Introduction
As a result of persistent antigen exposure, which
occurs in chronic viral infection and cancer, T cells progressively
lost their effector functions, entering a state called ‘exhaustion’
(1-3).
Exhausted T cells are characterized by the overexpression of
multiple inhibitory receptors (immunological checkpoints), such as
programmed death-1 (PD-1, also known as CD279) (4-6).
After binding its ligand PD-L1, PD-1 attenuates antigen-specific T
cell response, suppressing the tumor-killing activity of T cells
(7,8). Functional restoration of exhausted T
cells by antibodies masking PD-1 on immune effector cells or PD-L1
on tumor cells has produced promising results and has become a
significant breakthrough in cancer immunotherapy (9-11).
Therapeutic benefits from PD-1/PD-L1 axis blockade have been
achieved in patients with an expanding scope of malignancies
(12-15).
Antibody-based immune checkpoint blockade therapy, however, is
expensive to manufacture and administer, and also has several
disadvantages such as the lack of oral bioavailability, and a long
half-life, which causes difficulties when attempting to remove them
from the body, particularly in cases of serious adverse events
(16,17). Novel modalities utilizing small
molecules targeting the same pathways are thus highly anticipated.
The development of small molecules toward these targets, however,
is hampered by the limited structural information on these immune
checkpoint proteins (18).
PD-L1 is an immune co-regulatory molecule belonging
to the B7 family (19). While PD-1
is predominantly expressed on activated T cells, PD-L1 is expressed
on the surface of both cancer and immune cells. By triggering an
inhibitory signal towards the T cell receptor-mediated activation,
PD-L1 suppresses the proliferation, activation and infiltration of
cytotoxic T-lymphocytes, consequently facilitating tumor immune
escape and cancer progression (20-22).
Furthermore, PD-L1 has been reported to be aberrantly overexpressed
in numerous types of tumor cell, including melanoma, ovarian and
lung cancers, and patients with high PD-L1 expression are
associated with unfavorable prognosis and significant risk of
cancer-specific mortality (23-25).
Therefore, pharmaceutical inhibition of PD-L1 expression represents
an alternative approach to currently available cancer
immunotherapy.
Brefelamide is an aromatic amide isolated from
Dictyostelium cellular slim molds. A previous study revealed that
brefelamide inhibits the proliferation of human-derived 1321N1
astrocytoma cells in response to growth factors (26), and the anti-proliferative effect was
associated with a reduction of phosphorylation of extracellular
signal-regulated kinase (ERK), AKT and c-jun-N-terminal kinase
(27). More recently, our previously
published study demonstrated that brefelamide inhibited osteopontin
(OPN) expression and OPN-mediated cell invasion of human lung
adenocarcinoma-derived A549 cells (28). In the present study amide analogues
of brefelamide were found to suppress PD-L1 expression in different
cancer cell lines and mitigated PD-1/PD-L1-mediated exhaustion of
Jurkat T-lymphocytes co-cultured with A549 cells, and the Hippo
pathway is possibly involved in the inhibition of PD-L1 by the
amide analogues, which is mediated by a putative binding site for
transcriptional co-activator with PDZ (TAZ)/Yes-associated protein
(YAP)-TEA domain (TEAD) on PD-L1 promoter.
Materials and methods
Plasmids
For construction of the reporter vector pPD-L1-luc,
the promoter sequence from -2094 to +54 was amplified by PCR using
human genomic DNA as a template with the following primers:
Forward, 5'-ataggtaccACTGCTCTTCTCCCATCTCA-3' and reverse
5'-ataccatggtggctttaccaacagtaccggattgccaagcttAAGCTGCGCAGAACTGGGGC-3'.
The amplified products were digested with Kpn I and Nco
I, and subsequently ligated with pGL3-basic (Promega
Corporation) which has previously digested with the same enzymes.
pmTREPD-L1-luc and pdTREPD-L1-luc are identical to pPD-L1-luc,
except for the mutation or deletion introduced to disrupt the TEAD
responsive element (TRE), respectively, which were generated using
PCR site-directed mutagenesis. Briefly, using pPD-L1-luc as the
template, the proximal part of the PD-L1 promoter sequence was
amplified using 5' primer containing either the mutation
(5'-TGAAAGCTTCCGCCGATTTCACCGAAGGTCTCCTTTCTCCAACGCCCGGCAAACTGGATTTG-3'
for pmTREPD-L1-luc) or deletion
(5'-TGAAAGCTTCCGCCGATTTCACCGAAGGTCGCCCGGCAAACTGGATTTG-3' for
pdTREPD-L1-luc) and the aforementioned reverse primer. The
resultant products were then digested with HindIII and NcoI and
inserted into pPD-L1-luc, which was previously digested with the
same enzymes. The sequence of the PCR-manipulated regions and the
presence of the expected mutations or deletions were confirmed by
nucleotide sequencing.
Cells
The human lung adenocarcinoma-derived A549 cells and
the PC-3 human prostate cancer cell line were purchased from
American Type Culture Collection and maintained in Dulbecco's
modified Eagle's medium (Invitrogen: Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (FBS) and 50 U/ml
penicillin and streptomycin at 37˚C in a humidified incubator with
5% CO2. The THP-1 human leukemia monocytic cell line and
the Jurkat T-lymphocytes were maintained in RPMI1640 supplemented
with 10% FBS. The cell line, A549/PD-L1-luc, was established by
co-transfection of A549 cells with pPD-L1-luc and pPUR (Clontech
Laboratories, Inc.), followed by selection in the presence of 1
µg/ml puromycin (Sigma-Aldrich; Merck KGaA).
Cytokines, reagents and
antibodies
Recombinant human interferon (IFN)-γ was purchased
from PeproTech, phorbol 12-myristate 13-acetate (PMA), and
phytohemagglutinin (PHA) were purchased from Sigma-Aldrich (Merck
KGaA), and Roche Diagnostics, respectively. Anti-human PD-1
antibody and recombinant PD-L1 were purchased from R&D Systems,
Inc.
