Introduction
Pulmonary inflammation occurs in severe disorders,
including in acute lung injury (ALI), where it results in diffuse
alveolar damage, which can lead to hypoxemia and respiratory
failure (1). ALI is estimated to
occur, with a high incidence rate, in the pediatric population
worldwide (2). The genetic and
environmental factors associated with ALI in children have been
previously reported (3). However, an
effective and specific treatment for pediatric ALI is not currently
available.
In recent years, accumulating evidence has indicated
that inflammation serves an important role in ALI in children
(4). Pediatric ALI is characterized
by the infiltration of inflammatory cells, including mast cells,
eosinophils, lymphocytes, basophils and monocytes. An increased
production of interleukin (IL)-1, IL-6 or tumor necrosis factor
(TNF)-α by inflammatory macrophages has been previously reported in
children with ALI (5). As an
important transmembrane pattern-recognition receptor of the innate
immune system, toll-like receptor 4 (TLR4) was identified in a
variety of inflammatory conditions, including pneumonia (6). The activation of TLR4 was associated
with the expression of pro-inflammatory cytokines and the
activation of NF-κB signaling pathways (7). Furthermore, the expression of TLR4 was
positively correlated with TNF-α and IL-6 expression (8).
Recently, as a novel member of the IL-12 family,
IL-35 has been identified to be associated with pulmonary disease
(9). IL-35 is a heterodimer that is
composed of Epstein-Barr (EB) virus-induced gene 3 and p35; the
structure is homologous to that of IL-12 due to the similar β
chains of EB12 (10). High
expression of IL-35 was indicated in non-stimulated mouse T
regulatory cells, and the expression of IL-35 was upregulated in
human non-T cells, including microvascular endothelia cells, aortic
smooth muscle cells and epithelial cells, following stimulation
with TNF-α (11). The expression of
IL-35, at the protein and mRNA levels, was decreased in individuals
with allergic asthma (12). In
vivo, the high expression of IL-35 was reported to decrease the
number of inflammatory cells and the levels of IL-4, IL-5, IL-13
and IL-17 in a mouse model of asthma (13). However, the precise mechanism by
which IL-35 regulates ALI in children is unclear. In the present
study, the expression of IL-35 was investigated in vivo and
in vitro. The potential mechanisms and signaling pathways
associated with the expression of IL-35 were studied. The results
of the present study demonstrated that IL-35 expression was
downregulated in vivo and in vitro, and resulted in
the activation of the TLR4/NF-κB signaling pathways. The present
study provides a theoretical foundation for the targeting
inflammation in the pathogenesis of ALI in children.
Materials and methods
Animal model
All experiments in the present study were performed
in accordance with the Guidelines on Animal Experiments from The
Committee of Medical Ethics, The National Health Department of
China. The animal experiments were approved by the Institutional
Animal Care and Use Committee of Jilin University (Changchun,
China) (KT201902017). The ALI mouse model was set up according to a
previous study (14). The mice (6–8
weeks old; 25–35 g) were housed in isolator cages and received food
and water ad libitum. The laboratory temperature was 24±1°C,
and relative humidity was 40–80% with a 12-h light/dark cycle. A
total of 20 normal male BALB/c juvenile mice were randomly divided
into two groups (n=10/group): Control group and the model group.
The model group was treated with 0.5 mg/kg lipopolysaccharide (LPS)
and the control group was given an equivalent amount of 0.9% NaCl
solution with 8 h.
Histology
For euthanasia, the mouse home cage was placed in a
22-l transparent polycarbonate euthanasia chamber (Shanghai Yuyan
Instruments Co., Ltd.). The euthanasia chamber was covered with an
acrylic lid with ports for gas inlet and outlet. The chamber air
was replaced with CO2 at 30%/min. Death was confirmed
when the blood pressure and heart rate had reached 0, according to
telemetry.
CO2 treatment ensured that the animals
were anaesthetized prior to euthanasia and all efforts were made to
minimize their suffering. The mice were sacrificed and the lungs
were exposed and removed. In a series of experimental steps, lung
samples were sectioned into 4–6 µm slices, fixed in 4% formalin for
12 h at room temperature and embedded in paraffin for histological
analysis. Hematoxylin and eosin staining was performed at room
temperature (Hematoxylin for 3–5 min and eosin for 1 min) and was
used to assess morphology and inflammation. The results were
detected by light microscopy with ×100 magnification.
