Introduction
CHARGE syndrome is a rare, autosomal dominant
disorder characterized by multiple congenital anomalies. According
to Orphanet, CHARGE syndrome Online Mendelian Inheritance of Man
entry no. 214800; https://www.orpha.net/consor/cgi-bin/) has a
prevalence of 1-9/100,000. CHARGE is an acronym for the most common
symptoms observed in patients with the condition: Coloboma, heart
defects, atresia of the choanae, retarded growth and/or
development, genital hypoplasia and ear abnormalities (1). However, other clinical manifestations
have also been identified in diagnosed cases. Patients with CHARGE
syndrome exhibit complex symptoms that differ from one case to
another, indicating the variable clinical expression of the
disorder. Cranial nerve (CN) dysfunction, external ear anomalies,
semicircular canal anomalies, coloboma, choanal atresia, genital
hypoplasia, congenital cardiovascular defects, cleft lip and/or
palate and anosmia are identified in >50% of patients. Motor
retardation, swallowing and severe feeding problems, and
intellectual disability are also frequent among patients with
CHARGE syndrome (2).
The identification of clinical features is important
for the diagnosis of patients with suspected CHARGE syndrome
(3). Sanlaville and Verloes
(4) divided the clinical aspects of
CHARGE syndrome into major and minor criteria, but the highly
variable clinical expression and the lack of obligatory elements
have continued to pose major diagnostic problems. Major and minor
clinical criteria have been refined and updated (4,5), and a
new checklist organized by body system and age has been developed
to aid the diagnosis of CHARGE syndrome (6). Recently, a novel genetic approach has
changed the importance of clinical criteria for the diagnosis of
the disorder. Hale et al (7)
suggested that difficulties regarding the clinical diagnosis of
CHARGE syndrome may be overcome by including the pathogenic
chromodomain helicase DNA binding protein 7 (CHD7) variant as a
major diagnostic criterion. Accordingly, a pathogenic CHD7 variant
status plus one major feature would be sufficient for the diagnosis
of the disorder (7,8). It is important for each novel mutation
that extends the spectrum of pathogenic mutations in the CHD7 gene
to be included in the databases, to facilitate the diagnosis and
estimate the prevalence of the disorder (3). Since the publication of the new
criteria in 2016, only a few case reports discussing patients with
CHARGE syndrome with a pathogenic mutation in the CHD7 gene,
previously considered as atypical CHARGE, have been published
(9,10). By understanding the different
genotypes of CHARGE syndrome, it was possible to make clear
genotype-phenotype associations for all CHD7 variants.
In the present study, a case with a
plurimalformative syndrome, whose etiology was identified by
clinical whole-exome-sequencing (WES) analysis, was described.
According to the criteria of Hale et al (7), the patient was diagnosed with CHARGE
syndrome due to the presence of the pathogenic CHD7 variant. By
reporting a novel case of CHARGE syndrome, the present study
emphasizes the importance of the new criteria, which may encourage
clinicians to perform extensive genetic testing, e.g., WES, for an
accurate diagnosis of the disorder.
Materials and methods
Clinical and paraclinical
evaluation
Physical examination of the patient was followed by:
Ear, nose and throat assessment, audiogram, thoracic computed
tomography (CT) and abdominal and heart Doppler
ultrasonography.
Genetic analysis
Genomic DNA was extracted from peripheral venous
blood, as previously described by Miller et al (11). Clinical WES was used to detect an
exonic pathogenic variant that may explain the phenotype of the
patient. Gene sequencing was performed by the Invitae Laboratory.
Genomic DNA was enriched for targeted regions using a
hybridization-based protocol and sequenced using Illumina
technology (Illumina, Inc.). The average coverage depth of the WES
of all targeted regions was ≥50X. Reads were aligned to a reference
sequence Genome Reference Consortium human build 37 (GRCh37)
(12) and sequence changes were
identified and interpreted in the context of a single clinically
relevant transcript. Exonic deletions and duplications were
identified using an algorithm that determined the copy number at
each target. Subsequently, the biological and clinical implications
of each variant were identified. All reported pathogenic or likely
pathogenic variants that had potential medical implications for the
patient were verified by orthogonal technologies. Single molecule
real-time (SMRT) sequencing technology (Pacific Biosciences, Inc.)
was used to further verify the results of the WES analysis. Using a
combination of public tools and databases, a comprehensive search
of phenotype-genotype associations was performed. Disruptive
variants, including variants that caused premature truncation
events, interfered with canonical splice sites or resulted in
initiator, frameshift or multi-exon deletions that were detected in
genes associated with Mendelian conditions that corresponded to the
patient's presentation, were reported.
Subsequently, trio-WES analysis was performed on
samples obtained from the parents of the patient, which identified
the mutation as a de novo variant that was only present in
the patient.
The variants that were identified as potentially
explaining the clinical features of the patient were reported
according to the guidelines established by the American College of
Genetics and Genomics (13).
Sequence variants with a minor allele frequency of >0.05 in the
Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/) and the
NHLBI Grand Opportunity Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) were
excluded.
Results
Case presentation
The present study described the case of a
16-month-old female who was evaluated in the Louis Țurcanu
Emergency Hospital for Children and in the Pediatric Cardiology
Department of Pius Brînzeu Emergency County Clinical Hospital,
Timisoara, Romania, in February 2019, for dysmorphic features and
multiple congenital defects in order to establish a diagnosis and
to aid in the management of a future pregnancy. The patient was
born post-term via normal spontaneous delivery. The parents
(mother, 33 years; father, 34 years) were non-consanguineous and
appeared healthy. The patient had no siblings. Ultrasonography
performed during the pregnancy did not detect any pathological
changes in the patient. Anthropometric measurements at birth were
within the normal ranges: Body weight, 3,630 g; body height, 52 cm;
and cranial perimeter, 35 cm. The Apgar scores of the patient were
5/6/10 and the family history of the patient was unremarkable.