Chemicals
Amide analogues of brefelamide were synthesized by
maintaining the position of the benzene rings, the carbonyl groups
and the amino groups contained in brefelamide for the purpose of
focusing on the suppressive effect of brefelamide on PD-L1
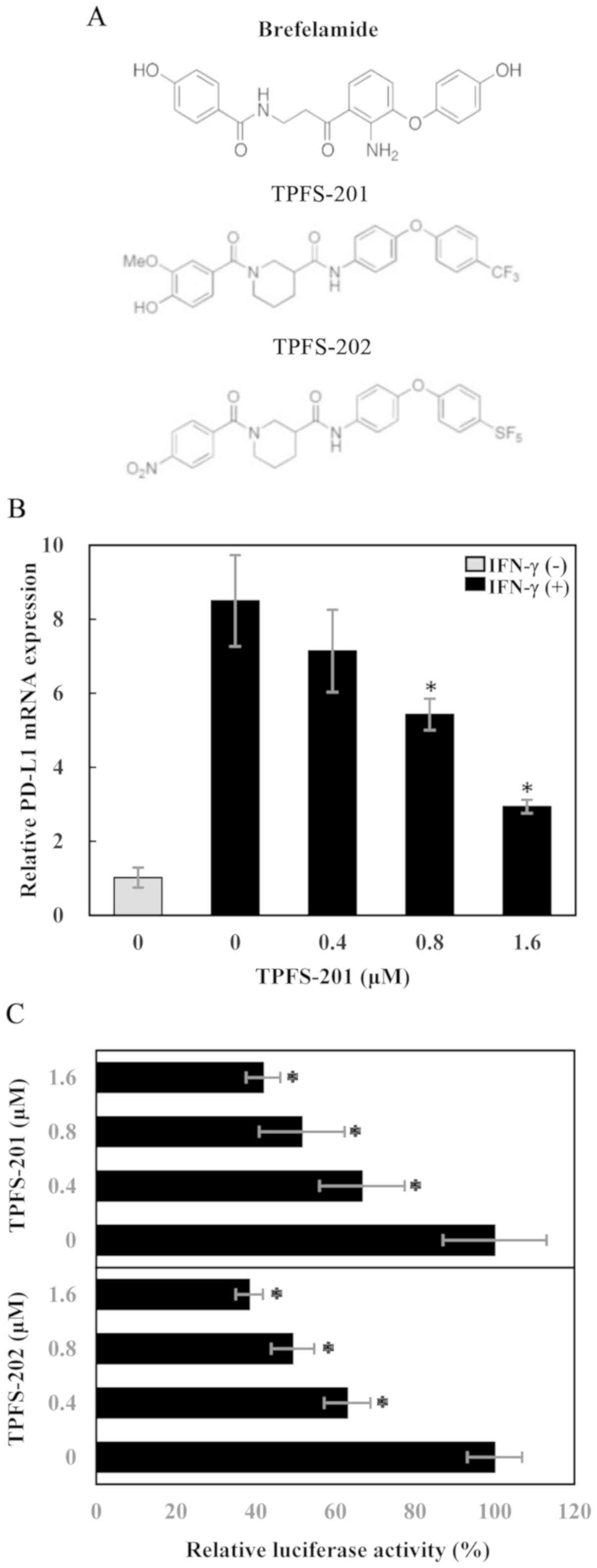
expression. TPFS-201 and TPFS-202 (Fig.
1A) are the representative amide analogues. They were dissolved
in DMSO at a concentration of 50 mmol/l and store at -20˚C.
Aliquots of the stock solution were subsequently diluted to the
indicated concentration before treatment of the cells.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells were lysed with TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and total RNA was
extracted according to the manufacturer's instructions. After
treatment with RNase-free DNase (Promega Corporation), the DNA-free
RNA (250 ng) was used for synthesis of the first-strand cDNA at
42˚C for 60 min using MMLV reverse transcriptase (Invitrogen;
Thermo Fisher Scientific, Inc.). RT-qPCR was performed using Power
SYBR Green PCR Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) at 95˚C for 15 s and at 60˚C for 1 min, for 40
cycles, in a 96-well format on StepOnePlus™ Real-Time PCR Systems
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The following
primer sequences were used: PD-L1 forward,
5'-GGCATTTGCTGAACGCAT-3', and reverse, 5'-CAATTAGTGCAGCCAGGT-3';
GAPDH forward, 5'-TGATGACATCAAGAAGGTGG-3', and reverse,
5'-TCCTTGGAGGCCATGTGGGC-3'.
Flow cytometric analysis
Surface expression of PD-L1 was assessed by flow
cytometry. Briefly, cell suspensions were prepared and washed in
fluorescence-activated cell sorter buffer consisting of
phosphate-buffered saline containing 1% bovine serum albumin. After
blocking with normal mouse serum, cells were incubated with
fluorochrome (PC7) labeled anti-CD274 antibody (cat. no. A78884;
Beckman Coulter, Inc.) or mouse IgG1 isotype control (cat. no.
737662; Beckman Coulter, Inc.) in the dark for 30 min, in an ice
bath. After fixation, the fluorescent signals from cells were
acquired on a Cell Sorter SH800 flow cytometer (Sony Europe B.V.)
and the data was analyzed using FlowJo software (BD Biosciences).
To determine the surface expression of PD-L1 in mouse cells, PE
labeled anti-mouse CD274 antibody (cat. no. 12-5982-82) or rat
IgG2a κ (cat. no. 12-4321-80; both Thermo Fisher Scientific, Inc.)
was used.
Luciferase assay
A549/PD-L1-luc cells were cultured for 48 h at
various concentrations of TPFS compounds. The cells were harvested,
and the cell lysates were prepared for the luciferase assay with
the Luciferase Reporter Assay System (Promega Corporation)
according to the manufacturer's protocol. Luciferase activities
were measured using a GloMax® 20/20 Luminometer (Promega
Corporation). The protein concentration in the cell lysate was
determined with Pierce BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.) according to the manufacture's protocol. The
luciferase values were normalized to protein content.
T cell apoptosis assays
T cell apoptosis assays were performed as described
previously (29). Briefly, A549
cells were treated with interferon (IFN)-γ alone or together with
TPFS-201 for 48 h. Jurkat T cells were activated with 0.25 µg/ml
PHA and 12.5 ng/ml PMA for 48 h to induce PD-1 expression. IFN-γ
and/or TPFS-201-treated A549 cells were co-cultured with PHA-
PMA-activated Jurkat cells at a ratio of 10:1. After 24 h,
apoptosis in Jurkat cells was measured using the
Caspase-Glo® 3/7 Assay Systems (Promega Corporation). In
parallel experiments, Jurkat T cells were pre-incubated with 10
µg/ml anti-PD-1 antibody (R&D Systems) or mouse IgG isotype
control for 3 h prior to co-culturing with IFN-γ-stimulated A549
cells.
T cell activation bioassay
Jurkat T cells were transfected with an interleukin
(IL)-2 reporter plasmid, pIL2-luc, using TransIT-Jurkat
Transfection Reagent (Mirus Bio LLC). The cells were stimulated
with 0.25 µg/ml PHA and 12.5 ng/ml PMA to initiate PD-1 expression,
24 h after transfection. A549 cells were treated with IFN-γ alone
or together with TPFS-201 for 48 h. IFN-γ- and/or TPFS-201-treated
A549 cells were co-cultured with PHA- and PMA-activated Jurkat T
cells at an effector-to-target ratio of 10:1. In a parallel
experiment, PHA- and PMA-activated Jurkat T cells were preincubated
with 10 µg/ml anti-PD-1 for 3 h before co-culturing with
IFN-γ-stimulated A549 cells. In a further experiment, IFN-γ- and
TPFS-treated A549 cells were co-cultured with Jurkat T cells in the
presence of 10 µg/ml recombinant human PD-L1/B7-H1 Fc (R&D
Systems). Jurkat T cells were collected 24 h later, and activities
of luciferase driven by IL-2 promoter were measured.