Cell culture
Murine RAW264.7 macrophages (The Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences) were plated
in culture dishes and cultured in DMEM (high glucose; Hyclone; GE
Healthcare Life Sciences) with 10% FBS (Hyclone; GE Healthcare Life
Sciences) and 1% antibiotic solution at 37°C in a humidified
atmosphere of 5% CO2. The medium was replaced every 2
days and cells were subcultured at 37°C in a humidified atmosphere
of 5% CO2 or subjected to subsequent experimental
procedures when the culture reached a confluence of 70–80%.
Cloning of IL-35 into a lentiviral
vector and transfection
Murine IL-35 (mIL-35) cDNA from murine RAW264.7
macrophages was obtained using the TIANscript RT kits according to
the manufacturer's instructions (Tiangen Biotech Co., Ltd.) and
amplified using the following primers: Forward,
5′-CGCGGATCCCTGAGATCACCGGTAGGAGG-3′; and reverse,
5′-TCCCCCCGGGGAGCTAGCTTTAGGCGG-3′. PrimeSTAR HS DNA Polymerase
(Takara Bio, Inc.) was used for the PCR. The reaction conditions
were as follows: A total of 30 cycles of pre-denaturation at 98°C
for 30 sec, denaturation at 98°C for 10 sec, annealing at 50°C for
15 sec, extension at 72°C for 1 min followed by a final extension
at 72°C for 7 min. PCR products were recovered using the Agarose
gel DNA Recovery kit (Tiangen Biotech Co., Ltd). The recovered PCR
products were digested using restriction endonuclease (Takara Bio,
Inc.) and subcloned into a lentiviral vector-pMD2.G (Beijing
Huayueyang Co., Ltd.) by T4 DNA ligase (Takara Bio, Inc.) to be
used as an IL-35 agomir (agomir-IL-35). The empty lentiviral vector
was used as a control (agomir-NC). Following 24 h, the murine
RAW264.7 macrophages were transfected using
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.)
with agomir-IL-35 or agomir-NC. Murine RAW264.7 macrophages were
plated on 24-well plate at 500 µl/well at density of
5×104−10×104 cells/ml in medium and
propagated to 80% confluency at the time of transfection. In this
present study, the Opti-MEM reduced serum medium (Thermo Fisher
Scientific, Inc.) contained 500 ng agomir-IL-35 or agomir-NC.
Agomir-IL-35 or agomir-NC was complexed with 2 µl of Lipofectamine
3000 with 1.5 µl of P3000 as described in the manufacturer's
protocol, in the Opti-MEM reduced serum medium. The medium
containing the lentivirus was replaced with complete medium 24 h
after transfection. Following treatment with 1 µg/ml LPS for 24 h,
the murine RAW264.7 macrophages were collected and used for
subsequent experiments. The murine RAW264.7 macrophages were
divided into five groups: Control (non-LPS-induced cells),
agomir-NC, LPS, agomir-LPS, agomir-IL-35 + LPS and
agomir-IL-35.
Measurement of IL-6 and TNF-α activity
in murine RAW264.7 macrophages
The cellular activities of IL-6 and TNF-α were
determined using ELISAs kits (IL-6, cat. no. 431304; TNF-α, cat.
no. 430907) according to the manufacturers' instructions
(BioLegend, Inc.). All samples were assayed six times.
Western blotting
Nuclear and cytosolic proteins were extracted using
Nuclear and Cytoplasmic Protein Extraction kit (cat. no. P0028;
Beyotime Institute of Biotechnology), according to the
manufacturer's instructions. Protein concentrations were quantified
using the bicinchoninic acid (BCA) assay kit (cat. no. P0011;
Beyotime Institute of Biotechnology). A total of 50 µg of each
sample was used for 10% SDS-PAGE and, the proteins were transferred
onto PVDF membranes using the wet transfer method. Membranes were
blocked in 5% milk for 1 h at room temperature. Subsequently, the
proteins were probed with the diluted primary antibodies anti-IL-35
(cat. no. ab133751), -TLR-4 (cat. no. ab13556), -NF-κB p65 (cat.
no. ab207297), -NF-κB p50 (cat. no. ab14059), -NF-κB inhibitor-α
(IκBα) (cat. no. ab7217), -β-actin (cat. no. ab8226) and -histone
H3 (cat. no. ab1791; all 1:1,000; all from Abcam), at 4°C
overnight. Following three 8 min PBST washes, the membranes were
incubated with goat-anti-rabbit IgG secondary antibody (cat. no.