Medical history
The patient displayed facial dysmorphism from birth,
which is the reason for further investigation performed by the
neonatology and pediatric services. At birth, severe dysphagia was
reported; therefore, enteral nutrition was the recommended feeding
route. The cause of dysphagia was investigated and neuromuscular
dysphagia, tumors and hemangioma were excluded as causes of the
condition. In time, the patient's condition improved and at 3
months of age, the patient was fed orally. Thoracic and abdominal
CT scans were performed, revealing an aberrant right subclavian
artery (arteria lusoria) and a horseshoe kidney. The patient was
not receiving any medication at the time of presentation.
Clinical evaluation
At 16 months of age, the anthropometric measurements
of the patient upon first physical examination in the Genetics
Department were as follows: Body height, 75 cm; body weight, 8,730
g; and cranial perimeter, 45.5 cm (in the normal range) (14). Upon physical examination at 21 months
of age, the following anthropometric measurements were recorded:
Body height, 78 cm; body weight, 10 kg (in the normal range); and
cranial perimeter, 46.6 cm (in the normal range) (14). At 16 and 21 months of age, the
patient was fed only with liquids and semisolids.
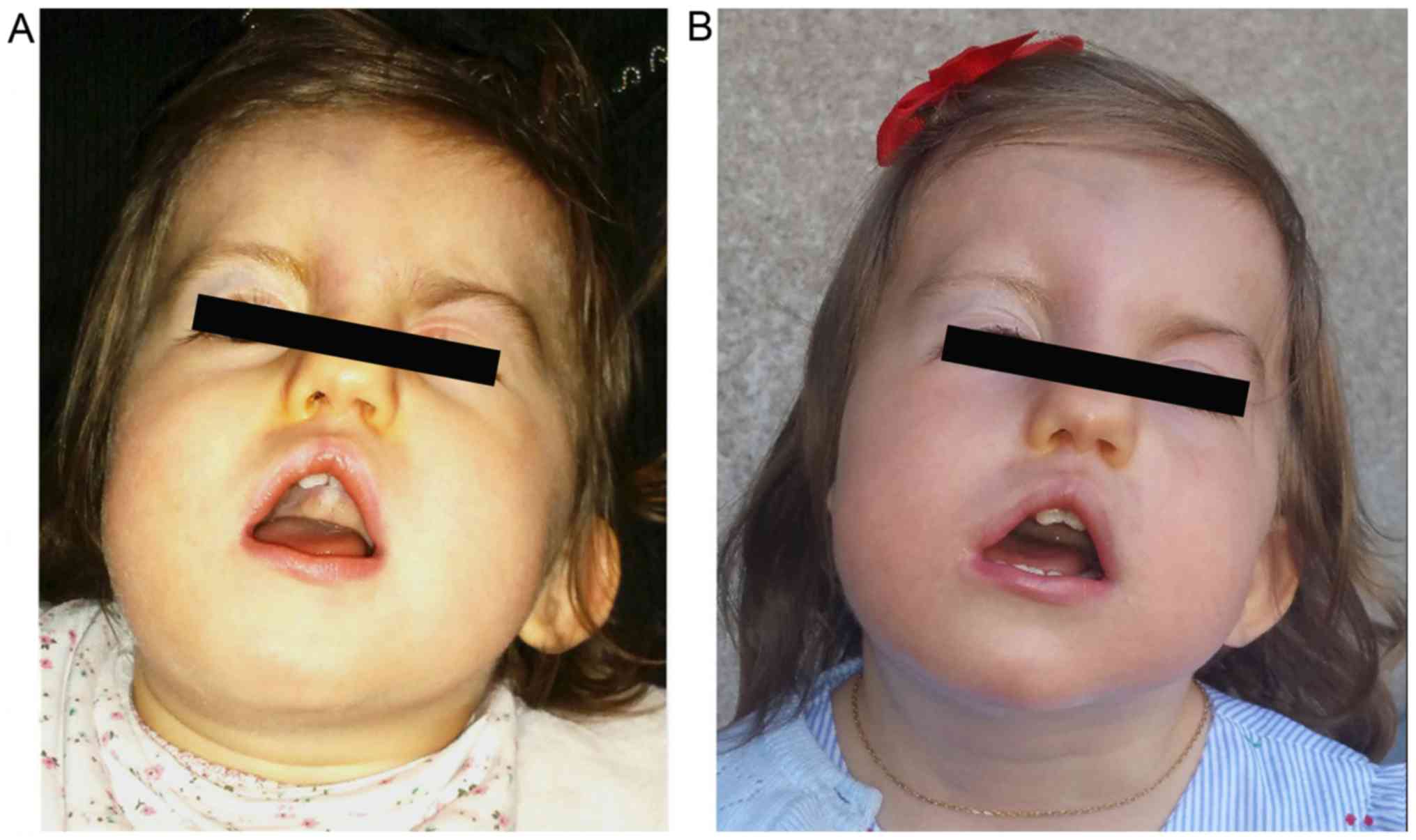
The patient displayed dysmorphic facial features,
which included square face, craniofacial asymmetry, prominent
forehead, wide nasal ridge, high palate, triangular-shaped open
mouth, low-set left ear, asymmetry of the shape and size of the
ears, abnormality of the pinna cartilage of the outer ear, ear lobe
agenesis on the right side and unilateral left blepharoptosis with
associated upper eyelid contraction (Figs. 1 and 2). Ophthalmic examination revealed
choroidal coloboma.
