Introduction
Hepatocellular carcinoma (HCC) is the third leading
cause of cancer-associated mortality worldwide and is highly
associated with hepatitis C and B infection, as well as alcohol
abuse (1). In East Asia and
sub-Saharan Africa, chronic hepatitis B infection is a vital factor
in the pathogenesis of liver cancer (2). To date, numerous studies have
identified hepatitis B virus (HBV)-associated factors, including
hepatitis B e antigen and the hepatitis B surface antigen, as key
predictors of HBV-associated HCC development in patients with
hepatitis B infection (3). In
addition, the viral load of HBV, duration of infection and severity
of liver disease have been linked to the risk of developing
cirrhosis (2,4); however, the mechanisms of
HBV-associated HCC development remain to be fully elucidated, which
poses challenges for the clinical treatment of this disease.
Post-operative recurrence is the most imperative issue to overcome
(5). Bioinformatics analyses may be
applied to determine the potential mechanisms underlying the
development of HBV-associated HCC. Furthermore, predictive
biomarkers may improve the prediction of patient prognosis and the
understanding of the molecular mechanisms underlying this disease
(6).
α-Fetoprotein, associated with gene duplications
(7), was the sole biomarker reported
in randomized controlled trials (8).
As only a small number of biomarkers have been identified,
developments in the treatment and diagnosis of HCC, as well as
prognostic evaluation, are restricted. With recent advances in
high-throughput technologies, an increasing number of gene chips
are available for the detection of differentially expressed genes
(DEGs); thus, bioinformatics analyses of associated data are
frequently being performed (9).
Based on these novel techniques, databases of vast core gene data
have been produced, including the National Center for Biotechnology
Information (NCBI) Gene Expression Omnibus (GEO) and the Cancer
Genome Atlas (10). Analysis of
these freely available gene expression data may lead to
identification of DEGs in tumor vs. normal tissue, which may
provide novel insight into the development of cancers. Jiao et
al (11) reported that
upregulated eukaryotic translation initiation factor 2B subunit ε
(EIF2B5) expression was associated with HCC development based on
GSE54236 and GSE76427 microarray data, indicating that EIF2B5 may
be employed as a novel biomarker for patients with liver
cancer.
In the present study, the gene expression dataset
GSE121248, including hepatitis B-induced HCC and adjacent normal
samples, was subjected to bioinformatics analysis. The DEGs were
screened and the functions and associated pathways in
HBV-associated HCC were further evaluated. The core genes
identified in the present study may be considered as potential
novel targets for the treatment of HBV-associated HCC. The present
results may also improve the understanding of the development and
recurrence of this disease, and provide a basis for developments
regarding its clinical treatment.
Materials and methods
Data sourcing and identification of
DEGs
The GSE121248 microarray dataset was acquired and
downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/), on the basis of the
GPL570 platform analyzed by the Affymetrix Human Genome U133 Plus
2.0 Array, and included the data of 70 HBV-associated HCC tumor
samples and 37 adjacent normal tissue samples. Simultaneously, the
Series Matrix File of GSE121248 was downloaded. In order to obtain
more meaningful targets for the clinical application, |log fold
change (FC)|>1(12) was set as
the limit and GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) was used.
Genes were identified as significant DEGs based on adjusted
P<0.05(13), which was applied to
reduce the false-positive rate.
Gene Ontology (GO) functional and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis of DEGs
GO analysis in the categories cellular component,
molecular function and biological process (14) was performed for the functional
annotation of genes. KEGG was used to determine the biological
pathways associated with the DEGs (15). The Database for Annotation,
Visualization and Integrated Discovery (DAVID; http://david.ncifcrf.gov) (16), a free online biological database, was
employed to perform GO and KEGG analyses with the Bingo plug-in to
obtain systematic and comprehensive information for all DEGs.
P<0.05 was considered to indicate a statistically significant
difference.
Construction of the protein-protein
interaction (PPI) network
To investigate the mechanism underlying the
development of HCC, the Search Tool for the Retrieval of
Interacting Genes (https://string-db.org/), a free online tool that may
be employed to evaluate and construct a PPI network of target genes
and DEGs, including downregulated and upregulated genes, was used.
Subsequently, Cytoscape software (17) was utilized to select hub genes in the
PPI network. In addition, DAVID was used to perform GO and KEGG
analyses to obtain additional information on the hub genes.
Association of hub genes with clinical
outcomes and diagnostic value
Overall survival (OS) and disease-free survival
(DFS) analyses of hub genes were performed using the Gene
Expression Profiling Interactive Analysis (GEPIA) web server
(http://gepia.cancer-pku.cn/); log-rank
P<0.05 was considered to indicate statistical significance.
Immunohistochemical data for three hub genes with the highest
degrees in patients with or without HCC were then downloaded from
the Human Protein Atlas (HPA; https://www.proteinatlas.org/). In addition, GraphPad
prism 7 software (GraphPad Software, Inc.) was used to generate
receiver operating characteristic (ROC) curves to determine the
diagnostic value of the 15 hub genes in HCC.
Results
Identification of DEGs in
HBV-associated HCC
The dataset comprised 107 samples, including 70
tumor samples and 37 adjacent normal tissue samples. The GEO2R tool
was used to identify the DEGs, with adjusted P<0.05 and
|logFC|>1 selected as the cut-off criteria. A total of 1,153
DEGs, comprising 376 downregulated and 777 upregulated genes, were
retrieved by analyzing the GSE121248 dataset. All DEGs were
included in the subsequent analysis.
GO functional and KEGG pathway
enrichment analyses of DEGs
After the DEGs were identified, DAVID was employed
to perform GO functional and KEGG pathway enrichment analyses of
the DEGs. The data of the DEGs were analyzed with the tool; in the
GO analysis, the functional terms in three different categories,
namely cellular component, biological process and molecular
function enriched by the DEGs were determined. In summary, the DEGs
were significantly accumulated in cellular component terms
including ‘protein binding’ (GO:0005515), ‘cytoplasm’ (GO:0005737),
‘extracellular exosome’ (GO:0070062) and ‘cytosol’ (GO:005829).