TA130015; 1:5,000; OriGene Technologies, Inc.), goat-anti-mouse IgG
secondary antibody (cat. no. ab205719; 1:5,000; Abcam) or goat
anti-chicken IgY secondary antibody (cat. no. ab6877; 1:5,000;
Abcam) for 1 h at room temperature. An enhanced chemiluminescence
reagent (cat. no. WBKLS0500; Pierce; Thermo Fisher Scientific,
Inc.) was used to visualize the protein bands. ImageJ version 1.38
(National Institutes of Health) software was used for densitometry
analysis of the appropriate lanes.
Statistical analysis
Experimental results are presented as the mean ±
SEM. Using the SSPS 17.0 (SPSS, Inc.) statistical analysis software
for data analysis, two groups were analyzed using independent
Student's t-test, whereas multiple groups were compared using a
one-way ANOVA test with Bonferroni. P<0.05 was considered to
indicate a statistically significant difference.
Results
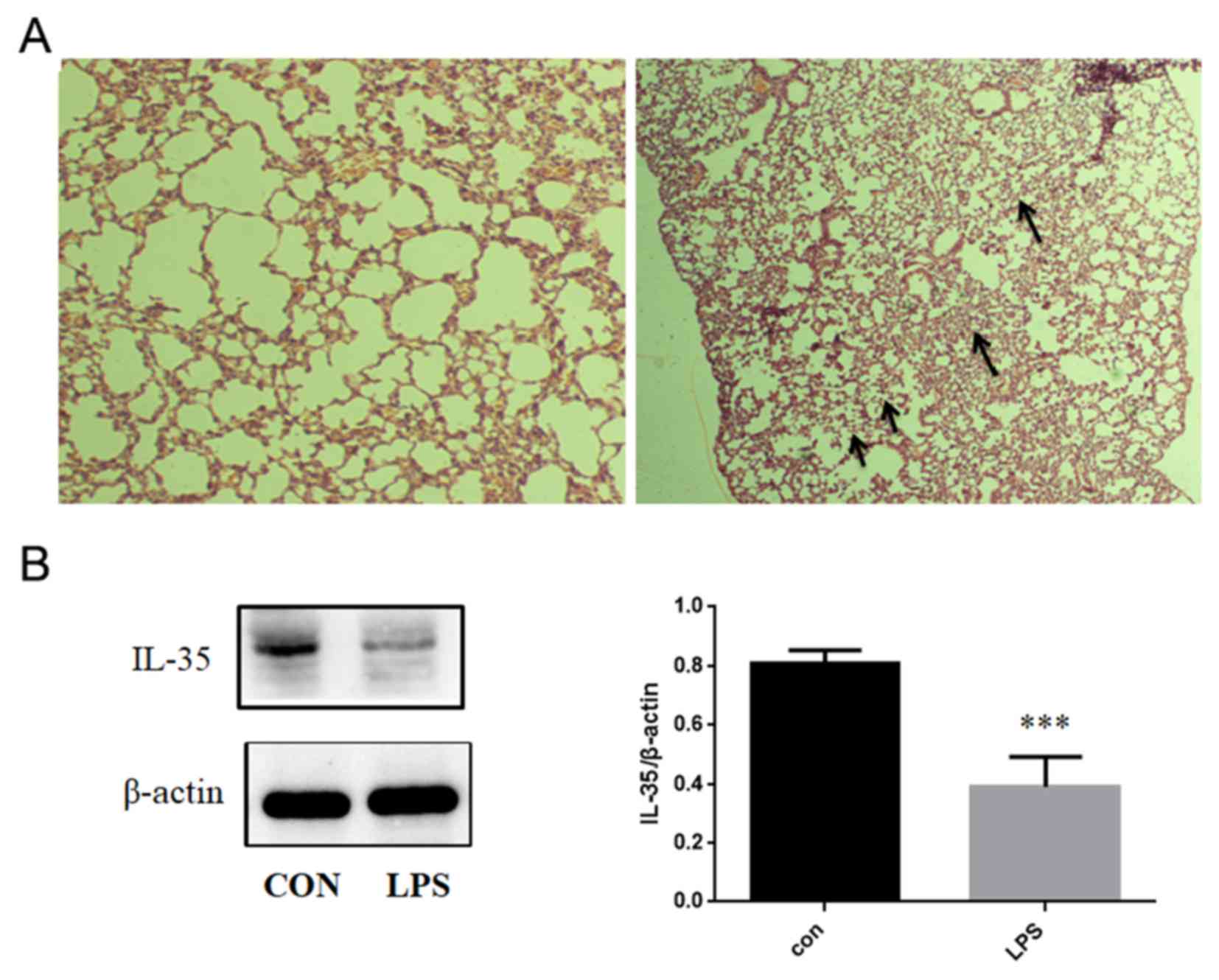
IL-35 is downregulated in mice treated
with LPS
In the present study, LPS was used to induce an ALI
model. The lungs from LPS-treated mice revealed the infiltration of
neutrophils, thickening of the alveolar wall, edema and hemorrhage
(Fig. 1A). The expression of IL-35
was determined using western blotting following the different
treatments. The expression of IL-35 in mice treated with LPS was
significantly lower compared with the control group (Fig. 1B).