Statistical analysis
Experimental data are presented as the mean ±
standard deviation from three or more independent experiments. All
P-values were determined using one-way ANOVA followed by Dunnett's
multiple comparisons test. Data were analyzed using Statcel 4
software (OMS, Inc.) P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibition of PD-L1 expression by
TPFS-201 and TPFS-202
While the regulation of PD-L1 expression is yet to
be elucidated, transcription of PD-L1 has been reported to be
activated in response to different signaling pathways including
EGF-induced PI3K/AKT and RTK/Ras/MAPK signaling (30-32).
Considering that multiple target genes downstream of these pathways
were downregulated in microarray-based gene profiling analysis of
brefelamide-treated A549 cells (28), it was hypothesized whether
brefelamide or its amide analogues could suppress PD-L1 expression
in tumor cells, thereby promoting T cell-mediated antitumor
immunity. To address this issue, the effect of brefelamide amide
analogues on PD-L1 mRNA expression was investigated. Following
culturing in the presence of IFN-γ alone or in combination with
TPFS-201, RNA from PC-3 was extracted and the PD-L1 mRNA level was
determined using RT-qPCR. As shown in Fig. 1B, IFN-γ stimulation markedly
increased PD-L1 mRNA level in PC-3 cells, and interestingly,
treatment with TPFS-201 dose-dependently reduced IFN-γ-induced
PD-L1 mRNA expression, while basal PD-L1 expression levels were not
affected. To further characterize the effect of TPFS-201 on PD-L1
transcription, an experimental system was created, using the A549
cell line stably transfected with pPD-L1-luc, in which the
luciferase gene is under the control of human PD-L1 promoter. After
treatment for 48 h, with increasing concentration of TPFS-201 or
TPFS-202, luciferase expression in A549/PD-L1-luc cells was
suppressed in a concentration-dependent manner (Fig. 1C), confirming a transcriptional
repression of PD-L1 by the amide analogues of brefelamide. The
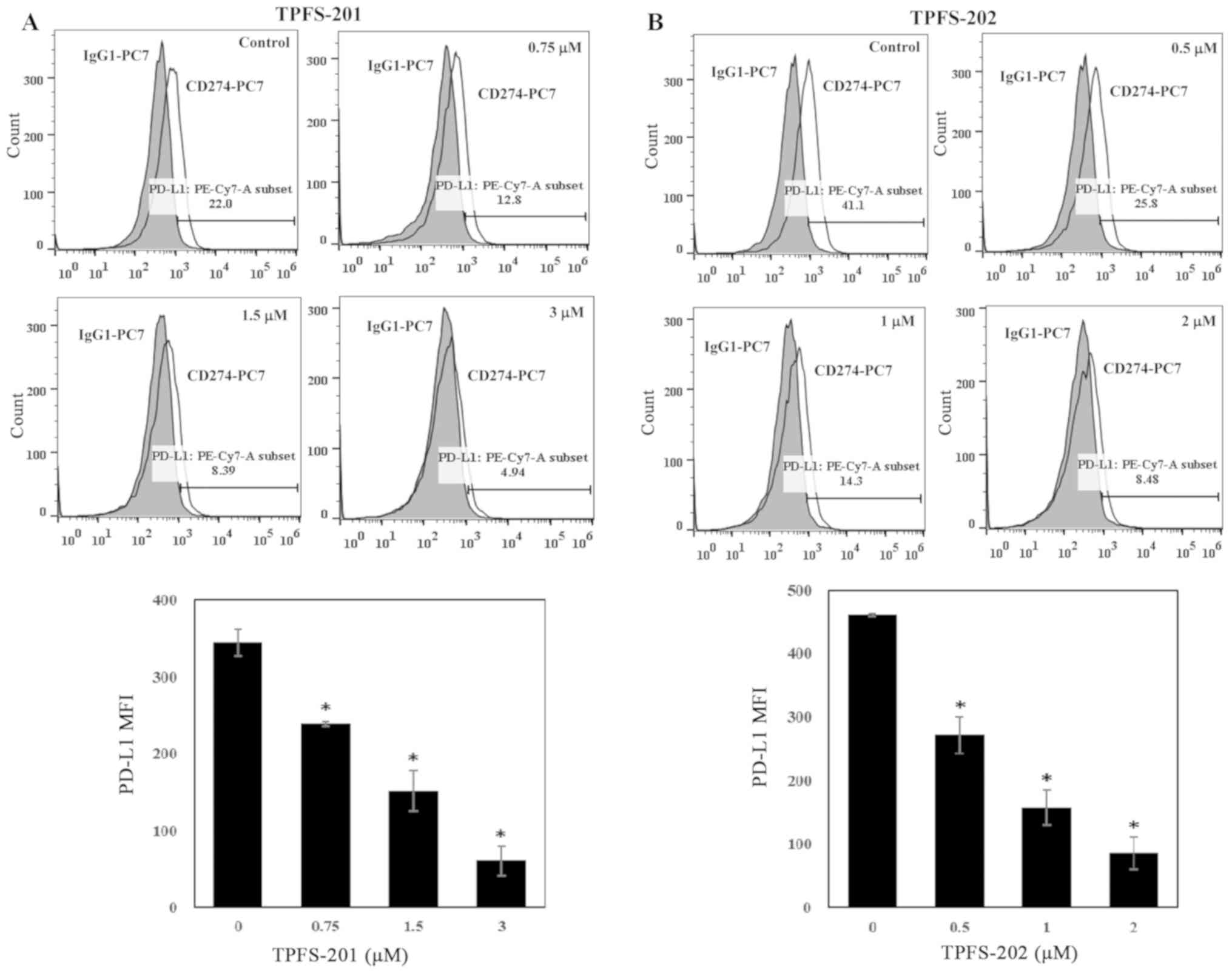
consistent change in PD-L1 expression was also identified in the
flow cytometric analysis, in which there was a 31, 56 and 83%
decrease in the median fluorescence intensity of THP-1 cells
treated with TPFS-201 at the concentration of 0.75, 1.5 and 3 µM,
respectively (Fig. 2A), and a 41, 66
and 81% decrease in THP-1 cells treated with TPFS-202 at the
concentration of 0.5, 1 and 2 µM, respectively (Fig. 2B). The IC50 values
calculated from dose-response curves were 0.93 and 0.67 µM for
TPFS-201 and TPFS-202, respectively (Table SI). These results suggest that
inhibition of PD-L1 by amide analogues of brefelamide is not cell
type specific.