The patient also presented with fixed head
retroflexion, congenital craniofacial dysostosis, facial palsy
(left hemi-face), general facial hypotonia, facial grimacing,
abnormal conjugate eye movement and Marcus Gunn jaw winking
synkinesis. The aforementioned symptoms, together with the left
blepharoptosis, conferred a peculiar facial expression. Cranial
nerves (CN) VII, VIII, IX and X were affected. The patient suffered
from profound deafness.
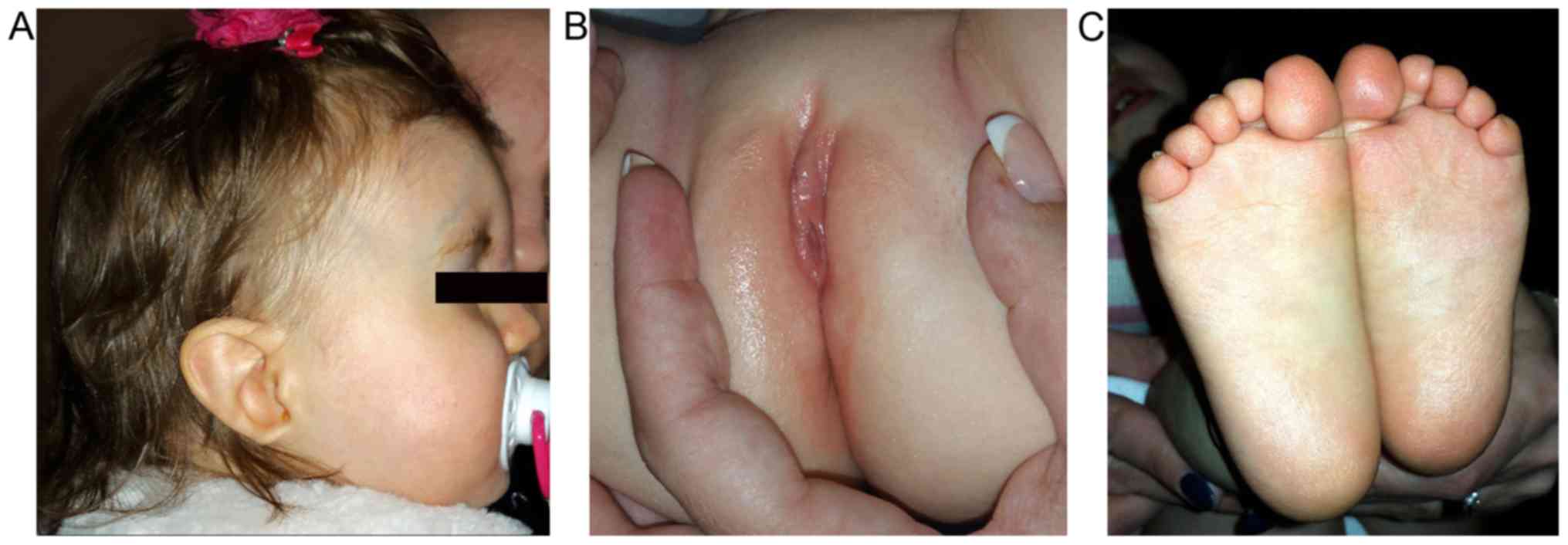
Further anomalies that were identified by systemic
examination included labia minora hypoplasia, abnormality of the
plantar skin and abnormal plantar dermatoglyphics (Fig. 2). In addition, the patient was only
able to walk with support.
Nasal endoscopic exam revealed that the patient did
not have choanal atresia, while audiogram revealed severe bilateral
hearing impairment. The patient did not speak, but she smiled and
was able to cooperate. The left auditory nerve was not visualized
on cranial CT.
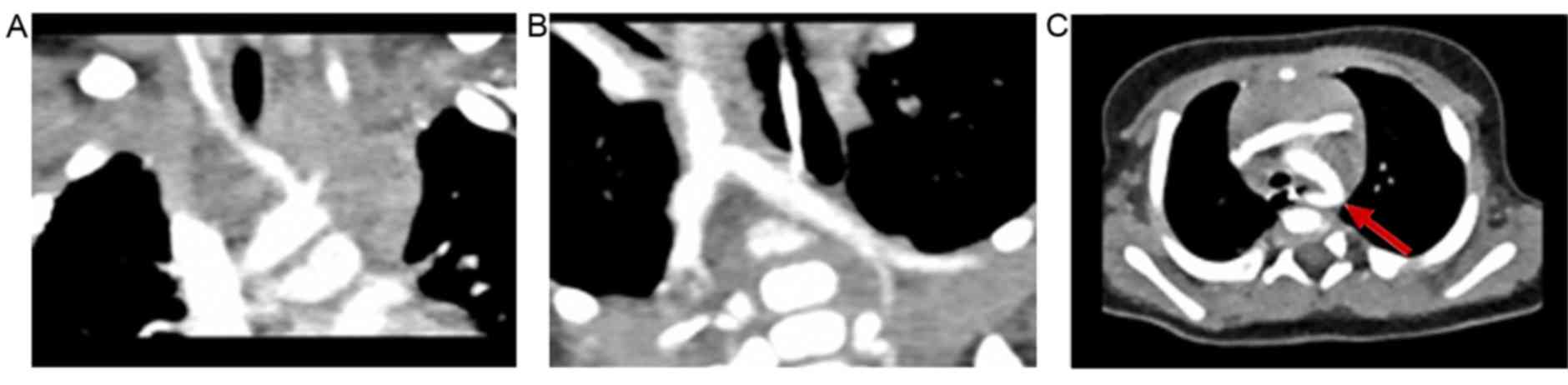
Thoracic CT identified an aberrant origin of the
right subclavian artery (arteria lusoria) without obstructive
manifestations of the esophagus or trachea (Fig. 3).