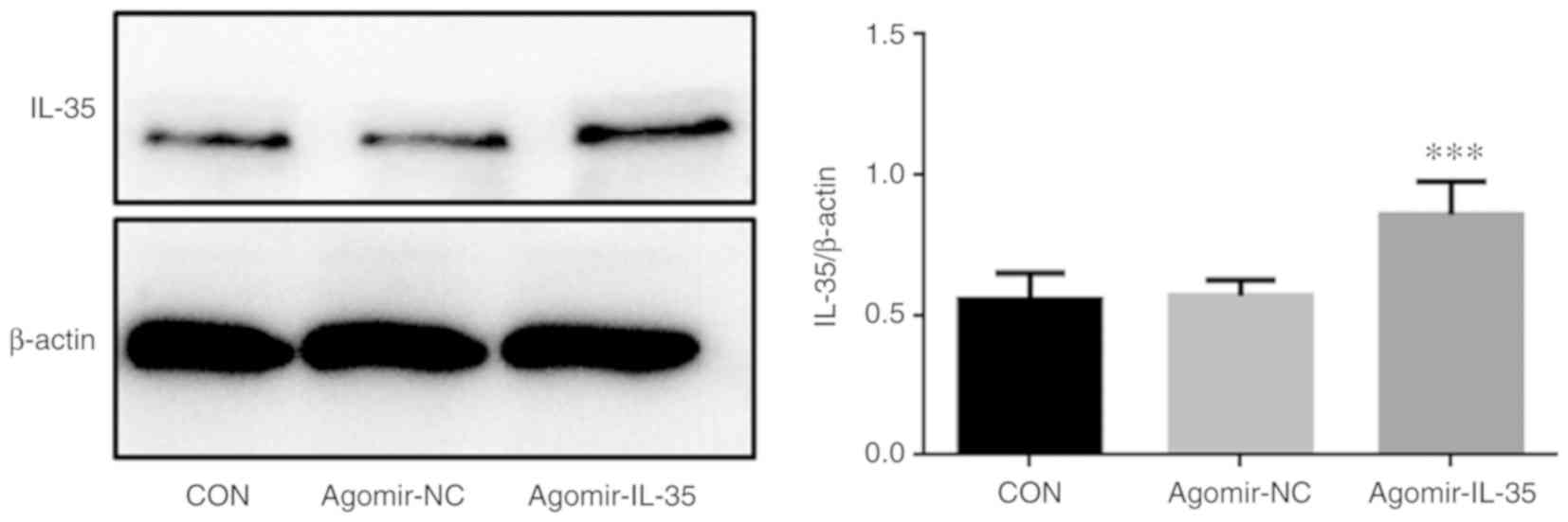
IL-35 expression is upregulated in
murine RAW264.7 macrophages
Following treatment with 1 µg/ml LPS for 24 h, the
murine RAW264.7 macrophages were transfected with agomir-IL35 or
agomir-NC. Western blotting indicated upregulated expression of
IL-35 in agomir-IL-35 murine RAW264.7 macrophages (Fig. 2).
