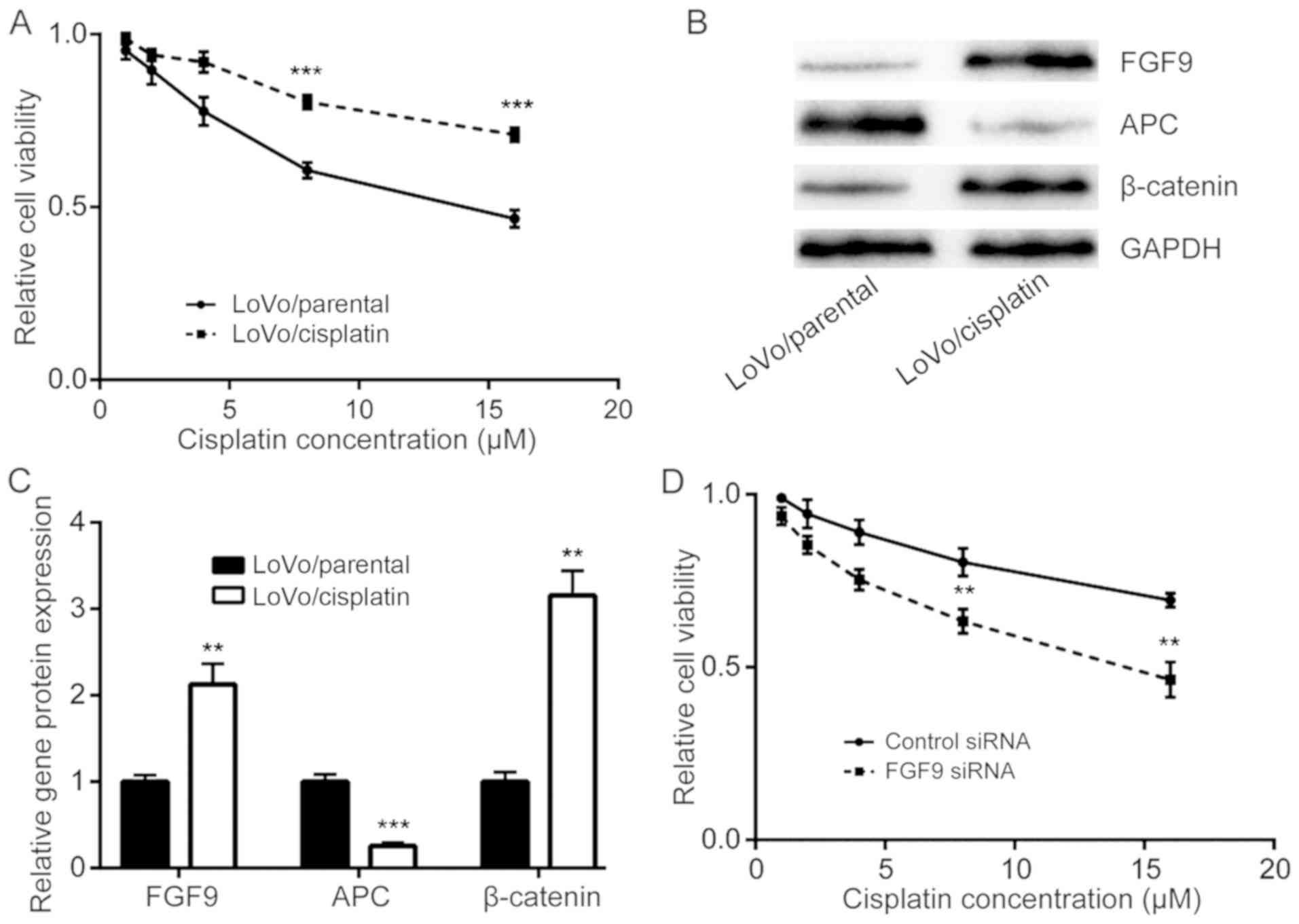
FGF9 overexpression is responsible for
development of cisplatin resistance
The role of FGF9 in the development of cisplatin
resistance in colorectal cancer cells was investigated. Via
continuous exposure of LoVo cells with either DMSO or cisplatin,
cisplatin resistant LoVo cells (LoVo/cisplatin) and parental LoVo
cells (LoVo/parental) were established. Cell viability assays
demonstrated that LoVo/cisplatin cells were less sensitive to
cisplatin treatment compared with LoVo/parental cells (Fig. 5A). Western blot analysis determined
that FGF9 and β-catenin protein expression was increased and APC
protein expression was decreased in LoVo/cisplatin cells, compared
with LoVo/parental cells (Fig. 5B and
C). Notably, silencing of FGF9, enhanced cisplatin sensitivity
of LoVo/cisplatin cells (Fig. 5D),
which suggested that the FGF9/Wnt signaling pathway has a key role
in cisplatin resistance development in colorectal cancer cells.