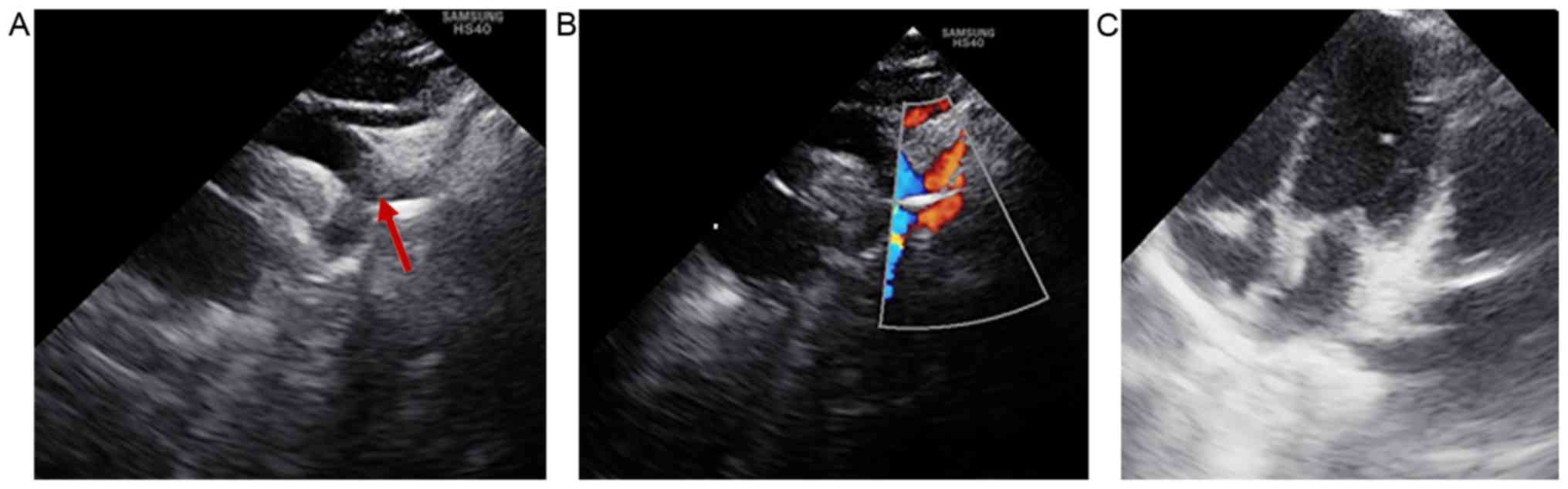
Heart ultrasonography revealed that the patient did
not display any intracardiac structural abnormalities; therefore,
the only cardiac defect identified that was associated with CHARGE
syndrome was the aberrant origin of the right subclavian artery
(Fig. 4).
The patient did not receive any medication. After
receiving kinesiotherapy for 2 h daily, the patient was able to
maintain equilibrium on the feet and walk short distances without
support and retroflexion of the head and left palpebral ptosis were
reduced. Cardiology follow-up for evaluation of the cardiac status
was recommended. A cochlear implant was scheduled.
Mutation
A de novo pathogenic variant, c.4379_4380del
(p.Ile1460Argfs*15), was identified in exon 19 of the
CHD7 gene. The variant consisted of one thymine deletion from
position 4,379 and one adenine deletion from position 4,380
(rs398124319). The mutation led to a reading frameshift starting
from isoleucine at codon 1,460, which resulted in a premature
translational stop signal (p.Ile1460Argfs*15) and was
hypothesized to result in an absent or disrupted protein product.