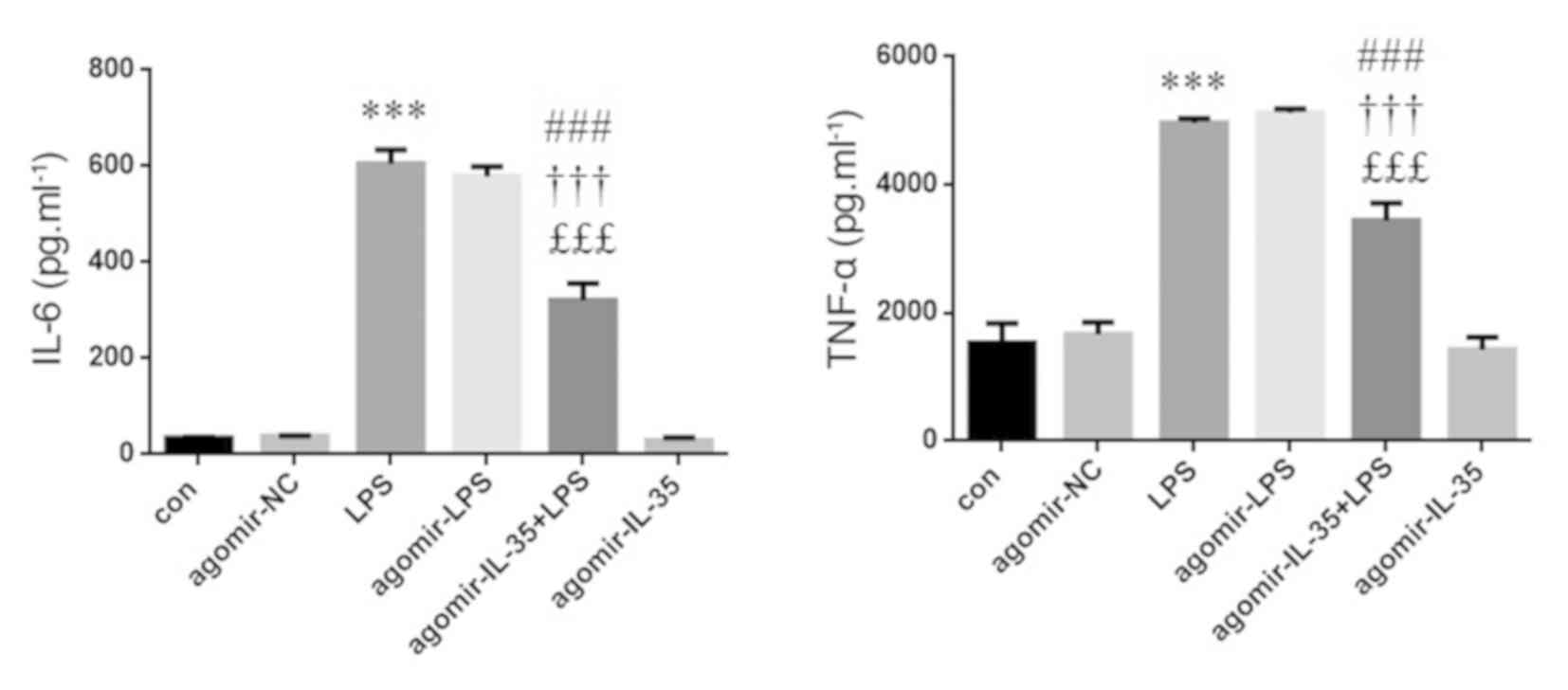
Expression levels of IL-6 and TNF-α
are downregulated in murine RAW264.7 macrophages transfected with
agomir-IL-35
The levels of IL-6 and TNF-α were evaluated using
ELISA. The levels of IL-6 and TNF-α were increased in murine
RAW264.7 macrophages following treatment with LPS compared to
control, whereas IL-6 and TNF-α expression was decreased in
agomir-IL-35 + LPS-treated murine RAW264.7 macrophages compared to
LPS or agomir-LPS (Fig. 3).
IL-35 attenuates inflammation via the
TLR4/NF-κB signaling pathways in murine RAW264.7 macrophages
The expression of TLR4, NF-κB p65 and NF-κB p50 was
evaluated using western blot analysis. It was demonstrated that the
expression of TLR4, and the degradation of IκBα, was increased in
murine RAW264.7 macrophages following treatment with LPS compared
to control (Fig. 1A). In contrast,
the expression of TLR4, and the degradation of IκBα, was decreased
in agomir-IL-35 + LPS-treated murine RAW264.7 macrophages compared
to LPS and agomir-LPS treated RAW264.7 macrophages (Fig. 4A). The data demonstrated that the
expression of IL-35 inhibited the translocation of NF-κB p65 and
NF-κB p50 from the cytoplasm to the nucleus in murine RAW264.7
macrophages treated with LPS (Fig. 4B
and C).