Effect of TPFS compounds on
PD-1/PD-L1-mediated immunosuppression
By engaging PD-1 on T cells, tumor-expressed PD-L1
inactivates the anti-tumor response by suppressing T cell
proliferation, promoting T cell apoptosis and inhibiting cytokine
production. The therapeutic potential of TPFS-201 and TPFS-202 in
restoring anti-cancer immune response by targeting PD-L1 expression
was subsequently explored. Co-culture experiment of A549 cells and
Jurkat T cells was conducted to investigate if TPFS
compound-mediated PD-L1 inhibition could alleviate the function
loss of co-cultured Jurkat T cells. Apoptosis and IL-2-directed
reporter gene expression (as an indicator of IL-2 production) in
co-cultured Jurkat T cells were measured to monitor T cell
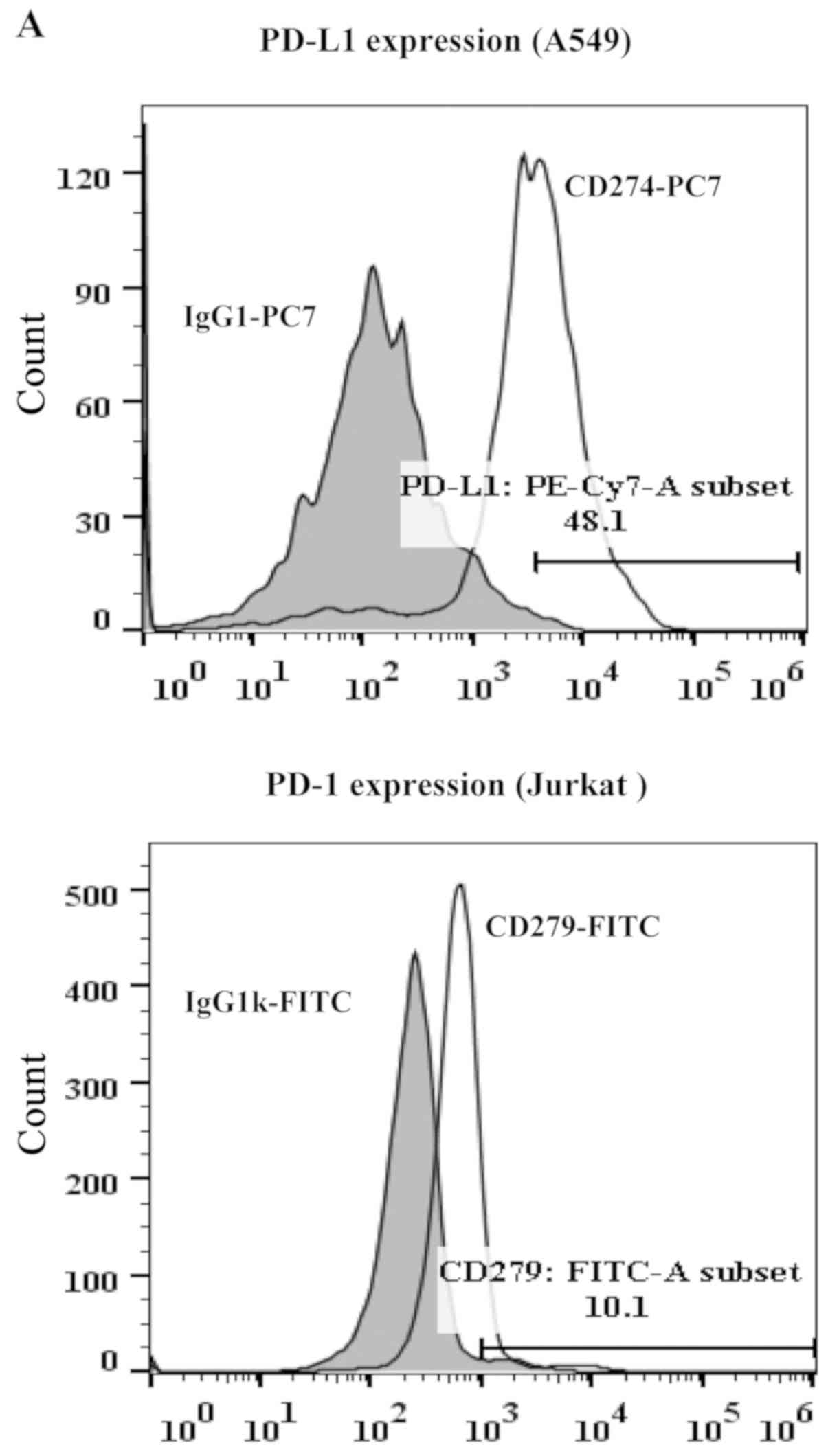
function. Prior to co-culture, A549 cells were stimulated with
IFN-γ to induce PD-L1 expression, and Jurkat cells were activated
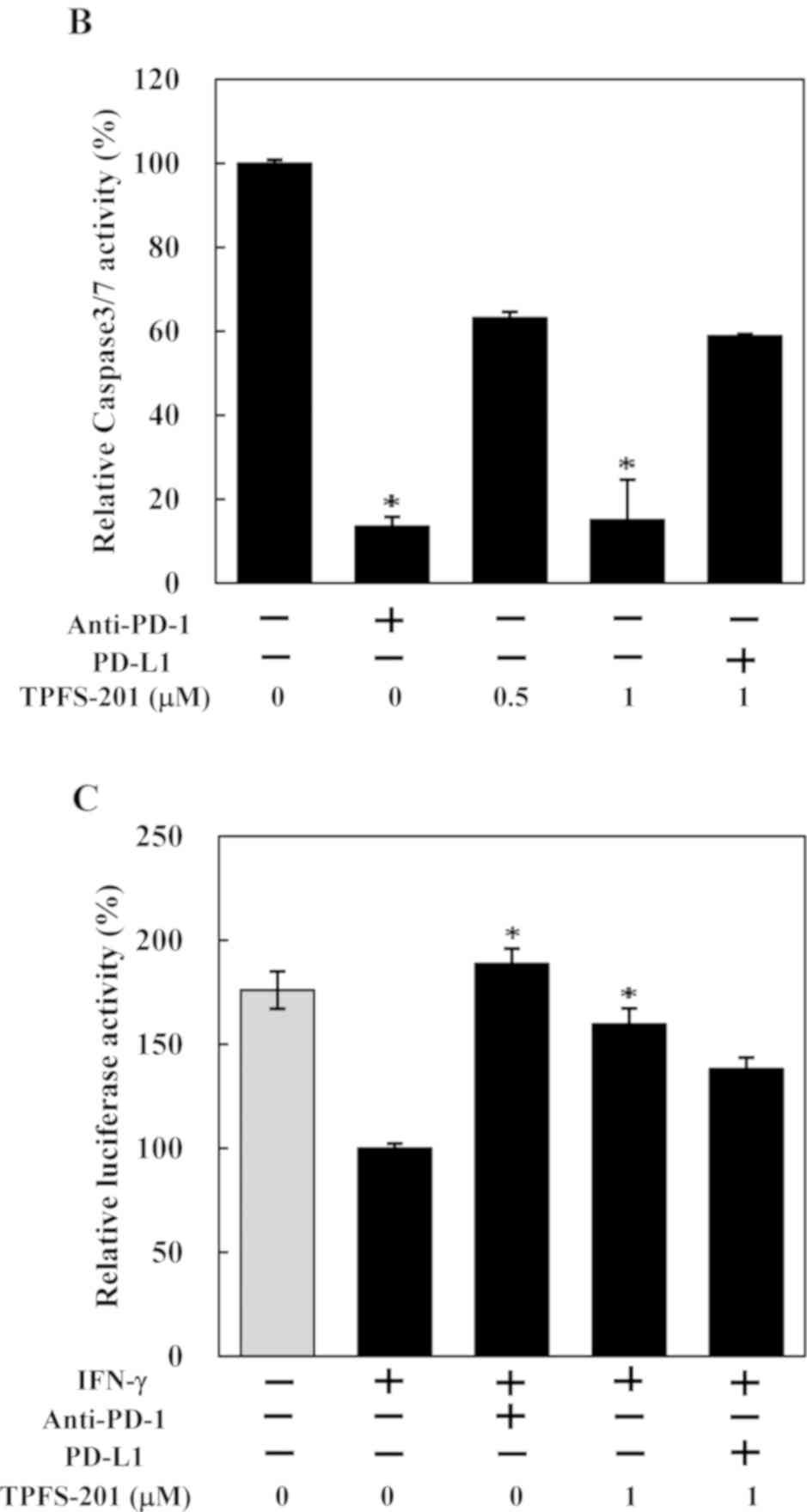
with PMA and PHA to induce PD-1 expression (Fig. 3A). As shown in Fig. 3B, A549 cells induced apoptosis in
co-cultured Jurkat T cells, and pre-incubation of Jurkat T cells
with anti-PD-1 antibody disrupting PD-1/PD-L1 interaction prior to
co-culture, almost fully abrogated apoptosis in Jurkat T-cells,
indicating apoptosis was mediated by PD-1/PD-L1 interaction.