Specifically, upregulated genes were mainly enriched in cellular
component terms, including ‘extracellular region’ (GO:0005576),
‘extracellular exosome’ (GO:0070062) and ‘extracellular space’
(GO:0005615), while downregulated genes were mainly enriched in
biological process terms, including ‘cell division’ (GO:0051301)
and ‘mitotic nuclear division’ (GO:0007067). In Table I, the top 10 GO terms of the
upregulated and downregulated genes are listed according to their
P-value from the lowest to the highest value.
 | Table IGO analysis of differentially
expressed genes. |
Table I
GO analysis of differentially
expressed genes.
| A, Upregulated
genes |
|---|
| Category | Term | Count | P-value | FDR |
|---|
|
GOTERM_CC_DIRECT |
GO:0005576-extracellular region | 119 |
3.20x10-20 |
4.34x10-17 |
|
GOTERM_CC_DIRECT |
GO:0070062-extracellular exosome | 166 |
1.25x10-18 |
1.69x10-15 |
|
GOTERM_CC_DIRECT |
GO:0005615-extracellular space | 99 |
2.59x10-16 |
3.00x10-13 |
|
GOTERM_CC_DIRECT | GO:0072562-blood
microparticle | 29 |
1.31x10-14 |
1.78x10-11 |
|
GOTERM_BP_DIRECT |
GO:0055114-oxidation-reduction
process | 58 |
4.38x10-14 |
7.78x10-11 |
|
GOTERM_MF_DIRECT |
GO:0016705-oxidoreductase activity, acting
on paired donors, with incorporation or reduction of molecular
oxygen | 18 |
5.79x10-13 |
9.05x10-10 |
|
GOTERM_BP_DIRECT |
GO:0019373-epoxygenase P450 pathway | 12 |
6.99x10-13 |
1.24x10-9 |
|
GOTERM_MF_DIRECT | GO:0020037-heme
binding | 25 |
3.51x10-12 |
5.48x10-9 |
|
GOTERM_MF_DIRECT | GO:0019825-oxygen
binding | 16 |
4.49x10-12 |
7.01x10-9 |
|
GOTERM_CC_DIRECT |
GO:0031090-organelle membrane | 20 |
1.06x10-11 |
1.44x10-8 |
| B, Downregulated
genes |
| Category | Term | Count | P-value | FDR |
|
GOTERM_BP_DIRECT | GO:0051301-cell
division | 38 |
7.08x10-20 |
1.18x10-16 |
|
GOTERM_BP_DIRECT | GO:0007067-mitotic
nuclear division | 29 |
6.27x10-16 |
1.11x10-12 |
|
GOTERM_CC_DIRECT |
GO:0000777-condensed chromosome
kinetochore | 16 |
4.44x10-12 |
5.98x10-9 |
|
GOTERM_BP_DIRECT | GO:0007062-sister
chromatid cohesion | 17 |
9.20x10-12 |
1.53x10-8 |
|
GOTERM_CC_DIRECT |
GO:0030496-midbody | 18 |
1.46x10-11 |
1.97x10-8 |
|
GOTERM_BP_DIRECT | GO:0000082-G1/S
transition of mitotic cell cycle | 16 |
9.34x10-11 |
1.55x10-7 |
|
GOTERM_CC_DIRECT |
GO:0000775-chromosome, centromeric
region | 12 |
9.48x10-10 |
1.28x10-6 |
|
GOTERM_MF_DIRECT | GO:0005515-protein
binding | 189 |
5.91x10-9 |
8.35x10-6 |
|
GOTERM_BP_DIRECT | GO:0000281-mitotic
cytokinesis | 9 |
1.37x10-8 |
2.28x10-5 |
|
GOTERM_CC_DIRECT |
GO:0000776-kinetochore | 12 |
4.49x10-8 |
6.04x10-5 |
Furthermore, KEGG pathway analysis revealed that the
DEGs were mainly involved in ‘metabolic pathways’ [Homo
sapiens (hsa)01100], ‘chemical carcinogenesis’ (hsa05204) and
‘fatty acid degradation’ (hsa00071). In addition, the upregulated
genes were mainly enriched in ‘retinol metabolism’ (hsa00830),
which is associated with the development of diabetes (18). Downregulated genes were accumulated
in the pathways of ‘cell cycle’ (hsa04110) and ‘extracellular
matrix-receptor interaction’ (hsa04512), as well as ‘p53 signaling
pathway’ (hsa04115) (Table II).
| Table IIKEGG analysis of differentially