Discussion
Overexpression of FGF9 has been reported in
colorectal cancer and is associated with poor clinical outcome in
patients (29). A recent study
demonstrated that FGF9-overexpressing colorectal cancer cells
exhibited anti-epidermal growth factor receptor therapeutic
properties via overactivation of FGFR signaling (30). To date, the role of FGF9 in mediating
chemotherapy resistance in colorectal cancer remains not fully
understood. The present study determined that FGF9 was a regulator
of cisplatin sensitivity in colorectal cancer cells and a potential
molecular mechanism was identified.
Several mechanisms have been identified to promote
FGF9 expression in colorectal cancer including gene amplification,
hypoxia stimulation and non-coding RNA elevation (18,30,31). The
present study observed significant elevation of FGF9 in colorectal
tumor tissues compared with normal tissues, which is consistent
with a previous study (18). In
addition, higher expression of FGF9 was associated with late-stage
colorectal cancer compared with early-stage, suggesting that FGF9
may have a pivotal role in progression of colorectal cancer. FGF9
promotes colorectal cancer cell proliferation, migration and
invasion (15). The present study
determined that overexpression of FGF9 attenuated the cytotoxic
effect of cisplatin in LoVo cells, whilst silencing of FGF9
enhanced the cytotoxic effect of cisplatin. Furthermore, FGF9
overexpression reduced cisplatin-induced cell apoptosis whilst FGF9
knockdown enhanced the apoptotic effect of cisplatin treatment.
Notably, FGF9 expression was significantly elevated in the
established cisplatin-resistant LoVo cells compared with parental
LoVo cells. Silencing of FGF9 sensitized LoVo/cisplatin cells to
cisplatin treatment. Taken together, these findings suggested that
FGF9 was a mediator of cisplatin resistance in colorectal cancer
cells, and might be a biomarker and treatment target for colorectal
cancer.
It is well-known that the Wnt/β-catenin signaling
pathway has a critical role in progression of colorectal cancer
(28). In particular, activation of
the Wnt/β-catenin signaling pathway is essential for maintenance of
colorectal cancer cell stemness which contributes to development of
chemotherapy resistance (27,32). The
transcriptional control exerted by β-catenin is under stringent
negative regulation mediated by various interacting factors, such
as APC (21). It has been reported
that FGF9 activates the Wnt/β-catenin signaling pathway in
epithelial cells (33), and FGF9 has
been identified as a target gene of Wnt/β-catenin signaling
(34). The present study observed
that FGF9 overexpression decreased APC expression and elevated
β-catenin expression in LoVo cells. By contrast, FGF9 silencing
increased APC expression and decreased β-catenin levels. In
LoVo/cisplatin cells, an increase in APC expression and reduction
of β-catenin expression was demonstrated. Taken together, these
findings suggested that FGF9 might promote cisplatin resistance via
activation of the Wnt/β-catenin signaling pathway in colorectal
cancer cells.