Notably, decreases in apoptosis was also observed in Jurkat T cells
co-cultured with A549 cells pretreated with TPFS-201. Furthermore,
apoptosis inhibition in co-cultured Jurkat T cells by TPFS-201 was
negated to some extent by the addition of recombinant human PD-L1
protein, suggesting that TPFS-201 inhibited effector (Jurkat T)
cells apoptosis, at least partially, through downregulation of
PD-L1 expression on co-cultured target (A549) cells. In addition,
co-cultivation with IFN-γ-stimulated A549 cells inhibited IL-2-luc
expression in Jurkat reporter cells, which was restored by
pre-incubating with a PD-1 blocking antibody (Fig. 3C). Treatment of A549 cells with
TPFS-201 also restored IL-2-luc expression in Jurkat T cells
co-cultured with these cells, albeit less efficiently. Moreover,
TPFS-201-mediated restoration of IL-2-luc expression was less
evident when co-cultured in the presence of recombinant human PD-L1
protein. Together, these results indicate that TPFS-201-mediated
PD-L1 downregulation in cancer cells could partially reverse the
functional loss of co-cultured Jurkat T cells, suggesting a
therapeutic potential of the TPFS compound for cancer
immunotherapy.
Species-specific inhibition of PD-L1
by TPFS-202
Following the characterized of the inhibitory effect
of the brefelamide amide analogues on PD-L1 in vitro, an
in vivo evaluation of the efficacy of these compounds as
novel immunotherapeutics was subsequently considered. One concern
in utilizing a mouse model for drug development, as a prediction of
human immunology, is the divergence in the transcriptional programs
between the immune systems of the two species, despite a
significant similarity in the expression of immune-related genes
between the two species (33-35).
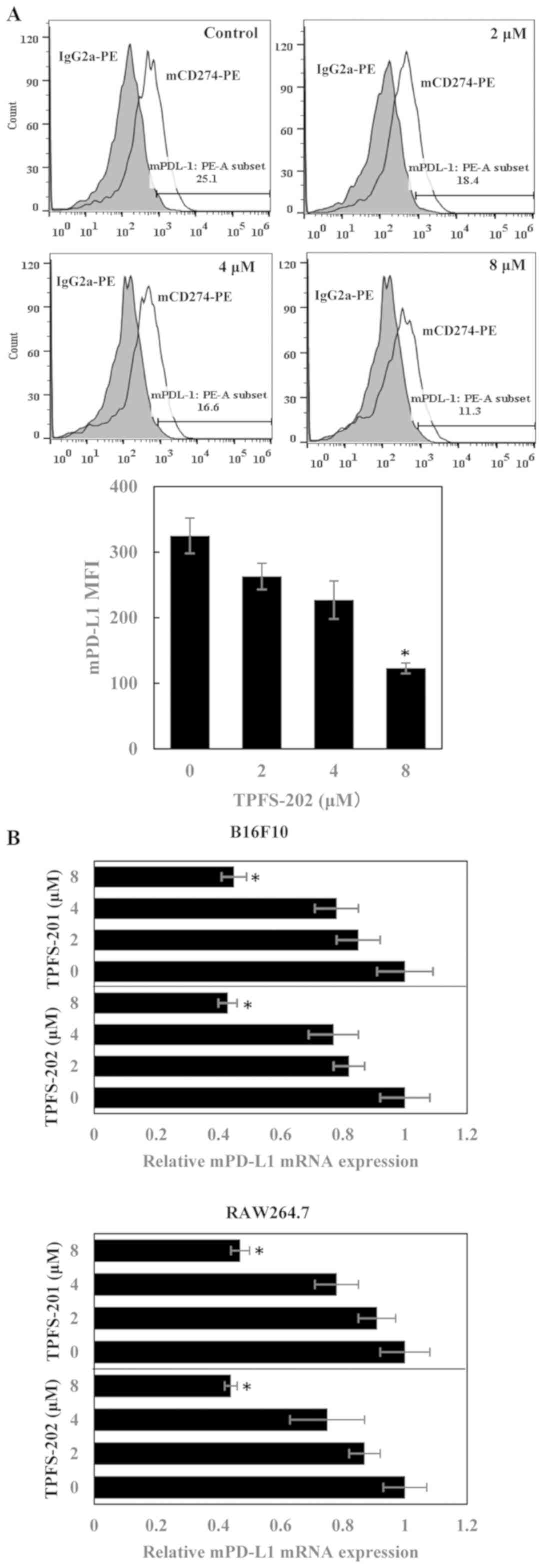
To explore the feasibility of using immunocompetent syngeneic mouse
model for this purpose, it was investigated whether TPFS-202 could
also suppresses PD-L1 expression in mouse cancer cell lines. As
expected, the B16F10 mouse melanoma cell line efficiently expressed
PD-L1 in response to mouse IFN-γ stimulation. However, treatment
with TPFS-202 only slightly inhibited PD-L1 expression on B16F10
cells (Fig. 4A), with an
IC50 value of 6.2 µM for TPFS-202, which is 9-fold
higher compared with the corresponding value for THP-1 cells.