The analysis identified a monoallelic deletion in the CHD7 gene,
NM_017780.3 (CHD7, c.4379_4380del), which was also identified by
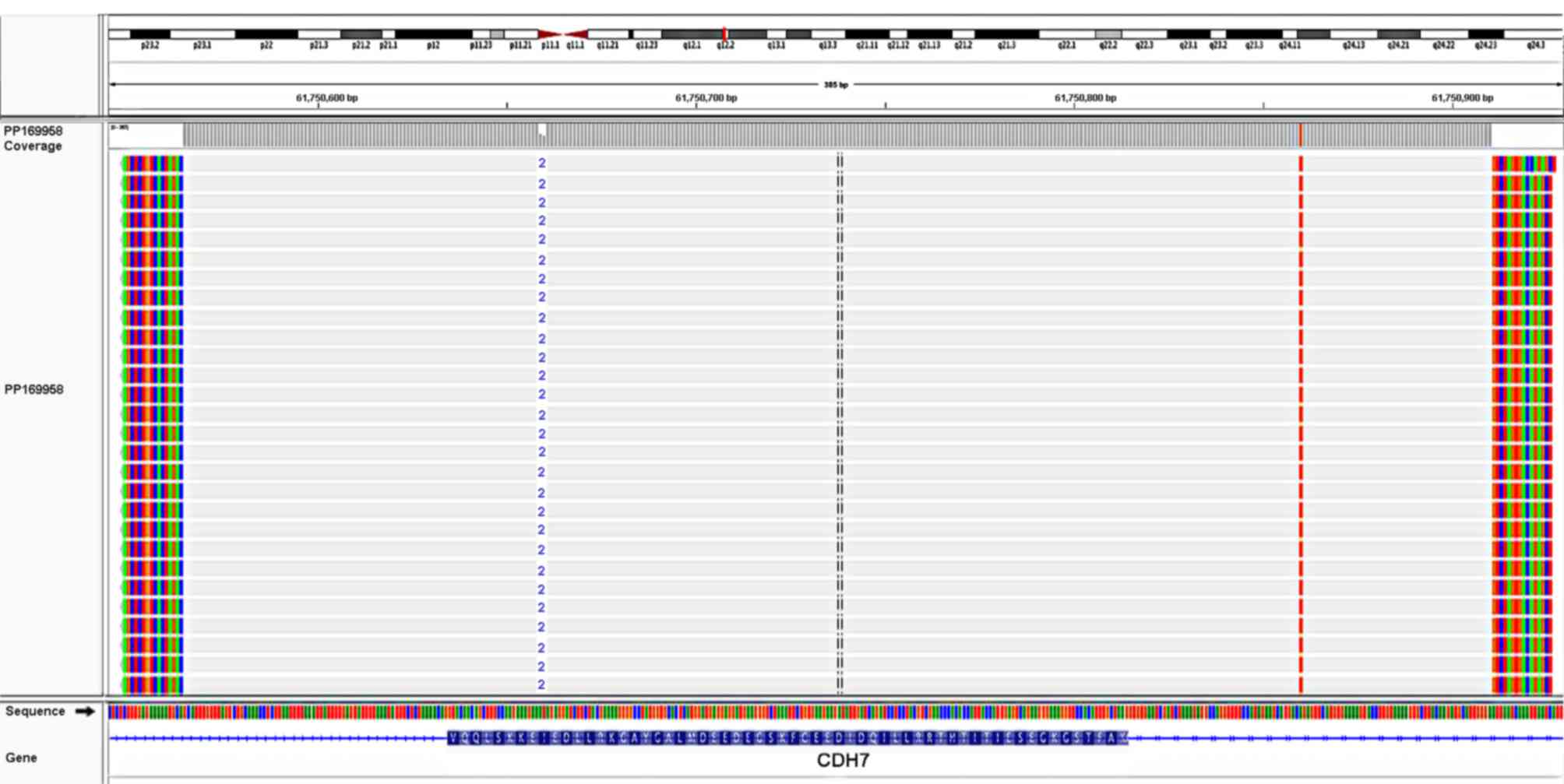
SMRT sequencing (Fig. 5).
The mutation was present in the Human Gene Mutation
Database (http://www.hgmd.org) and the Clinical
Variance database (http://www.ncbi.nlm.nih.gov/clinvar). The heterozygous
frameshift mutation was not detected in the parents of the patient,
suggesting that it was a de novo mutation.
Discussion
CHARGE syndrome is a variable disorder that affects
a number of different parts of the body and is characterized by a
non-random cluster of congenital anomalies and dysmorphisms, with
significant variation between patients (15). The diagnosis relies on a set of major
and minor clinical criteria (4,5).
According to the criteria of Sanlaville and Verloes (4), a patient must display the following:
Three major criteria, or two major and two minor criteria for
diagnosis with the typical form; two major and one minor criterion
for diagnosis with the partial form; two major but no minor
criterion, or one major and two minor criteria for diagnosis with
the atypical form (4). Hale et
al (7), proposed an update to
the clinical diagnostic criteria, suggesting that a pathogenic CHD7
variant status should be considered as a major diagnostic
criterion, which, when present together with one other major
criterion, is sufficient for the diagnosis of typical CHARGE
syndrome.
The majority of patients with CHARGE syndrome
display distinctive facial features, including a square-shaped face
and facial asymmetry, which were also observed in the case
described in the present study (3).
Patients with CHARGE syndrome also display ear abnormalities, which
may contribute to hearing problems. The case reported in the
present study displayed abnormalities of the shape and position of
the bilateral ears, as well as ear lobe agenesis on the right
side.
The central nervous system is also frequently
affected in patients with CHARGE syndrome, with CN dysfunction
reported in >90% of cases. Different clinical manifestations
occur when >1 CN is dysfunctional. In the case reported in the
present study, CN VII, VIII, IX and X were affected. The patient
suffered from profound deafness and the cranial CT indicated that
the patient had no left auditory nerve. Of note, visual and hearing
impairments, as well as intellectual disability, are frequently
observed in patients with CHARGE syndrome (16). Unilateral left facial palsy was first
reported at birth in the patient; however, at the time of the
present study, it had not been determined whether the patient had a
full or diminished sense of smell, or complete anosmia, coordinated
by CN I.
The minor characteristics of CHARGE syndrome include
slow growth starting in late infancy, delayed sitting and
unsupported walking, all of which were present in the case reported
in the present study. The patient also displayed congenital
cardiovascular defects, including an aberrant origin of the right
subclavian artery (arteria lusoria), a horseshoe kidney, VII and
VIII CN palsy and coloboma, which is a major criterion. The
presence of one major and three minor criteria suggested that the
patient had atypical CHARGE syndrome, according to the Sanlaville
and Verloes criteria (3).
Congenital heart defects are present in 3/4 of
patients with CHARGE syndrome (17).
The types of congenital heart defect are variable, from conotruncal
and atrioventricular septal defects to aortic arch anomalies. Among
the congenital aortic arch anomalies, the aberrant origin of the
right subclavian artery is the most common (18).