expressed genes (top 5 according to P-value). |
Table II
KEGG analysis of differentially
expressed genes (top 5 according to P-value).
| A, Upregulated
genes |
|---|
| Term | Count | P-value |
|---|
| hsa01100:Metabolic
pathways | 107 |
2.83x10-14 |
| hsa05204:Chemical
carcinogenesis | 21 |
9.07x10-11 |
| hsa00830:Retinol
metabolism | 17 |
7.29x10-9 |
| hsa04610:Complement
and coagulation cascades | 17 |
2.35x10-8 |
| hsa00980:Metabolism
of xenobiotics by cytochrome P450 | 17 |
6.78x10-8 |
| B, Downregulated
genes |
| Term | Count | P-value |
| hsa04110:Cell
cycle | 21 |
6.49x10-15 |
|
hsa04512:ECM-receptor interaction | 11 |
1.44x10-6 |
| hsa04115:p53
signaling pathway | 9 |
1.28x10-5 |
| hsa05200:Pathways
in cancer | 20 |
2.06x10-5 |
| hsa03030:DNA
replication | 7 |
2.36x10-5 |
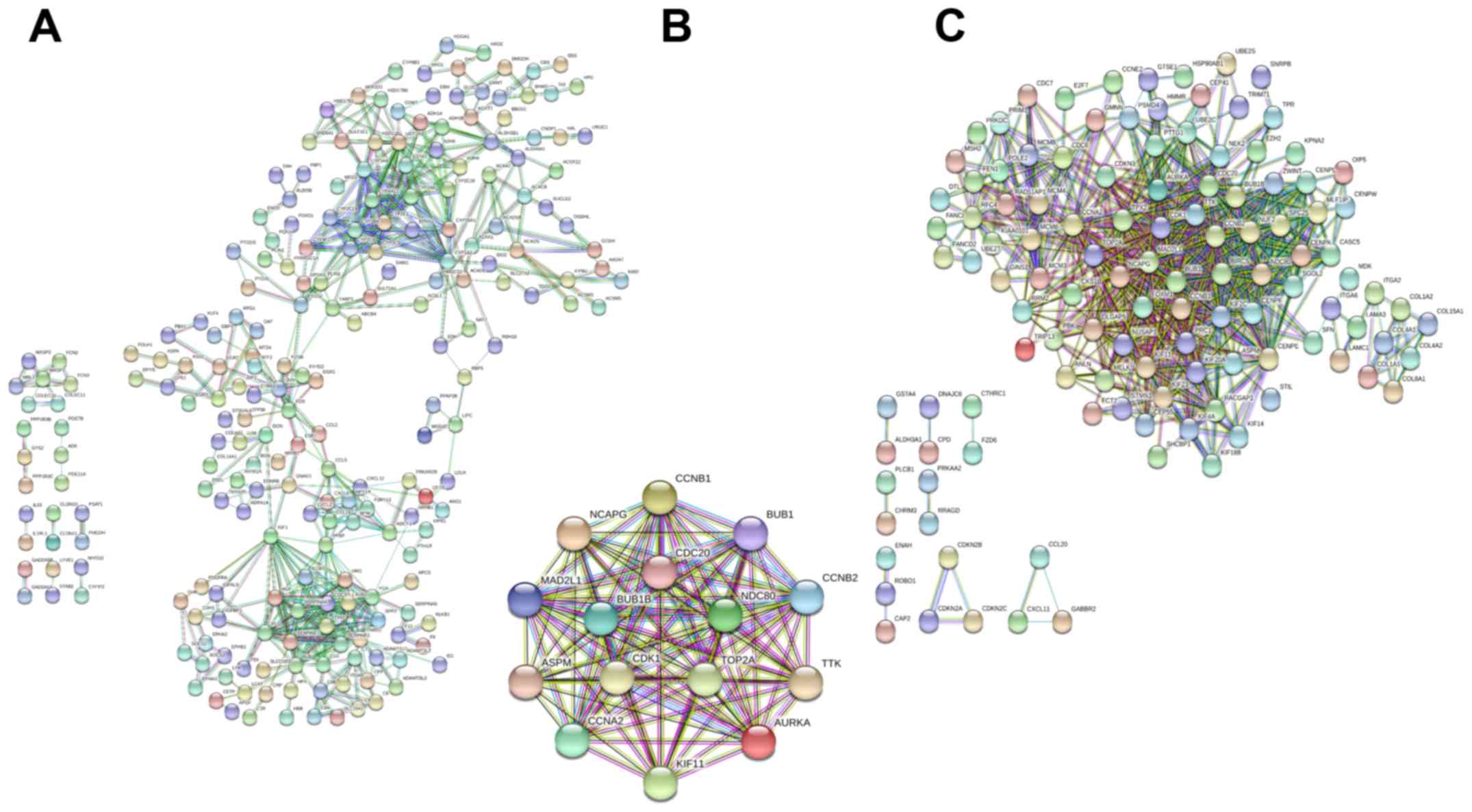
PPI network analysis of DEGs
As the minimum interaction score required, 0.900 was
applied. The PPI network of all the DEGs contained 677 nodes and
1,118 edges (enrichment P<1.0x10-16). Cytoscape
software was then employed to identify the top 15 hub genes that
may be associated with HBV-associated HCC according to their high
degree of connectivity from high to low based on the results of PPI
network. These genes included abnormal spindle-like
microcephaly-associated protein (ASPM), aurora kinase A (AURKA),
budding uninhibited by benzimidazoles (BUB)1, BUB1B, cyclin A2
(CCNA2), CCNB1, CCNB2, cell division cycle 20 (CDC20),
cyclin-dependent kinase 1 (CDK1), kinesin family member 11 (KIF11),
mitotic arrest deficient 2 like 1 (MAD2L1), non-SMC condensin I
complex subunit G (NCAPG), NDC80 kinetochore complex component
(NDC80), DNA topoisomerase II α (TOP2A) and TTK protein kinase
(TTK) (Table III). To further
investigate the hub genes, a heatmap and box plot of the expression
of the top 15 hub genes in HBV-associated HCC tumor samples and
adjacent normal tissue samples were generated using GraphPad prism
7 software (Fig. 1). The PPI
networks of upregulated and downregulated genes, as well as hub
genes, are presented in Fig. 2.
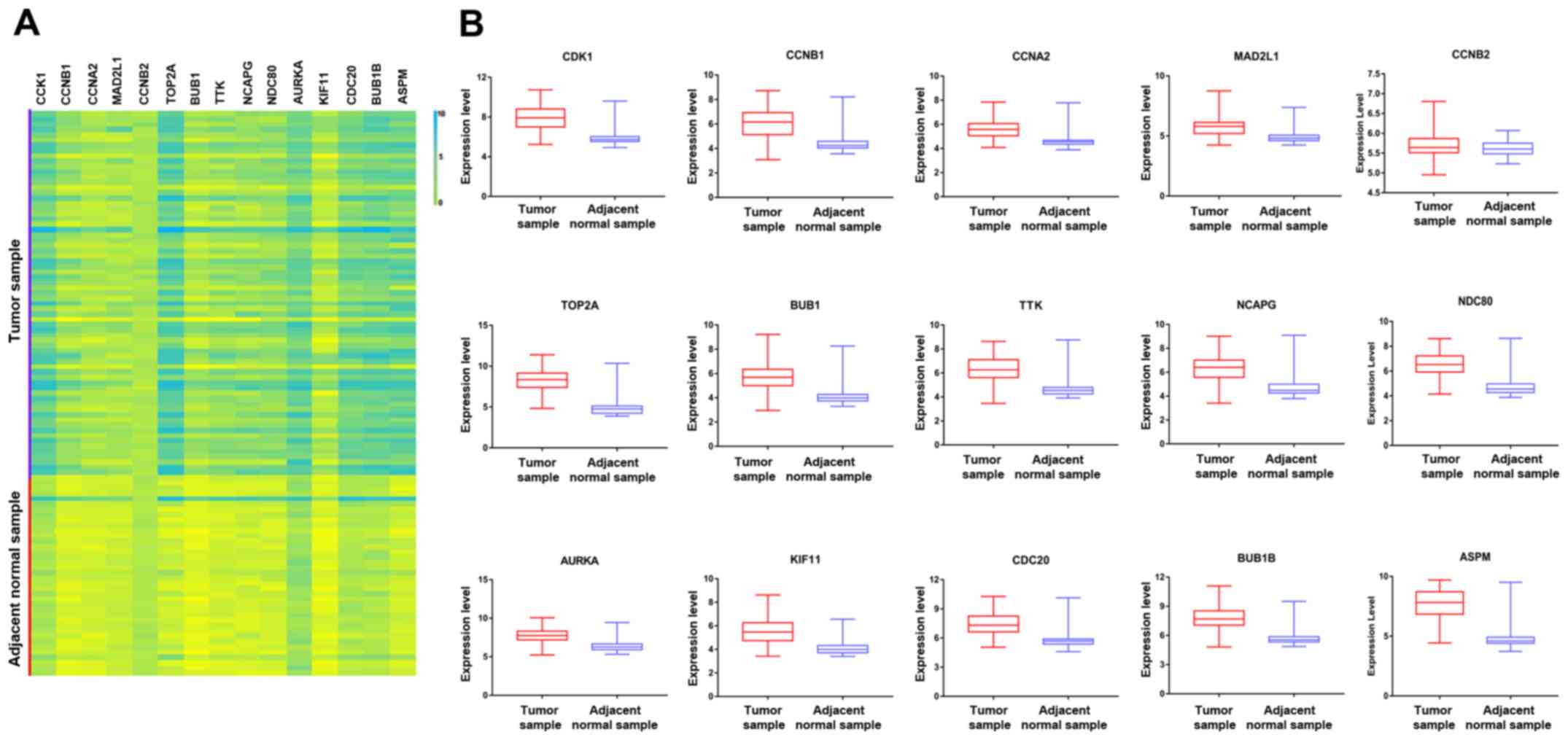 | Figure 1(A) Volcano map of 15 hub genes in
the protein-protein interaction network according to the highest
degree. (B) Expression of various genes in the tumor samples and
adjacent normal samples, including ASPM (P<0.0001), AURKA
(P<0.0001), BUB1 (P<0.0001), BUB1B (P<0.0001), CCNA2
(P<0.0001), CCNB1 (P<0.0001), CCNB2 (P=0.1067), CDC20
(P<0.0001), CDK1 (P<0.0001), KIF11 (P<0.0001), MAD2L1
(P<0.0001), NCAPG (P<0.0001), NDC80 (P<0.0001), TOP2A
(P<0.0001) and TTK (P<0.0001). CDK1, cyclin-dependent kinase
1; CCNA2, cyclin A2; MAD2L1, mitotic arrest deficient 2 like 1;
TOP2A, DNA topoisomerase IIα; BUB1, budding uninhibited by
benzimidazoles 1; TTK, TTK protein kinase; NCAPG, non-SMC condensin
I complex subunit G; NDC80, NDC80 kinetochore complex component;
AURKA, aurora kinase A; KIF11, kinesin family member 11; CDC2, cell
division cycle 20; ASPM, abnormal spindle microtubule assembly;
Adj, adjacent. |
| Table IIITop 15 hub genes in the
protein-protein interaction network ranked by degree. |
Table III
Top 15 hub genes in the
protein-protein interaction network ranked by degree.