Consistently, RT-qPCR results revealed that treatment with TPFS-201
or TPFS-202 only marginally inhibited PD-L1 mRNA expression in
B16F10 and RAW264.7 cells, a murine macrophage cell line (Fig. 4B). The IC50 values of
TPFS-201 and TPFS-202 in B16F10 and RAW264.7 cells were 6.4 and 6.1
µM and 6.6 and 6.2 µM, respectively (Table SI). Accordingly, the results show
that inhibition of PD-L1 by TPFS compounds is species-specific.
Involvement of Hippo signaling in
TPFS-202-mediated PD-L1 suppression
While hypothesizing the molecular basis underlying
the observed divergence between human and mouse cells, a study by
Janse van Rensburg et al (29) identified PD-L1 as a novel member in
the Hippo pathway-regulated gene network in human breast and lung
cancer cell lines, and transcriptional regulation of PD-L1 by TAZ
and YAP was not conserved in mouse cell lines. In view of this
publication, it was postulated that the distinct effect of TPFS
compounds on PD-L1 expression in human and mouse cell lines may be
associated with a different regulation mechanism of PD-L1 by TAZ in
these two species. Reporter vectors, in which the TRE was
substituted (pmTREPD-L1-luc) or deleted (pdTREPD-L1-luc) to create
a mutant PD-L1 promoter, were constructed to investigate this
hypothesis (Fig. 5), and A549 cells
stably transfected with these constructs were established.
Consistent with data shown in Fig.
1C, treatment with TPFS-202 dose-dependently inhibited
luciferase expression in cells with wild-type pPD-L1-luc plasmid,
and disruption of TRE in the two mutant constructs, especially in
pdTREPD-L1-luc, markedly abrogated the inhibition mediated by
TPFS-202. This suggests that inhibition of PD-L1 by TPFS-202 is
mediated, at least partially, by Hippo-TAZ/YAP signaling, although
another pathway may also be involved in the observed inhibition of
PD-L1.
Discussion
Immune checkpoint blockade has emerged as an
effective treatment option for a wide range of tumor types,
however, the objective tumor response is still limited to a
fraction of cases and tumor types. Furthermore, monoclonal antibody
(mAb)-based checkpoint inhibitors are associated with unique
immune-related adverse events and high costs (36,37).
Therefore, there is an urgent requirement to explore alternative
modalities based on non-mAb-based therapeutics, including small
molecules. In the present study, the feasibility and therapeutic
potential of amide analogues of brefelamide as small molecule
immune checkpoint inhibitors by suppressing PD-L1 expression on
tumor cells was investigated. The data presented in the current
study revealed inhibitory effects of TPFS-201 and TPFS-202 on
promoter activity, endogenous mRNA and PD-L1 surface protein
expression, and inhibition of PD-L1 by the TPFS compounds
consequently restored T cell activity, as evidenced by diminished
apoptosis and increased IL-2 production from Jurkat T cells
co-cultured with TPFS compound-treated A549 cells.
The majority of orthologous transcription factors
play conserved roles in both human and mouse, however, some
immune-related transcription regulation appear to be species
specific (38,39). A part of this regulation divergence
in humans and mice is attributable to the differential expression
of the transcriptional regulators in both species. Indeed, the
expression pattern of transcriptional regulator, even master
regulator, was found to be only partially conserved between humans
and mice. Another possible mechanism for this difference in
regulation between species is based on cis-regulatory elements
being enriched in one species but not in the other (40,41). In
this regard, a recent study by Janse van Rensburg et al
(29) has revealed species-specific
regulation in the TAZ transcription program, and a couple of
TAZ-regulated genes, including PD-L1 in human cells, were weakly
responsive to TAZ overexpression in mouse cells, possibly due to
difference in the cis-regulatory element of the promoter. In view
of this distinction, it is not surprising that TPFS-mediated PD-L1
inhibition was blunted in mice cell lines. Considering the distinct
regulation of PD-L1 by TAZ in the two species, it is reasonable to
assume that PD-L1 inhibition by TPFS compounds may involve
Hippo-TAZ signaling. The reporter assay performed in the present
study revealed that disruption of the putative TAZ/TEAD-binding
motif markedly abrogated the inhibition of PD-L1 by TPFS-202
(Fig. 5), suggesting a role for the
Hippo-TAZ pathway in TPFS-mediated PD-L1 inhibition.
Increasing evidence has demonstrated YAP/TAZ-driven
tumorigenesis in multiple types of solid tumors and highlighted a
link between YAP/TAZ activation and cancer cell stemness,
proliferation, chemoresistance and metastasis (42,43).
Thus, YAP/TAZ are emerging as promising therapeutic targets in
malignant diseases. Further investigation of the effect of the
brefelamide amide analogues on other downstream targets of Hippo
signaling, besides PD-L1, may be warranted to determine if the
compounds TPFS-201 and TPFS-202 are bona fide negative regulators
of Hippo signaling. If confirmed, the anti-YAP/TAZ activities of
these compounds have potential clinical relevance, and it may be
worthy for further development of brefelamide amide analogues as
promising therapeutic candidates for the malignancies associated
with ablated YAP/TAZ activation.
Compared with those observed in pdTREPD-L1-luc, the
inhibition effect of TPFS-202 was less attenuated by the
substitution of TEAD responsive element in pmTREPD-L1-luc. While it
is well known that transcription factors (TFs) bind to
cis-regulatory elements with specific sequence preference, DNA
structure is emerging as another important determinant of TF-DNA
binding specificity. The mechanism in which TEAD/TAZ utilizes to
recognize their cognate DNA in the PD-L1 promoter has not been
fully elucidated, however, it is possible that the TEAD/TAZ-DNA
interaction may be affected, not only by the primary nucleotide
sequence, but by the structural features of the TEAD-binding sites
in the PD-L1 promoter. The DNA structure crucial for TEAD-binding
may be disrupted to a lesser extent in pmTREPD-L1-luc constructs
compared with that in pdTREPD-L1-luc, which may account for their
difference in respond to treatment with TFPS compounds.
The regulation of PD-L1 expression is complex and
multiple transcription regulators are involved. In addition to
IFN-γ, a well-characterized stimulus for PD-L1 expression, recent
evidence has linked multiple cell-intrinsic oncogenic pathways to
PD-L1 expression in cancer cells. Among them are epidermal growth
factor receptor (EGFR), mitogen-activated protein kinase kinase
(MEK)-ERK and mitogen-activated protein kinase p38, the
transcription factor MYC, and the kinase AKT (44-46).
Thus, in addition to Hippo-TAZ/YAP signaling, the impact of the
compounds TPFS-201 and TPFS-202 on these pathways may also
contribute to the PD-L1 inhibition observed in the present study.
Consistent with this hypothesis, it was found that disruption of
the putative TRE did not fully abolished the PD-L1 inhibition by
TPFS-202, the retained inhibition may partially be attributable to
the effect of TPFS-202 on the oncogenic pathways aforementioned.