The patient presented with severe dysphagia
immediately after birth with unknown causes, which spontaneously
remitted after several months. The thoracic CT displayed the
aberrant right subclavian artery, which may have been the cause of
dysphagia. However, at the age of 16 months, the patient did not
have any dysphagia; therefore, surgical intervention was not
required and only cardiac monitoring was recommended.
A horseshoe kidney without impaired renal function
was identified in the patient. Urinary system anomalies are present
in 10-40% of patients with CHARGE syndrome (19). Other minor abnormalities, such as
hypoplastic labia minora, also cause differential diagnostic
problems with Meier-Gorlin syndrome type 1 or 3, Prader-Willi
syndrome, Schinzel-Giedion syndrome, mixed gonadal dysgenesis
(20,21). Several clinical features of these
syndromes overlap with CHARGE syndrome, but they also have
distinctive signs. All these syndromes were excluded based on
clinical findings and on the presence of pathogenic mutation in the
CHD7 gene found in the patient in the present case report.
The patient received follow-up assessments for 6
months and the clinical re-evaluation performed after 6 months
revealed an important improvement. A skeletal survey and a
dual-energy X-ray absorptiometry scan for osteoporosis were
recommended for the patient (5,22). The
patient cooperated, smiled and responded to non-verbal stimuli. By
investigating the range of developmental abilities observed in
patients with CHARGE syndrome, as measured by the Adaptive Behavior
Evaluation Scale, researchers concluded that the age at which the
patient was able to walk was the best predictor of scores (23). The IQ of patients with CHARGE
syndrome ranges from near-normal to severe retardation. The
behavioral phenotype typically consists of low-level adaptive
behavioral skills (e.g., social skills, seeking of attention from
others, externalizing problems) and symptoms of autism-like
spectrum disorder. Data regarding a unique behavioral phenotype for
patients with CHARGE syndrome are emerging (5).
The CHD7 gene is associated with autosomal-dominant
CHARGE syndrome (24). Regarding
genotype-phenotype correlations, missense mutations are associated
with milder phenotypes and variants leading to a premature stop
codon are associated with more severe phenotypes (2,25,26). The
case described in the present study displayed a severe phenotype
and a pathogenic variant (c.4379_4380del) was identified. The
mutation resulted in a premature translational stop signal
(p.Ile1460Argfs*15) in exon 19 of the CHD7 gene. Several
studies have reported that frameshift mutations are the most common
type of mutation in patients with CHARGE syndrome, with a frequency
of 34-73% (8,27,28).
Clinical WES analysis takes into account information regarding the
target genes associated with the major symptoms, as well as the
verification of the genes indicated by the American College of
Medical Genetics (13,29,30).
Compared to classical sequencing, WES analysis has a shorter
analysis time, lower costs and offers a more precise diagnosis. In
the present study, Trio-WES analysis was performed, which allowed
the de novo mutation to be identified, as neither of the
patient's parents displayed the mutation in the CDH7 gene. In a
large study, parental DNA profiles were assessed and all pathogenic
variants were identified as de novo mutations, except for
one case (8). An intragenic deletion
was identified in two brothers, but was not identified in the blood
samples of the parents. It was hypothesized that one parent had
germinal mosaicism (8). Considering
this possibility, prenatal diagnosis should be recommended for each
future pregnancy (31,32). De novo mutations are more
frequent than inherited mutations and the majority are frameshift
mutations (33-35).
Although rare, pathogenic variants may be inherited from a mildly
affected parent; therefore, parents should be carefully examined
(3,4). An analysis of a French cohort suggested
that 66.38% of patients with CHARGE syndrome had different
pathogenic variants and 90% of these variants were novel (8). It is estimated that 74% of patients
with CHARGE syndrome have congenital heart defects due to a CHD7
mutation (25). Congenital heart
defects are more frequent in patients with truncating variants of
the gene (80%) compared with patients with missense or splice-site
variants (58%) (25). Aortic arch
anomalies, frequently including an aberrant subclavian artery or
right aortic arch, were reported in 14% of cases in a previous
study (18). In the present study,
the patient was the first case of reported CHARGE syndrome in
Romania presenting with a truncating CHD7 mutation and an aberrant
subclavian right artery. Another case with an unspecified mutation
in the CHD7 gene has also been reported in Romania (36).
The variant identified in the present study,
c.4379_4380del in the CHD7 gene, was not present in population
databases (Exome Aggregation Consortium: No frequency) and appeared
to have not been reported in the literature. ClinVar contains an
entry for this variant (variation ID: 95789) that is considered to
be a pathogenic and germinal mutation. Over 500 mutations in the
CHD7 gene have been reported (28),
but only one case had the mutation (https://www.ncbi.nlm.nih.gov/clinvar/variation/95789/)
that was identified in the present study and it has not been
reported in a published scientific journal associated with CHARGE
syndrome. The rare variant c.4379_4380del in the CHD7 gene is
consistent with a loss-of-function variant, which is known to be
pathogenic (37), and
haploinsufficiency is the most likely pathogenic mechanism
(38).
CHD7 mutations cause inactivation of remodeling
activity of varying degrees. Remodeling of nucleosomes is a key
function of CHD7 during developmental processes, which may explain
why CHD7 mutations cause developmental defects but do not prevent
the development of pregnancy (39-42).