| Gene symbol | Degree |
|---|
| CDK1 | 90 |
| CCNB1 | 79 |
| CCNA2 | 75 |
| MAD2L1 | 73 |
| CCNB2 | 70 |
| TOP2A | 70 |
| BUB1 | 69 |
| TTK | 67 |
| NCAPG | 67 |
| NDC80 | 66 |
| AURKA | 66 |
| KIF11 | 66 |
| CDC20 | 65 |
| BUB1B | 63 |
| ASPM | 63 |
Survival analyses of hub genes
The GEPIA online database, a web-based tool, was
designed to rapidly obtain customizable functionalities (19). Utilizing this tool, OS (Fig. 3) and the DFS (Fig. 4) curves were obtained and the
log-rank P-values of the 15 hub genes were determined. The results
indicated that all of the hub genes except CCNB2 were significantly
associated with OS. Subsequently, the series matrix data downloaded
from the GEO database were analyzed and ROC curves were generated
to gain a comprehensive understanding of the predictive value of
the hub genes. The results demonstrated that all the hub genes were
able to efficiently distinguish HCC tissues from normal tissues
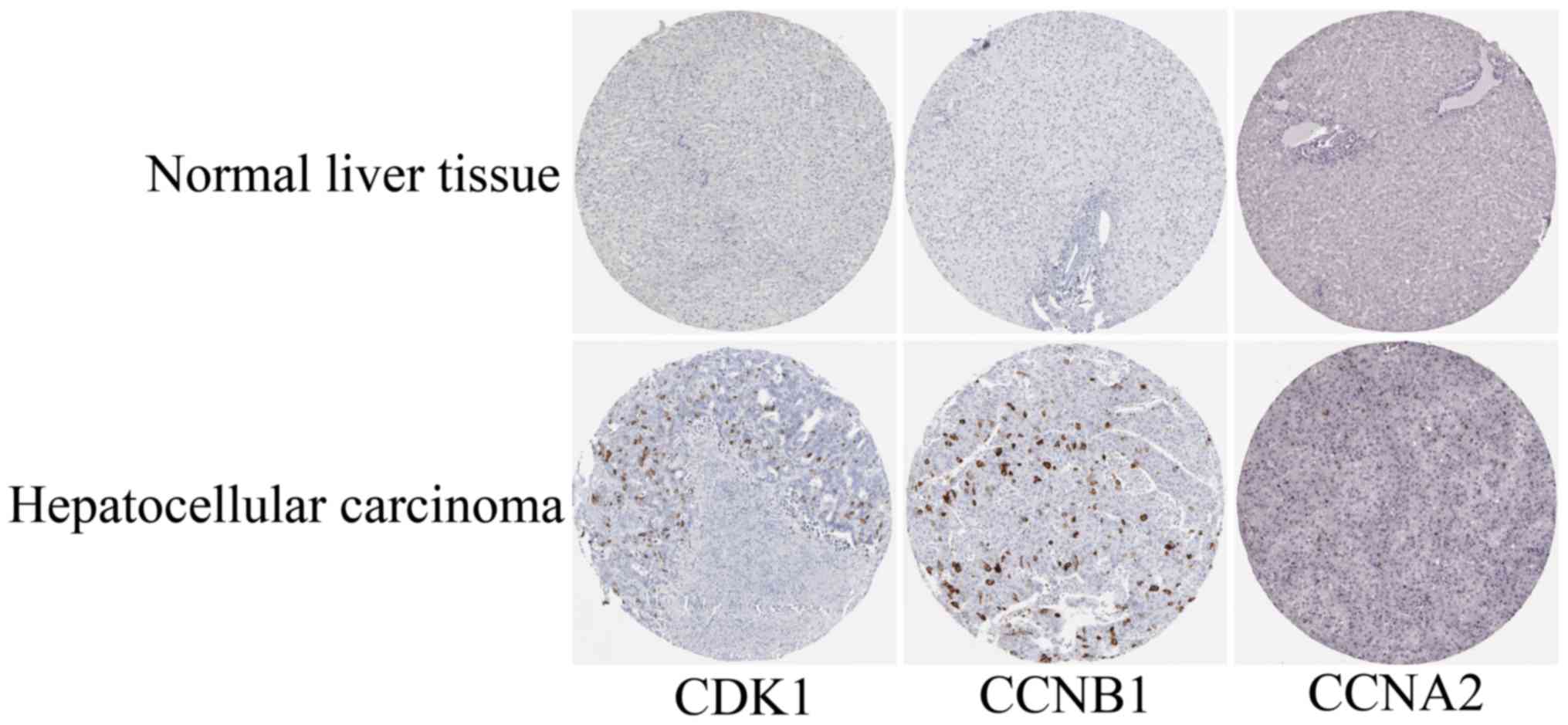
(Fig. 5). In addition, the top three
genes were selected based on the degree of connectivity in the PPI
network for analysis of the immunohistochemical data in the HPA.