In conclusion, the present study determined that
FGF9 was an important mediator of cisplatin resistance in
colorectal cancer cells via regulation of Wnt/β-catenin signaling,
which suggested that FGF9 may be a potential biomarker and
treatment target for colorectal cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of materials and data
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Author's contributions
ZZ, YZ, XQ and YW carried out the experiments and
analyzed the data. JF conceived the experiments, supervised the
present study and prepared the manuscript.
Ethics approval and consent to
participate
Experiments involving patient tissues were approved
by the Ethics Committee of the Shanghai Eighth People's Hospital.
Written consent was provided by all enrolled patients.
Patient consent for publication
Not applicable.
Competing of interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Florea AM and Büsselberg D: Cisplatin as
an anti-tumor drug: Cellular mechanisms of activity, drug
resistance and induced side effects. Cancers (Basel). 3:1351–1371.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giacchetti S, Perpoint B, Zidani R, Le
Bail N, Faggiuolo R, Focan C, Chollet P, Llory JF, Letourneau Y,
Coudert B, et al: Phase III multicenter randomized trial of
oxaliplatin added to chronomodulated fluorouracil-leucovorin as
first-line treatment of metastatic colorectal cancer. J Clin Oncol.
18:136–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Englinger B, Mair M, Miklos W, Pirker C,
Mohr T, van Schoonhoven S, Lötsch D, Körner W, Ferk F, Knasmüller
S, et al: Loss of CUL4A expression is underlying cisplatin
hypersensitivity in colorectal carcinoma cells with acquired
trabectedin resistance. Br J Cancer. 116:489–500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen MH, Gootenberg J, Keegan P and
Pazdur R: FDA drug approval summary: Bevacizumab plus FOLFOX4 as
second-line treatment of colorectal cancer. Oncologist. 12:356–361.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arnold D, Prager GW, Quintela A, Stein A,
Moreno Vera S, Mounedji N and Taieb J: Beyond second-line therapy
in patients with metastatic colorectal cancer: A systematic review.
Ann Oncol. 29:835–856. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stintzing S: Management of colorectal
cancer. F1000Prime Rep. 6:1082014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ellman MB, Yan D, Ahmadinia K, Chen D, An
HS and Im HJ: Fibroblast growth factor control of cartilage
homeostasis. J Cell Biochem. 114:735–742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ornitz DM and Itoh N: The Fibroblast
Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol.
4:215–266. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dieci MV, Arnedos M, Andre F and Soria JC:
Fibroblast growth factor receptor inhibitors as a cancer treatment:
From a biologic rationale to medical perspectives. Cancer Discov.
3:264–279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sato T, Oshima T, Yoshihara K, Yamamoto N,
Yamada R, Nagano Y, Fujii S, Kunisaki C, Shiozawa M, Akaike M, et
al: Overexpression of the fibroblast growth factor receptor-1 gene
correlates with liver metastasis in colorectal cancer. Oncol Rep.
21:211–216. 2009.PubMed/NCBI
|
|
12
|
Ornitz DM, Xu J, Colvin JS, McEwen DG,
MacArthur CA, Coulier F, Gao G and Goldfarb M: Receptor specificity
of the fibroblast growth factor family. J Biol Chem.
271:15292–15297. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song J and Slack JM: XFGF-9: A new
fibroblast growth factor from Xenopus embryos. Dev Dyn.
206:427–436. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohgino K, Soejima K, Yasuda H, Hayashi Y,
Hamamoto J, Naoki K, Arai D, Ishioka K, Sato T, Terai H, et al:
Expression of fibroblast growth factor 9 is associated with poor
prognosis in patients with resected non-small cell lung cancer.
Lung Cancer. 83:90–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deng M, Tang HL, Lu XH, Liu MY, Lu XM, Gu
YX, Liu JF and He ZM: miR-26a suppresses tumor growth and
metastasis by targeting FGF9 in gastric cancer. PLoS One.