To the best of our knowledge, the present study
reported the first case of CHARGE syndrome in Romania that
displayed an aberrant right subclavian artery and a horseshoe
kidney, and was clearly diagnosed on the basis of the pathogenic
mutation in the CDH7 gene. The frameshift mutation resulted in the
formation of a premature stop codon leading to the inactivation of
the CHD7 protein. CHARGE syndrome is a rare disease; therefore,
every newly reported case improves the understanding of the
spectrum of mutations in the CDH7 gene and extends the knowledge of
genotype-phenotype correlations.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
CG and DS contributed to the conceptualization of
the study, literature search as well as writing, editing and
critically reviewing the manuscript; CG, AL and FB performed data
analysis; EVG and MAV performed data analysis, critical review and
supervision. MP treated the patient in the clinic throughout the
admission and provided patient data. All authors have read and
approved the final manuscript for publication.
Ethics approval and consent to
participate
The study described in this article was carried out
in accordance with the Code of Ethics of the World Medical
Association (Declaration of Helsinki) for experiments involving
humans and Uniform Requirements for manuscripts submitted to
biomedical journals. The study was approved by the Local Ethics
Commission for Scientific Research of Pius Brînzeu Emergency County
Clinical Hospital Timisoara, Romania; (approval no.
172/05.08.2019). The parents of the patient provided written
informed consent regarding genetic testing and participation in the
study.
Patient consent for publication
The parents of the patient provided written informed
consent regarding the publication of the medical data and images of
the case.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pagon RA, Graham JM Jr, Zonana J and Yong
SL: Coloboma, congenital heart disease, and choanal atresia with
multiple anomalies: CHARGE association. J Pediatr. 99:223–227.
1981.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Basson MA and van Ravenswaaij-Arts C:
Functional insights into chromatin remodelling from studies on
CHARGE syndrome. Trends Genet. 31:600–611. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hsu P, Ma A, Wilson M, Williams G, Curotta
J, Munns CF and Mehr S: CHARGE syndrome: A review. J Paediatr Child
Health. 50:504–511. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sanlaville D and Verloes A: CHARGE
syndrome: An update. Eur J Hum Genet. 15:389–399. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Blake KD and Prasad C: CHARGE syndrome.
Orphanet J Rare Dis. 1(34)2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Trider CL, Arra-Robar A, van
Ravenswaaij-Arts C and Blake K: Developing a CHARGE syndrome
checklist: Health supervision across the lifespan (from head to
toe). Am J Med Genet A. 173:684–691. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hale CL, Niederriter AN, Green GE and
Martin DM: Atypical phenotypes associated with pathogenic CHD7
variants and a proposal for broadening CHARGE syndrome clinical
diagnostic criteria. Am J Med Genet A. 170A:344–354.
2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Legendre M, Abadie V, Attié-Bitach T,
Philip N, Busa T, Bonneau D, Colin E, Dollfus H, Lacombe D, Toutain
A, et al: Phenotype and genotype analysis of a French cohort of 119
patients with CHARGE syndrome. Am J Med Genet C Semin Med Genet.
175:417–430. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pranckėnienė L, Preikšaitienė E, Gueneau
L, Reymond A and Kučinskas V: De novo duplication in the
CHD7 gene associated with severe CHARGE syndrome. Genomics
Insights. 12(1178631019839010)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Katoh-Fukui Y, Yatsuga S, Shima H, Hattori
A, Nakamura A, Okamura K, Yanagi K, Iso M, Kaname T, Matsubara Y,
et al: An unclassified variant of CHD7 activates a cryptic splice
site in a patient with CHARGE syndrome. Hum Genome Var.
5(18006)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Miller SA, Dykes DD and Polesky HF: A
simple salting out procedure for extracting DNA from human
nucleated cells. Nucleic Acids Res. 16(1215)1988.PubMed/NCBI View Article : Google Scholar
|
|
12
|
https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/-.
Accessed in July 15, 2019.
|
|
13
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
Laboratory Quality Assurance Committee: Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Boia M, Iacob D, Manea A, Budișan C,
Enătescu I, Dima M and Costescu O: Creșterea şi dezvoltarea
postnatală. In: Noțiuni Practice de Puericultură. Babeş V (ed).
Timisoara. pp32–43. 2019.(In Romanian).
|
|
15
|
Delahaye A, Sznajer Y, Lyonnet S,
Elmaleh-Bergès M, Delpierre I, Audollent S, Wiener-Vacher S,
Mansbach AL, Amiel J, Baumann C, et al: Familial CHARGE syndrome
because of CHD7 mutation: Clinical intra- and interfamilial
variability. Clin Genet. 72:112–121. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hudson A, Trider CL and Blake K: CHARGE
Syndrome. Pediatr Rev. 38:56–59. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pierpont ME, Brueckner M, Chung WK, Garg
V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, et
al: American Heart Association Council on Cardiovascular Disease in
the Young; Council on Cardiovascular and Stroke Nursing; and
Council on Genomic and Precision Medicine: Genetic basis for
congenital heart disease: Revisited: A scientific statement from
the American Heart Association. Circulation. 138:e653–e711.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Corsten-Janssen N, van Ravenswaaij-Arts
CMA and Kapusta L: Congenital arch vessel anomalies in CHARGE
syndrome: A frequent feature with risk for co-morbidity. Int J
Cardiol Heart Vasc. 12:21–25. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Roider L, Abdelaziz A and Gaballah AH:
CHARGE syndrome with high bifurcation of the abdominal aorta and a
horseshoe kidney: A case report. J Vasc Interv Radiol.
29:1288–1290.e1. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Velea PI, Mogoi M, Dema A, David V, Gug C
and Paul C: Mixed gonadal dysgenesis associated with short stature
and gonadoblastoma. Case Rep Acta Endo Buc. 11:221–227. 2015.
|
|
21
|
https://www.ncbi.nlm.nih.gov/medgen/376558.