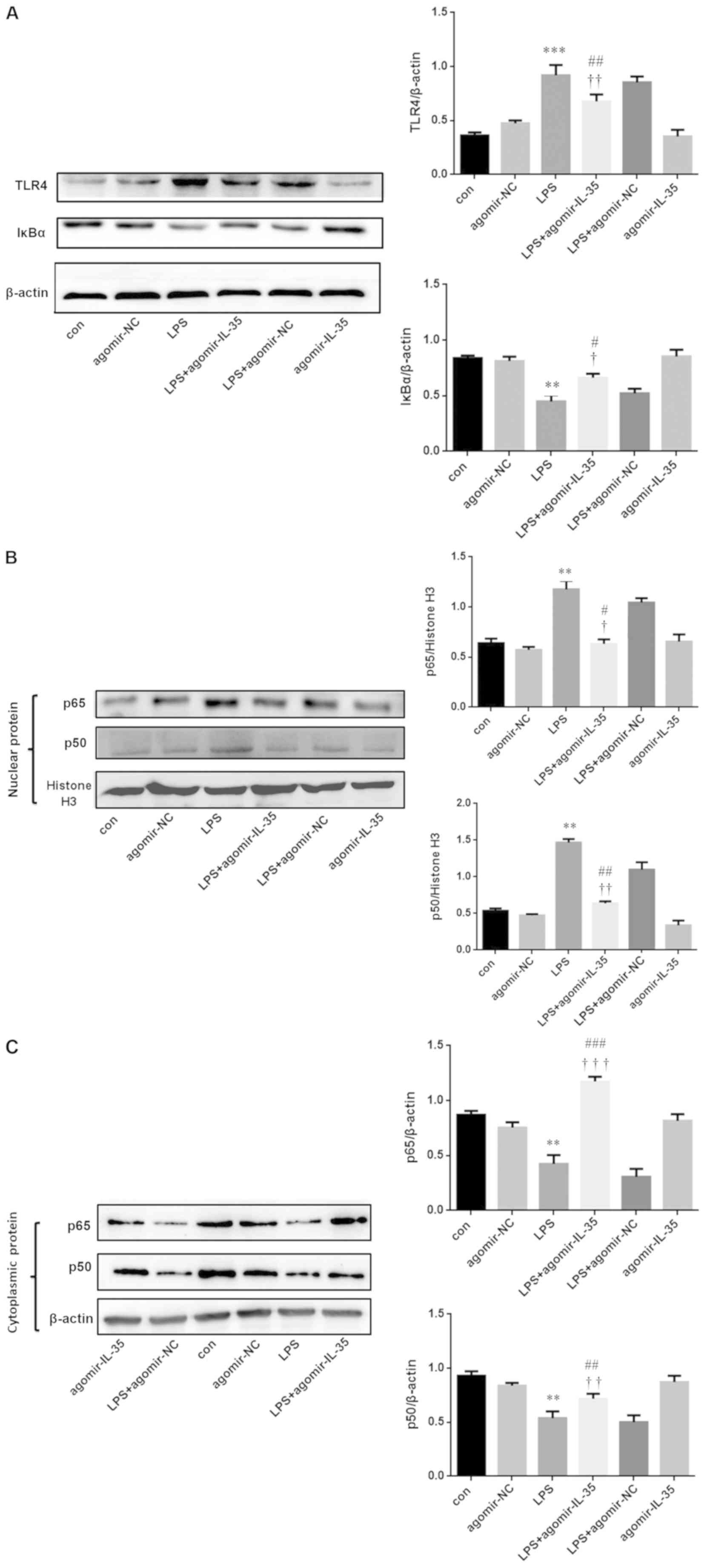 | Figure 4.Murine RAW264.7 macrophage expression
of TLR4/NF-кB signaling pathways TLR4 and IкBα p50 expression. (A)
The expression of TLR4 and IкBα in murine RAW264.7 macrophage. (B)
The expression of P65 and P50 in nuclear of murine RAW264.7
macrophage. (C) The expression of P65 and P50 in cytoplasmic of
murine RAW264.7 macrophage. All data are p as the mean ± SEM (n=3,
per group). **P<0.01, ***P<0.001 vs. con. #P<0.05,
##P<0.01, ###P<0.001 vs. LPS. †P<0.05, ††P<0.01,
†††P<0.001 vs. agomir-NC. IL-35, interleukin-35; LPS,
lipopolysaccharide; con, control; NC, negative control; TLR,
toll-like receptor. |
Discussion
ALI in children is one of the most common diseases
in pediatric medicine worldwide (15). A study demonstrated that inflammation
serves a key role in the multi-factorial pathogenesis of ALI in
children (16). Additionally, IL-35
has been reported to be associated with pediatric ALI by affecting
the number of inflammatory cells (9); however, the mechanism remains unclear.