Additionally, representative images indicated that the expression
of all three hub genes were upregulated in HCC tissues (Fig. 6).
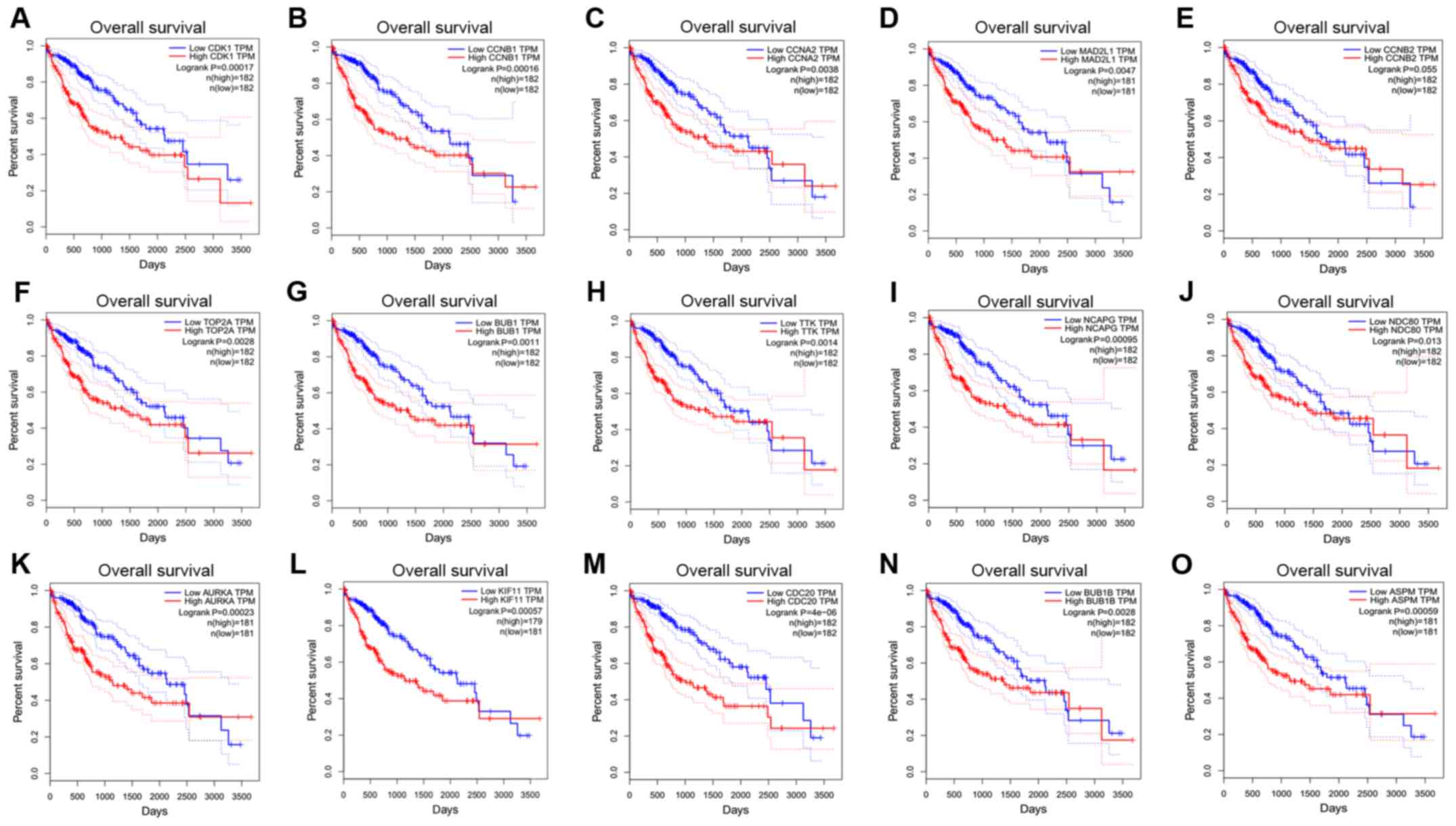 | Figure 3Overall survival curves for 15 hub
genes. (A) CDK1, (B) CCNB1, (C) CCNA2, (D) MAD2L1, (E) CCNB2, (F)
TOP2A, (G) BUB1, (H) TTK, (I) NCAPG, (J) NDC80, (K) AURKA, (L)
KIF11, (M) CDC20, (N) BUB1B and (O) ASPM. CDK1, cyclin-dependent
kinase 1; CCNA2, cyclin A2; MAD2L1, mitotic arrest deficient 2 like
1; TOP2A, DNA topoisomerase IIα; BUB1, budding uninhibited by
benzimidazoles 1; TTK, TTK protein kinase; NCAPG, non-SMC condensin
I complex subunit G; NDC80, NDC80 kinetochore complex component;
AURKA, aurora kinase A; KIF11, kinesin family member 11; CDC20,
cell division cycle 20; ASPM, abnormal spindle microtubule
assembly; TPM, transcripts per million. |
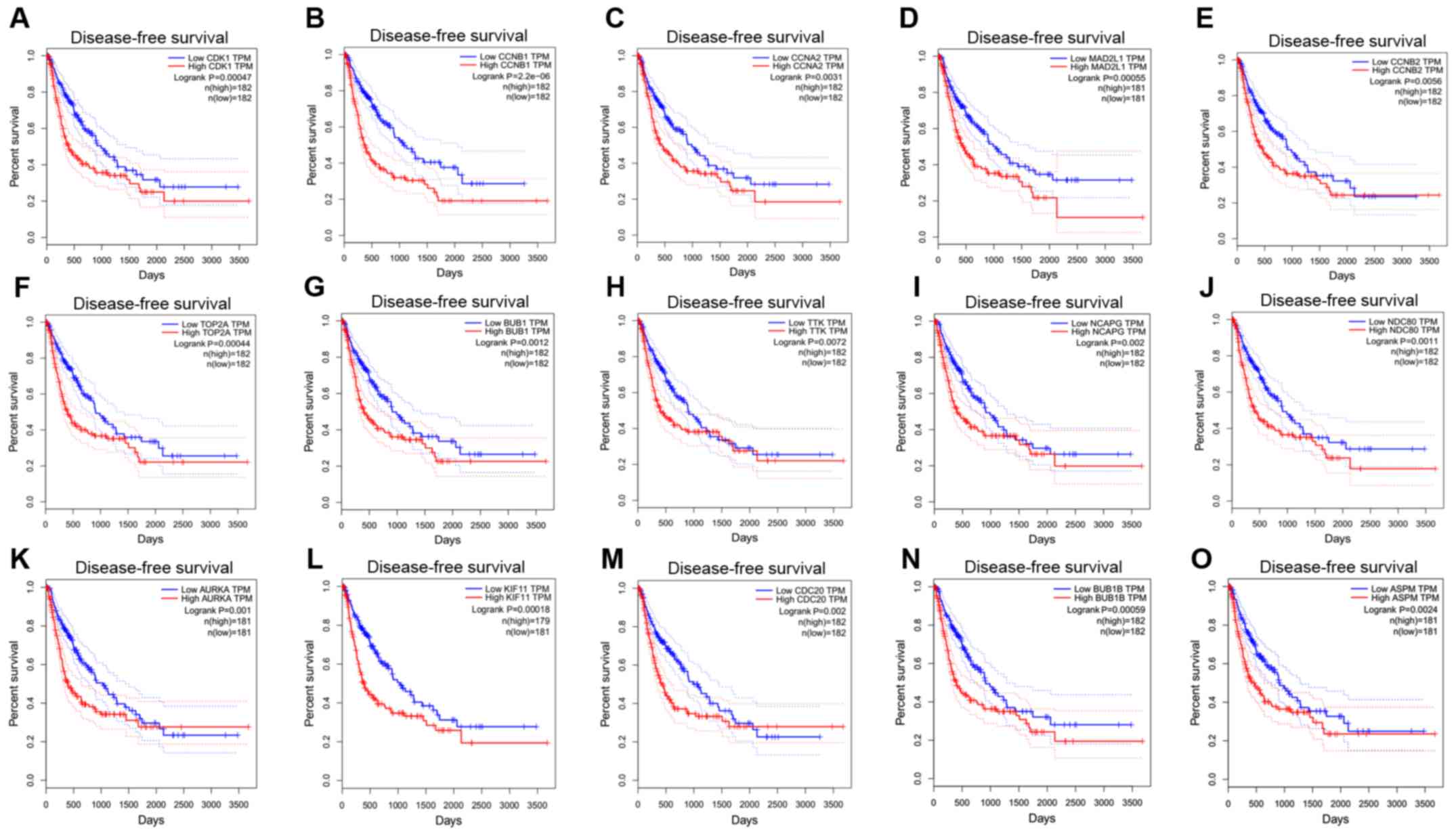 | Figure 4Disease-free survival curves of 15
hub genes. (A), CDK1, (B) CCNB1, (C) CCNA2, (D) MAD2L1, (E) CCNB2,
(F) TOP2A, (G) BUB1, (H) TTK, (I) NCAPG, (J) NDC80, (K) AURKA, (L)
KIF11, (M) CDC20, (N) BUB1B and (O) ASPM. CDK1, cyclin-dependent
kinase 1; CCNA2, cyclin A2; MAD2L1, mitotic arrest deficient 2 like
1; TOP2A, DNA topoisomerase IIα; BUB1, budding uninhibited by
benzimidazoles 1; TTK, TTK protein kinase; NCAPG, non-SMC condensin
I complex subunit G; NDC80, NDC80 kinetochore complex component;
AURKA, aurora kinase A; KIF11, kinesin family member 11; CDC20,
cell division cycle 20; ASPM, abnormal spindle microtubule
assembly; TPM, transcripts per million. |
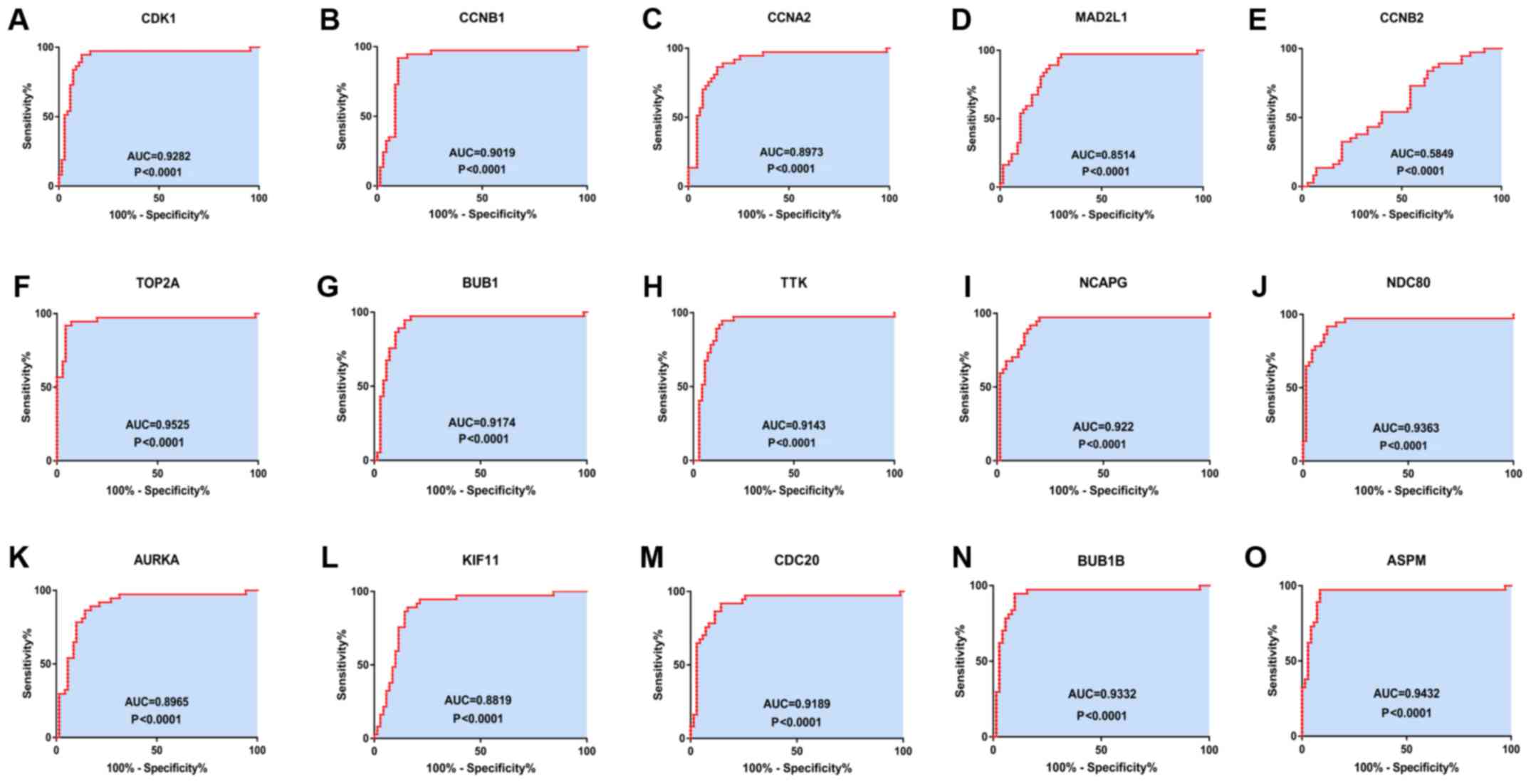 | Figure 5Receiver operating characteristics
curves for the 15 hub genes to identify HCC tissue from normal
tissue. (A) CDK1 (AUC=0.9282, P<0.0001), (B) CCNB1
(AUC=0.0.9019, P<0.0001), (C) CCNA2 (AUC=0.8973, P<0.0001),
(D) MAD2L1 (AUC=0.8514, P<0.0001), (E) CCNB2 (AUC=0.5849,
P=0.1496), (F) TOP2A (AUC=0.9525, P<0.0001), (G) BUB1
(AUC=0.9174, P<0.0001), (H) TTK (AUC=0.9143, P<0.0001), (I)
NCAPG (AUC=0.922, P<0.0001), (J) NDC80 (AUC=0.9363,
P<0.0001), (K) AURKA (AUC=0.8965, P<0.0001), (L) KIF11
(AUC=0.8819, P<0.0001), (M) CDC20 (AUC=0.9189, P<0.0001), (N)
BUB1B (AUC=0.9332, P<0.0001) and (O) ASPM (AUC=0.9432,
P<0.0001). CDK1, cyclin-dependent kinase 1; CCNA2, cyclin A2;