Accessed in March 5, 2020.
|
|
22
|
Cevei M, Cssepento C, Gasparik AI and
Stoicanescu D: Utility of risk factors in the current diagnosis of
osteoporosis. Osteoporos Int. 26(184)2015.
|
|
23
|
Salem-Hartshorne N and Jacob S: Adaptive
behavior in children with CHARGE syndrome. Am J Med Genet A.
133A:262–267. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
https://www.ncbi.nlm.nih.gov/medgen/?term=75567-.
Accessed in July 5, 2019.
|
|
25
|
Corsten-Janssen N, Kerstjens-Frederikse
WS, du Marchie Sarvaas GJ, Baardman ME, Bakker MK, Bergman JE, Hove
HD, Heimdal KR, Rustad CF, Hennekam RC, et al: The cardiac
phenotype in patients with a CHD7 mutation. Circ Cardiovasc Genet.
6:248–254. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bergman JE, Janssen N, van der Sloot AM,
de Walle HE, Schoots J, Rendtorff ND, Tranebjaerg L, Hoefsloot LH,
van Ravenswaaij-Arts CM and Hofstra RM: A novel classification
system to predict the pathogenic effects of CHD7 missense variants
in CHARGE syndrome. Hum Mutat. 33:1251–1260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zentner GE, Layman WS, Martin DM and
Scacheri PC: Molecular and phenotypic aspects of CHD7 mutation in
CHARGE syndrome. Am J Med Genet A. 152A:674–686. 2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Janssen N, Bergman JE, Swertz MA,
Tranebjaerg L, Lodahl M, Schoots J, Hofstra RM, van
Ravenswaaij-Arts CM and Hoefsloot LH: Mutation update on the CHD7
gene involved in CHARGE syndrome. Hum Mutat. 33:1149–1160.
2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gug C, Mihaescu A and Mozos I: Two
mutations in the thiazide-sensitive NaCl co-transporter gene in a
Romanian Gitelman syndrome patient: Case report. Ther Clin Risk
Manag. 14:149–155. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Gug C, Caba L, Mozos I, Stoian D, Atasie
D, Gug M and Gorduza EV: Rare splicing mutation in COL1A1 gene
identified by whole exomes sequencing in a patient with
osteogenesis imperfecta type I followed by prenatal diagnosis: A
case report and review of the literature. Gene. 741(144565): Epub
ahead of print. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Pauli S, Pieper L, Häberle J, Grzmil P,
Burfeind P, Steckel M, Lenz U and Michelmann HW: Proven germline
mosaicism in a father of two children with CHARGE syndrome. Clin
Genet. 75:473–479. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gug C, Huțanu D, Vaida M, Doroş G, Popa C,
Stroescu R, Furău G, Furău C, Grigoriță L and Mozos I: De
novo unbalanced translocation t(15;22)(q26.2;q12) with
velo-cardio-facial syndrome: A case report and review of the
literature. Exp Ther Med. 16:3589–3595. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jongmans MC, Admiraal RJ, van der Donk KP,
Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel
TJ, Veltman JA, et al: CHARGE syndrome: The phenotypic spectrum of
mutations in the CHD7 gene. J Med Genet. 43:306–314.
2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Martínez-Quintana E, Rodríguez-González F,
Garay-Sánchez P and Tugores A: Novel frameshift CHD7 mutation
related to CHARGE syndrome. Mol Syndromol. 5:36–40. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu YP, Shi LP and Zhu J: Atypical CHARGE
associated with a novel frameshift mutation of CHD7 in a Chinese
neonatal patient. BMC Pediatr. 18(203)2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Boer MC, Musat G and Budisteanu M: CHARGE
syndrome – a rare cause of nose and ear anomalies. A case report.
Rom J Rhinol. 3:45–48. 2013.
|
|
37
|
Marcos S, Sarfati J, Leroy C, Fouveaut C,
Parent P, Metz C, Wolczynski S, Gérard M, Bieth E, Kurtz F, et al:
The prevalence of CHD7 missense versus truncating mutations is
higher in patients with Kallmann syndrome than in typical CHARGE
patients. J Clin Endocrinol Metab. 99:E2138–2143. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kohmoto T, Shono M, Naruto T, Watanabe M,
Suga K, Nakagawa R, Kagami S, Masuda K and Imoto I: A novel
frameshift mutation of CHD7 in a Japanese patient with CHARGE
syndrome. Hum Genome Var. 3(16004)2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jurca A, Kinga K, Bembea M, Gug C and
Jurca C: Fanconi anemia with cleft palate. Rev Med Chir Soc Med Nat
Iasi. 118:1074–1077. 2014.PubMed/NCBI
|
|
40
|
Bouazoune K and Kingston RE: Chromatin
remodeling by the CHD7 protein is impaired by mutations that cause
human developmental disorders. Proc Natl Acad Sci USA.
109:19238–19243. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Gug C, Rațiu A, Navolan D, Drăgan I, Groza
IM, Păpurică M, Vaida MA, Mozoș I and Jurcă MC: The incidence and
spectrum of chromosomal anomalies in miscarriage samples: A
retrospective study of 330 cases. Cytogenet Genome Res.
158:171–183. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gug C, Burada F, Ioana M, Riza AL,
Moldovan M, Mozoș I, Rațiu A, Martiniuc V and Gorduza EV:
Polyploidy in first and second trimester pregnancies in Romania.
Clin Lab. 66:Epub ahead of print. 2020.PubMed/NCBI View Article : Google Scholar
|