Indeed, brefelamide was reported to inhibit phosphorylation of EGFR
and attenuated EGFR-mediated ERK signaling cascade (27).
In addition to a critical role in the tumor immune
evasion mechanism, PD-L1 has been reported to be involved in tumor
proliferation, stemness, metastasis, and chemoresistance in
multiple tumor types via tumor-intrinsic PD-L1 signaling (47-49),
therefore it is reasonable to assume that besides attenuation of
PD-1/PD-L1-mediated immunosuppression, the compounds TPFS-201 and
TPFS-202 may exert extra-immune anti-tumor effect by downregulation
of PD-L1 expression on tumor cells. Future work will be necessary
to investigate this hypothesis. If confirmed, the amide analogues
of brefelamide may be a promising therapeutic candidate used not
only as an invigorator of immune response but as a monotherapy or
in combination with conventional treatment to provide additive or
synergistic anticancer effects.
The data presented in the present study suggest the
effect of TPFS compounds on PD-L1 expression is species-specific.
The presence of such species-related differences suggests that the
predictive value of a conventional syngeneic mouse model is limited
in evaluating the efficacy and safety of a therapeutic approach
transcriptionally targeting PD-L1. In this regard, humanized mouse
models, which are derived by engrafting human hematopoietic stem
and progenitor cells into immunodeficient mice, may provide an
alternative approach for this purpose.
In conclusion, the data presented in the present
study indicate that the TPFS compounds suppressed PD-L1 expression
and partially restored PD-1/PD-L1-mediated immunosuppression. These
findings suggest the potential utility of brefelamide amide
analogues as small molecule immune checkpoint inhibitors, thereby
providing therapeutic alternatives, which can be used as
monotherapy or combination with mAb-based blockade.
Supplementary Material
IC50 (μM) values of
TPFS compounds.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ, OY, TH, YO and HK conceived and designed the
study. JZ, OY, SK and SM performed the experiments. JZ and OY wrote
the manuscript. All authors read and approved the manuscript.
Ethic approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zajac AJ, Blattman JN, Murali-Krishna K,
Sourdive DJ, Suresh M, Altman JD and Ahmed R: Viral immune evasion
due to persistence of activated T cells without effector function.
J Exp Med. 188:2205–2213. 1998.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gallimore A, Glithero A, Godkin A, Tissot
AC, Plückthun A, Elliott T, Hengartner H and Zinkernagel R:
Induction and exhaustion of lymphocytic choriomeningitis
virus-specific cytotoxic T lymphocytes visualized using soluble
tetrameric major histocompatibility complex class I-peptide
complexes. J Exp Med. 187:1383–1393. 1998.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fuller MJ, Khanolkar A, Tebo AE and Zajac
AJ: Maintenance, loss, and resurgence of T cell responses during
acute, protracted, and chronic viral infections. J Immunol.
172:4204–4214. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wherry EJ: T cell exhausion. Nat Immunol.
12:492–499. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Doering TA, Crawford A, Angelosanto JM,
Paley MA, Ziegler CG and Wherry EJ: Network and analysis reveals
centrally connected genes and pathways involved in CD8+ T cell
exhaustion versus memory. Immunity. 37:1130–1144. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Schietinger A and Greenberg PD: Tolerance
and exhaustion: Defining mechanisms of T cell dysfunction. Trends
Immunol. 35:51–60. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Butte MJ, Keir ME, Phamduy TB, Sharpe AH
and Freeman GJ: Programmed death-1ligand 1 interacts specifically
with the B7-1 costimulatory molecule to inhibit T cell responses.
Immunity. 27:111–122. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Freeman GJ, Long AJ, Iwai Y, Bourque K,
Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne
MC, et al: Engagement of the PD-1 immunoinhibitory receptor by a
novel B7 family member leads to negative regulation of lymphocyte
activation. J Exp Med. 192:1027–1034. 2000.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Couzin-Frankel J: Breakthrough of the year
2013. Cancer immunotherapy. Science. 342:1432–1433. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Butt AQ and Mills KH: Immunosuppressive
networks and checkpoints controlling antitumor immunity and their
blockade in the development of cancer immunotherapeutics and
vaccines. Oncogene. 33:4623–4631. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Merelli B, Massi D, Cattaneo L and Mandalà
M: Targeting the PD1/PD-L1 axis in melanoma: Biological rationale,
clinical challenges and opportunities. Crit Rev Oncol Hematol.
89:140–165. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Garon EB, Rizvi NA, Hui R, Leighl N,
Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L,
et al: Pembrolizumab for the treatment of non-small-cell lung
cancer. N Engl J Med. 372:2018–2028. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rosenberg JE, Hoffman-Censits J, Powles T,
van der Heijden MS, Balar AV, Necchi A, Dawson N, O'Donnell PH,
Balmanoukian A, Loriot Y, et al: Atezolizumab in patients with
locally advanced and metastatic urothelial carcinoma who have
progressed following treatment with platinum-based chemotherapy: A
single-arm, multicentre, phase 2 trial. Lancet. 387:1909–1920.
2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ansell SM, Lesokhin AM, Borrello I,
Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry
D, Freeman GJ, et al: PD-1 blockade with nivolumab in relapsed or
refractory Hodgkin's lymphoma. N Engl J Med. 372:311–319. 2015.
View Article : Google Scholar
|
|
15
|
Motzer RJ, Escudier B, McDermott DF,
George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G,
Plimack ER, et al: Nivolumab versus everolimus in advanced
renal-cell carcinoma. N Engl J Med. 373:1803–1813. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Caspi RR: Immunotherapy of autoimmunity
and cancer: The penalty for success. Nat Rev Immunol. 8:970–976.
2008.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Amos SM, Duong CP, Westwood JA, Ritchie
DS, Junghans RP, Darcy PK and Kershaw MH: Autoimmunity associated
with immunotherapy of cancer. Blood. 118:499–509. 2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su
HP, Mikami B, Okazaki T, Honjo T, Minato N and Garboczi DN: The
PD-1/PD-L1 complex resembles the antigen-binding Fv domains of
antibodies and T cell receptors. Pro Natl Acad Sci USA.
105:3011–3016. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Dong H, Zhu G, Tamada K and Chen L: B7-H1,
a third member of the B7 family, co-stimulates T-cell proliferation
and interleukin-10 secretion. Nat Med. 5:1365–1369. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Freeman GJ, Long AJ, Iwai Y, Bourque K,
Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne
MC, et al: Engagement of the PD-1 immunoinhibitory receptor by a
novel B7 family member leads to negative regulation of lymphocyte
activation. J Exp Med. 192:1027–1034. 2000.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Dong H, Strome SE, Salomao DR, Tamura H,
Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al:
Tumor-associated B7-H1 promotes T-cell apoptosis: A potential
mechanism of immune evasion. Nat Med. 8:793–800. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Ding H, Wu X and Gao W: PD-L1 is expressed
by human renal tubular epithelial cells and suppresses T cell
cytokine synthesis. Clin Immunol. 115:184–191. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yang CY, Lin MW, Chang YL, Wu CT and Yang
PC: Programmed cell death-ligand 1 expression in surgically
resected stage I pulmonary adenocarcinoma and its correlation with
driver mutations and clinical outcomes. Eur J Cancer. 50:1361–1369.