MAD2L1, mitotic arrest deficient 2 like 1; TOP2A, DNA topoisomerase
IIα; BUB1, budding uninhibited by benzimidazoles 1; TTK, TTK
protein kinase; NCAPG, non-SMC condensin I complex subunit G;
NDC80, NDC80 kinetochore complex component; AURKA, aurora kinase A;
KIF11, kinesin family member 11; CDC20, cell division cycle 20;
ASPM, abnormal spindle microtubule assembly; AUC, area under the
curve. |
Discussion
Previous studies have investigated biomarkers and
therapeutic targets for HBV-associated HCC (2); however, the markers currently available
are unsatisfactory. Thus, it is of importance to identify
additional targets for improving the therapeutic efficacy of
treatments for this disease. In the present study, the gene
expression data of the GSE121248 dataset including the gene
expression data of 107 samples comprising 70 HBV-associated HCC
tumor samples and 37 adjacent normal tissue samples were analyzed.
A total of 15 core genes were identified, namely CDK1, CCNB1,
CCNA2, MAD2L1, CCNB2, TOP2A, BUB1, TTK, NCAPG, NDC80, AURK1, KIF11,
CDC20, BUB1B and ASPM. The majority of these hub genes were
significantly associated with the development and progression of
HBV-associated HCC, among which CDK1, CCNB1 and CCNA2 were the top
three hub genes based on their degree in the PPI network.
To date, the association between the expression of
CDK1, CCNB1, CCNA2, KIF11 and CCNB2, and the development of HCC
deserves further investigation. Prevo and Pirovano (20) reported that CDK1 is able to mediate
the transition from G2 phase to mitosis via the CDK1/PI3K/β-catenin
signaling pathway. Haider et al (21) assessed the inhibition of CDK1 by the
kinase inhibitor compounds BA-12 and B1-14, which led to necrosis
and apoptosis of tumor cells without toxicity to adjacent normal
cells. In addition, the current study reported that CDK1 was
involved in the p53 signaling pathway. However, the true mechanism
underlying the correlation between CDK1 and HCC remains unclear
(22). Previous studies have
indicated that the CCNB1-Cdk1 complex is a key regulator of mitotic
entry (23,24). Chai et al (24) revealed that CCNB1 is highly expressed
in HCC and is closely associated with the poor prognosis of
patients with HCC, which was consistent with the present results.
This indicated the plausibility of CCNB1 as a potential therapeutic