The present study describes a cascade of events that link
TLR4/NF-κB activation to inflammation through the downregulation of
IL-35 in vivo and in vitro.
In the present study, an ALI model was established
using LPS. The lungs from LPS-treated mice exhibited signs of
infiltration by neutrophils, thickening of alveolar walls, edema
and hemorrhage. It was also demonstrated that the expression of
IL-35 was downregulated in mice treated with LPS.
IL-35 has been revealed to serve an
immunosuppressive role during infections, inflammation and in
autoimmune diseases (17). TNF-α,
which is a marker of clinical severity and airflow limitation, is
primarily secreted by macrophages. A previous report indicated that
the expression of TNF-α was increased in pulmonary diseases
(18), and the production of IL-6
was triggered by TNF-α. To investigate the effect of IL-35 on
inflammation-associated signaling cascades, IL-35 was overexpressed
in murine RAW264.7 macrophages. In the present study, the levels of
IL-6 and TNF-α were determined using ELISA. The levels of IL-6 and
TNF-α were increased in agomir-IL-35 + LPS-treated RAW264.7
macrophages compared with agomir-NC + LPS.
Previous studies have reported that TLR4 is critical
for the activation of NF-κB and the subsequent production of
pro-inflammatory cytokines that are implicated in a variety of
diseases (19–21). As a transcription factor, NF-κB
serves vital roles in numerous processes, including inflammation,
immunity and cell proliferation (22). NF-κB is an important signaling
pathway in the development of a number of inflammation-mediated
diseases, including pediatric bronchial asthma (23). p65 and p50 are members of the NF-κB
family. In the present study, the expression of TLR4 and the
degradation of IκBα were increased in murine RAW264.7 macrophages
treated with LPS, whereas the expression of TLR4 and the
degradation of IκBα were decreased in agomir-IL-35 + LPS murine
RAW264.7 macrophages. The present study demonstrated that IL-35
inhibited the translocation of NF-κB p65 and NF-κB p50 from the
cytoplasm to the nucleus in LPS-treated murine RAW264.7
macrophages. These results suggested that the upregulation of IL-35
leads to a decreased level of inflammation. Therefore, IL-35
affects inflammation through TLR4/NF-κB signaling pathways.
In conclusion, the present study revealed that
LPS-induced inflammation contributed to ALI. IL-35 has been
indicated to be an important target associated with ALI, that
influences inflammation via the TLR4/NF-κB signaling pathways.
Therefore, the inhibition of inflammation using IL-35 may provide a
novel therapeutic strategy to prevent ALI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XS guaranteed the integrity of the entire study. XS
and WP provided the study concepts. XS and WP were responsible for
the study design, the definition of intellectual content, and for
literature research. XX and YW performed experimental studies, and
were responsible for data analysis, statistical analysis,
manuscript preparation, manuscript editing and manuscript
review.
Ethics approval and consent to
participate
All experiments in the present study were performed
in accordance with the Guidelines on Animal Experiments from The
Committee of Medical Ethics, The National Health Department of
China. The animal work was approved by the institutional Animal
Care and Use Committee of Jilin University (Changchun, China;
approval no. KT201902017).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dong ZW and Yuan YF: Juglanin suppresses
fibrosis and inflammation response caused by LPS in acute lung
injury. Int J Mol Med. 41:3353–3365. 2018.PubMed/NCBI
|
|
2
|
Brun-Buisson C, Minelli C, Bertolini G,
Brazzi L, Pimentel J, Lewandowski K, Bion J, Romand JA, Villar J,
Thorsteinsson A, et al: Epidemiology and outcome of acute lung
injury in European intensive care units. Results from the ALIVE
study. Intensive Care Med. 30:51–61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gilliland FD, McConnell R, Peters J and
Gong H Jr: A theoretical basis for investigating ambient air
pollution and children's respiratory health. Environ Health
Perspect. 3:403–407. 1999. View Article : Google Scholar
|
|
4
|
Tang L, Zhang H, Wang C, Li H, Zhang Q and
Bai J: M2A and M2C macrophage subsets ameliorate inflammation and
fibroproliferation in acute lung injury through interleukin 10
pathway. Shock. 48:119–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McArdle MA, Finucane OM, Connaughton RM,
McMorrow AM and Roche HM: Mechanisms of obesity-induced
inflammation and insulin resistance: Insights into the emerging
role of nutritional strategies. Front Endocrinol (Lausanne).