2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M,
Nakajima Y, Zhou J, Li BZ, Shi YH, Xiao YS, et al: Overexpression
of PD-L1 significantly associates with tumor aggressiveness and
postoperative recurrence in human hepatocellular carcinoma. Clin
Cancer Res. 15:971–979. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hino R, Kabashima K, Kato Y, Yagi H,
Nakamura M, Honjo T, Okazaki T and Tokura Y: Tumor cell expression
of programmed cell death-1 ligand 1 is a prognostic factor for
malignant melanoma. Cancer. 116:1757–1766. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kikuchi H, Saito Y, Sekiya J, Okano Y,
Saito M, Nakahata N, Kubohara Y and Oshima Y: Isolation and
synthesis of a new aromatic compound, brefelamide, from
dictyostelium cellular slime molds and its inhibitory effect on the
proliferation of astrocytoma cells. J Org Chem. 70:8854–8858.
2005.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Honma S, Saito M, Kikuchi H, Saito Y,
OshimaY Nakahata N and Yoshida M: A reduction of epidermal growth
factor receptor is involved in brefelamide induced inhibition of
phosphorylation of ERK in human astrocytoma cells. Eur J Pharmacol.
616:38–42. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang J, Yamada O, Kida S, Matsushita Y,
Murase S, Hattori T, Kubohara Y, Kikuchi H and Oshima Y:
Identification of brefelamide as a novel inhibitor of osteopontin
that suppresses invasion of A549 lung cancer cells. Oncol Rep.
36:2357–2364. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Janse van Rensburg HJ, Azad T, Ling M, Hao
Y, Snetsinger B, Khanal P, Minassian LM, Graham CH, Rauh MJ and
Yang X: The hippo pathway component TAZ promotes immune evasion in
human cancer through PD-L1. Cancer Res. 78:1457–1470.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yamamoto R, Nishikori M, Tashima M, Sakai
T, Ichinohe T, Takaori-Kondo A, Ohmori K and Uchiyama T: B7-H1
expression is regulated by MEK/ERK signaling pathway in anaplastic
large cell lymphoma and Hodgkin lymphoma. Cancer Sci.
100:2093–2100. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J
and Hodi FS: The activation of MAPK in melanoma cells resistant to
BRAF inhibition promotes PD-L1 expression that is reversible by MEK
and PI3K inhibition. Clin Cancer Res. 19:598–609. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Atefi M, Avramis E, Lassen A, Wong DJ,
Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, et
al: Effects of MAPK and PI3K pathways on PD-L1 expression in
melanoma. Clin Cancer Res. 20:3446–3457. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mestas J and Hughes CC: Of mice and not
men: Differences between mouse and human immunology. J Immunol.
172:2731–2738. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Davis MM: A prescription for human
immunology. Immunity. 29:835–838. 2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hayday AC and Peakman M: The habitual,
diverse and surmountable obstacles to human immunology research.
Nat Immunol. 9:575–580. 2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Naidoo J, Page DB, Li BT, Connell LC,
Schindler K, Lacouture ME, Postow MA and Wolchok JD: Toxicities of
the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann
Oncol. 26:2375–2391. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Champiat S, Lambotte O, Barreau E, Belkhir
R, Berdelou A, Carbonnel F, Cauquil C, Chanson P, Collins M,
Durrbach A, et al: Management of immune checkpoint blockade
dysimmune toxicities: A collaborative position paper. Ann Oncol.
27:559–574. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shay T, Jojic V, Zuk O, Rothamel K,
Puyraimond-Zemmour D, Feng T, Wakamatsu E, Benoist C, Koller D and
Regev A: ImmGen Consortium: Conservation and divergence in the
transcriptional programs of the human and mouse immune systems.
Proc Natl Acad Sci USA. 110:2946–2951. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Diehl AG and Boyle AP: Conserved and
species-specific transcription factor co-binding patterns drive
divergent gene regulation in human and mouse. Nucleic Acids Res.
46:1878–1894. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Vierstra J, Rynes E, Sandstrom R, Zhang M,
Canfield T, Hansen RS, Stehling-Sun S, Sabo PJ, Byron R, Humbert R,
et al: Mouse regulatory DNA landscapes reveal global principles of
cis-regulatory evolution. Science. 346:1007–1012. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Odom DT, Dowell RD, Jacobsen ES, Gordon W,
Danford TW, MacIsaac KD, Rolfe PA, Conboy CM, Gifford DK and
Fraenkel E: Tissue-specific transcriptional regulation has diverged
significantly between human and mouse. Nat Genet. 39:730–732.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
42
|
Harvey KF, Zhang X and Thomas DM: The
Hippo pathway and human cancer. Nat Rev Cancer. 13:246–257.
2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Chen Q, Zhang N, Gray RS, Li H, Ewald AJ,
Zahnow CA and Pan D: A temporal requirement for Hippo signaling in
mammary gland differentiation, growth, and tumorigenesis. Genes
Dev. 28:432–437. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Rech AJ and Vonderheide RH: Dynamic
interplay of oncogenes and T cells induces PD-L1 in the tumor
microenvironment. Cancer Discov. 3:1330–1332. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Parsa AT, Waldron JS, Panner A, Crane CA,
Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et
al: Loss of tumor suppressor PTEN function increases B7-H1
expression and immunoresistance in glioma. Nat Med. 13:84–88.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
46
|
Casey SC, Tong L, Li Y, Do R, Walz S,
Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M and Felsher
DW: MYC regulates the antitumor immune response through CD47 and
PD-L1. Science. 352:227–231. 2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M,
Nakajima Y, Zhou J, Li BZ, Shi YH, Xiao YS, et al: Overexpression
of PD-L1 significantly associates with tumor aggressiveness and
postoperative recurrence in human hepatocellular carcinoma. Clin
Cancer Res. 15:971–979. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang P, Ma Y, Lv C, Huang M, Li M, Dong
B, Liu X, An G, Zhang W, Zhang J, et al: Upregulation of programmed
cell death ligand 1 promotes resistance response in non-small-cell
lung cancer patients treated with neo-adjuvant chemotherapy. Cancer
Sci. 107:1563–1571. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Gupta HB, Deng J, Clark CA, Drerup JM, Wu
B, Sareddy G, Hurez V, Vadlamudi R, Li R and Curiel TJ: Programmed
cell death ligand 1 (PD-L1) regulates tumor initiating cell (TIC)
generation by controlling the stemness gene Oct4 through mTORC1. J
Immunol. 200 (1 Supplement)(167)2018.
|