target for HCC. However, Weng et al (25) demonstrated that there was no
significant difference in CCNB1 expression between patients with
non-recurrent HCC and healthy subjects. The study concluded that
the role of CCNB1 overexpression in oncogenesis and the progression
of HCC remains unclear. Thus, further investigation is required to
determine the association between the expression of CCNB1 and the
development of HCC. Of note, CCNA2 was reported to be associated
with the formation of the HBV-CCNA2 chimeric transcript, which may
accelerate the cell cycle and result in tumor development (26). In the present study, after
constructing the CCNA2-associated Kaplan-Meier curves for OS and
DFS, which indicated a significant association between CCNA2
expression and the prognosis of HCC, it was concluded that high
expression levels of CCNA2 are associated with poor prognosis of
patients with HCC. Furthermore, the results of ROC curve analysis
demonstrated the ability of CCNA2 to distinguish HCC samples from
normal samples. KIF11 serves an essential role in centrosome and
chromosome dynamics in mitosis (27). To the best of our knowledge, the
association between the expression levels of KIF11 and the
development of HBV-associated HCC has remained elusive. In the GO
analysis of the present study, KIF11 was mainly enriched in the
terms ‘cytoplasm’, ‘cytosol’ and ‘cell division’, as well as
‘spindle formation’ and ‘carcinogenesis’; however, KEGG pathway
enrichment analysis did not suggest any association between a
particular pathway and KIF11. Furthermore, the Kaplan-Meier curves
for OS and DFS suggested that upregulated KIF11 expression was
linked to poor prognosis in HBV-associated HCC. Chen et al
(28) identified that the
overexpression of KIF11 was significantly associated with shorter
relapse-free survival times; however, the function of KIF11 and the
mechanism involved in HCC remains unknown. In addition, Li et
al (29) reported that
downregulated CCNB2 expression led to inhibition of the progression
of malignant neoplasms. Conversely, to the best of our knowledge,
the biological function of CCNB2 in HCC is largely unknown. In the
present study, low expression levels of CCNB2 were associated with
improved clinical outcomes of patients with HBV-associated HCC,
including DFS.