4:522013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai JP, Wang QW, Su Y, Gu LM, Zhao Y, Chen
XX, Chen C, Li WZ, Wang GF and Li KS: Emodin inhibition of
influenza A virus replication and influenza viral pneumonia via the
Nrf2, TLR4, p38/JNK and NF-kappaB pathways. Molecules.
22:E17542017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu J, Li L, Sun Y, Huang S, Tang J, Yu P
and Wang G: Altered molecular expression of the TLR4/NF-κB
signaling pathway in mammary tissue of Chinese Holstein cattle with
mastitis. PLoS One. 10:e01184582015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang J, Yang J, Ding JW, Chen LH, Wang YL,
Li S and Wu H: Sequential expression of TLR4 and its effects on the
myocardium of rats with myocardial ischemia-reperfusion injury.
Inflammation. 31:304–312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang S, Shan F, Zhang Y, Jiang L and
Cheng Z: Increased serum IL-17 and decreased serum IL-10 and IL-35
levels correlate with the progression of COPD. Int J Chron Obstruct
Pulmon Dis. 13:2483–2494. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gately MK, Desai BB, Wolitzky AG, Quinn
PM, Dwyer CM, Podlaski FJ, Familletti PC, Sinigaglia F, Chizonnite
R, Gubler U, et al: Regulation of human lymphocyte proliferation by
a heterodimeric cytokine, IL-12 (cytotoxic lymphocyte maturation
factor). J Immunol. 147:874–882. 1991.PubMed/NCBI
|
|
11
|
Li X, Mai J, Virtue A, Yin Y, Gong R, Sha
X, Gutchigian S, Frisch A, Hodge I, Jiang X, et al: IL-35 is a
novel responsive anti-inflammatory cytokine-a new system of
categorizing anti-inflammatory cytokines. PLoS One. 7:e336282012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang W, Li P and Yang J: Decreased
circulating interleukin-35 levels are related to
interleukin-4-producing CD8+ Tcells in patients with allergic
asthma. Iran J Allergy Asthma Immunol. 14:379–385. 2015.PubMed/NCBI
|
|
13
|
Li Y, Pan X, Peng X, Li S, Zhou Y, Zheng X
and Li M: Adenovirus-mediated interleukin-35 gene transfer
suppresses allergic airway inflammation in a murine model of
asthma. Inflamm Res. 64:767–774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu Q, Li R, Soromou LW, Chen N, Yuan X,
Sun G, Li B and Feng H: p-Synephrine suppresses
lipopolysaccharide-induced acute lung injury by inhibition of the
NF-κB signaling pathway. Inflamm Res. 63:429–439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thomas NJ, Jouvet P and Willson D: Acute
lung injury in children-kids really aren't just ‘little adults’.
Pediatr Crit Care Med. 14:429–432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Z, Chen Z, Li L, Zhou W and Zhu L:
Lack of SOCS3 increases LPS-induced murine acute lung injury
through modulation of Ly6C (+) macrophages? Respir Res. 18:2172017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Egwuagu CE, Yu CR, Sun L and Wang R:
Interleukin 35: Critical regulator of immunity and
lymphocyte-mediated diseases. Cytokine Growth Factor Rev.
26:587–93. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan X, Xu K and Li Y, Wang X, Peng X, Li M
and Li Y: Interleukin-35 expression protects against cigarette
smoke-induced lung inflammation in mice. Biomed Pharmacother.
110:727–732. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi W, Yang X, Ye N, Li S, Han Q, Huang J
and Wu B: TLR4 gene in the regulation of periodontitis and its
molecular mechanism. Exp Ther Med. 18:1961–1966. 2019.PubMed/NCBI
|
|
21
|
Zhu C, Bao NR, Chen S and Zhao JN: HBD-3
regulation of the immune response and the LPS/TLR4-mediated
signaling pathway. Exp Ther Med. 12:2150–2154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun W, Gao Y, Yu X, Yuan Y, Yi J, Zhang Z,
Cheng Y, Li Y, Peng X and Cha X: ‘Psoriasis 1’ reduces
psoriasis-like skin inflammation by inhibiting the VDR-mediated
nuclear NF-κB and STAT signaling pathways. Mol Med Rep.
18:2733–2743. 2018.PubMed/NCBI
|
|
23
|
Wanner C, Inzucchi SE, Lachin JM, Fitchett
D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC
and Zinman B; EMPA-REG OUTCOME Investigators, : Empagliflozin and
progression of kidney disease in Type 2 diabetes. N Engl J Med.
375:323–334. 2016. View Article : Google Scholar : PubMed/NCBI
|