8:e726622013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fillmore CM, Gupta PB, Rudnick JA,
Caballero S, Keller PJ, Lander ES and Kuperwasser C: Estrogen
expands breast cancer stem-like cells through paracrine FGF/Tbx3
signaling. Proc Natl Acad Sci USA. 107:21737–21742. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang Y, Jin C, Hamana T, Liu J, Wang C,
An L, McKeehan WL and Wang F: Overexpression of FGF9 in prostate
epithelial cells augments reactive stroma formation and promotes
prostate cancer progression. Int J Biol Sci. 11:948–960. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen TM, Shih YH, Tseng JT, Lai MC, Wu CH,
Li YH, Tsai SJ and Sun HS: Overexpression of FGF9 in colon cancer
cells is mediated by hypoxia-induced translational activation.
Nucleic Acids Res. 42:2932–2944. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arwert EN, Hoste E and Watt FM: Epithelial
stem cells, wound healing and cancer. Nat Rev Cancer. 12:170–180.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sierra J, Yoshida T, Joazeiro CA and Jones
KA: The APC tumor suppressor counteracts beta-catenin activation
and H3K4 methylation at Wnt target genes. Genes Dev. 20:586–600.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang C, Yu M, Xie X, Huang G, Peng Y, Ren
D, Lin M, Liu B, Liu M, Wang W and Kuang M: miR-217 targeting DKK1
promotes cancer stem cell properties via activation of the Wnt
signaling pathway in hepatocellular carcinoma. Oncol Rep.
38:2351–2359. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen B, Zhang D, Kuai J, Cheng M, Fang X
and Li G: Upregulation of miR-199a/b contributes to cisplatin
resistance via Wnt/β-catenin-ABCG2 signaling pathway in
ALDHA1+colorectal cancer stem cells. Tumour Biol.
39:10104283177151552017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Edge SB and Compton CC: The American Joint
Committee on Cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chikazawa N, Tanaka H, Tasaka T, Nakamura
M, Tanaka M, Onishi H and Katano M: Inhibition of Wnt signaling
pathway decreases chemotherapy-resistant side-population colon
cancer cells. Anticancer Res. 30:2041–2048. 2010.PubMed/NCBI
|
|
28
|
Schneikert J and Behrens J: The canonical
Wnt signalling pathway and its APC partner in colon cancer
development. Gut. 56:417–425. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Leushacke M, Sporle R, Bernemann C,
Brouwer-Lehmitz A, Fritzmann J, Theis M, Buchholz F, Herrmann BG
and Morkel M: An RNA interference phenotypic screen identifies a
role for FGF signals in colon cancer progression. PLoS One.
6:e233812011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizukami T, Togashi Y, Naruki S, Banno E,
Terashima M, de Velasco MA, Sakai K, Yoneshige A, Hayashi H, Fujita
Y, et al: Significance of FGF9 gene in resistance to anti-EGFR
therapies targeting colorectal cancer: A subset of colorectal
cancer patients with FGF9 upregulation may be resistant to
anti-EGFR therapies. Mol Carcinog. 56:106–117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hsiao KY, Lin YC, Gupta SK, Chang N, Yen
L, Sun HS and Tsai SJ: Noncoding effects of circular RNA CCDC66
promote colon cancer growth and metastasis. Cancer Res.
77:2339–2350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Takebe N, Harris PJ, Warren RQ and Ivy SP:
Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog
pathways. Nat Rev Clin Oncol. 8:97–106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng Z, Kang HY, Lee S, Kang SW, Goo B
and Cho SB: Up-regulation of fibroblast growth factor (FGF) 9
expression and FGF-WNT/β-catenin signaling in laser-induced wound
healing. Wound Repair Regen. 22:660–665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hendrix ND, Wu R, Kuick R, Schwartz DR,
Fearon ER and Cho KR: Fibroblast growth factor 9 has oncogenic
activity and is a downstream target of Wnt signaling in ovarian
endometrioid adenocarcinomas. Cancer Res. 66:1354–1362. 2006.
View Article : Google Scholar : PubMed/NCBI
|