The roles of MAD2L1, TOP2A, BUB1, TTK, NCAPG, NDC80,
AURKA, CDC20, BUB1B and ASPM in HCC have been well reported in
previous studies. As for MAD2L1, Li et al (30) identified this gene as a vital
mediator of pathways underlying chromosomal regulation in HCC. Yun
et al (31) demonstrated that
inhibition of MAD2L1 expression led to the suppression of HCC cell
proliferation, migration and invasion, suggesting that MAD2L1 may
serve as a potential target for the clinical treatment and
prognostic evaluation of patients with HCC. In agreement with a
previous study (32), TOP2A may be a
valuable prognostic marker and predictor of poor survival in
patients with HCC. Based on previous studies and the present
results, it was concluded that TOP2A may be a potential biomarker
linked to the development of HBV-associated HCC. BUB1 is a
serine/threonine kinase that binds centromeres during mitosis
(33), and has been associated with
the cell cycle and apoptosis (34),
as well as reduced OS in patients with HCC (35). The expression levels of protein
kinase human monopolar, also known as TTK, were previously reported
to be markedly increased in HBV-associated HCC (36); TTK was reported to predict poorer
outcomes for patients with HBV-associated HCC (37). The first TTK inhibitors to be
developed in combination with taxane chemotherapy have entered
phase 1 dose-escalation studies (38). NCAPG, which mediates the coiling
topology of individual chromatids (39), was proposed to be associated with
cell growth, proliferation and migration in HCC (40). NDC80 was reported to be associated
with the progression of HCC (41).
Of note, downregulation of NDC80 was able to suppress the
replication of HBV-associated HCC cells (42). Bound to MYC, AURKA, which has two
functional non-synonymous polymorphisms (lle31Phe and Val57lle),
was able to regulate tumor cell growth at the genetic level
(43). It has been reported that
AURKA lle31Phe enhances the proneness of HBV-infected individuals
to develop liver cancer (44). The
expression levels of CDC20 have been indicated to be linked to the
development of liver cancer and the prognosis of affected patients
(45). Thus, it may be a candidate
prognostic factor in patients with HBV-associated HCC (46). BUB1B was reported to be involved in
the regulation of the cell cycle of tumor cells (47). As for ASPM, its upregulation has been
suggested to serve as a novel marker for predicting the progression
of HCC, early tumor recurrence and poor prognosis (48).
Of note, the present study had certain limitations.
Data sourcing was performed using only one dataset, which may not
be sufficient to provide a convincing hypothesis. Analyses with
multiple datasets should be considered in the future. Hepatitis B
infection is considered an independent factor in the development of
HCC in the Cancer Genome Atlas database (TCGA) and therefore,
TCGA-based research should be conducted in the future. Furthermore,
the sample size was relatively small, which may result in
unavoidable bias. Finally, the gene chip used in the present study
did not contain any information on the pathological sections of
HBV-associated HCC, and therefore, it was not possible to determine
the association between HBV infection and HCC pathological grading
sample by sample. It is indicated that HBV infection, even if
temporary, may have a role in promoting the development of liver
cancer; at the same time, a longer duration of HBV infection may
accelerate the process of HCC (49).
However, to the best of our knowledge, no biomarker can predict the
association between HBV infection and the extent of HCC
pathological severity.
In conclusion, the present bioinformatics analysis
of a microarray dataset identified 15 core genes involved in the
development and progression of HBV-associated HCC. Assessment of
the association of these key genes with clinical outcomes indicated
that these genes may be potential therapeutic targets for HCC,
which may contribute to the development of treatments for HCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81471765 and
81771950).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on request.
Authors' contributions
CZ and BL designed the current study. YL, XG, LZ and
DZ acquired and analyzed the data. LZ and JM wrote and revised the
manuscript and analyzed the data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ghouri YA, Mian I and Rowe JH: Review of
hepatocellular carcinoma: Epidemiology, etiology, and
carcinogenesis. J Carcinog. 16(1)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Forner A, Reig M and Bruix J:
Hepatocellular carcinoma. Lancet. 391:1301–1314. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Geier A, Gartung C and Dietrich CG:
Hepatitis B e antigen and the risk of hepatocellular carcinoma. N
Engl J Med. 347:1721–1722. 2002.PubMed/NCBI View Article : Google Scholar
|
|
4
|
European Association for the Study of the
Liver. Electronic address: easloffice@easloffice.eu; European
Association for the Study of the Liver. EASL Clinical Practice
Guidelines: Management of hepatocellular carcinoma. J Hepatol.
69:182–236. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fu PY, Hu B, Ma XL, Yang ZF, Yu MC, Sun
HX, Huang A, Zhang X, Wang J, Hu ZQ, et al: New insight into BIRC3:
A novel prognostic indicator and a potential therapeutic target for
liver cancer. J Cell Biochem. 120:6035–6045. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Long J, Zhang L, Wan X, Lin J, Bai Y, Xu
W, Xiong J and Zhao H: A four-gene-based prognostic model predicts
overall survival in patients with hepatocellular carcinoma. J Cell
Mol Med. 22:5928–5938. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sauzay C, Petit A, Bourgeois AM, Barbare
JC, Chauffert B, Galmiche A and Houessinon A: Alpha-foetoprotein
(AFP): A multi-purpose marker in hepatocellular carcinoma. Clin
Chim Acta. 463:39–44. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tsuchiya N, Sawada Y, Endo I, Saito K,
Uemura Y and Nakatsura T: Biomarkers for the early diagnosis of
hepatocellular carcinoma. World J Gastroenterol. 21:10573–10583.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yan P, He Y, Xie K, Kong S and Zhao W: In
silico analyses for potential key genes associated with gastric
cancer. PeerJ. 6(e6092)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Teufel A: Bioinformatics and database
resources in hepatology. J Hepatol. 62:712–719. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jiao Y, Fu Z, Li Y, Meng L and Liu Y: High
EIF2B5 mRNA expression and its prognostic significance in liver
cancer: A study based on the TCGA and GEO database. Cancer Manag
Res. 10:6003–6014. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Shen S, Kong J, Qiu Y, Yang X, Wang W and
Yan L: Identification of core genes and outcomes in hepatocellular
carcinoma by bioinformatics analysis. J Cell Biochem.
120:10069–10081. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhou SL, Hu ZQ, Zhou ZJ, Dai Z, Wang Z,
Cao Y, Fan J, Huang XW and Zhou J: miR-28-5p-IL-34-macrophage
feedback loop modulates hepatocellular carcinoma metastasis.
Hepatology. 63:1560–1575. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Gaudet P, Skunca N, Hu JC and Dessimoz C:
Primer on the gene ontology. Methods Mol Biol. 1446:25–37.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wixon J and Kell D: The Kyoto encyclopedia
of genes and genomes-KEGG. Yeast. 17:48–55. 2000.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Baryshnikova A: Exploratory analysis of
biological networks through visualization, clustering, and
functional annotation in cytoscape. Cold Spring Harb Protoc 2016,
2016.
|
|
18
|
McClinton KJ, Aliani M, Kuny S, Sauvé Y
and Suh M: Differential effect of a carotenoid-rich diet on retina
function in non-diabetic and diabetic rats. Nutr Neurosci. 11:1–11.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Prevo R, Pirovano G, Puliyadi R, Herbert
KJ, Rodriguez-Berriguete G, O'Docherty A, Greaves W, McKenna WG and
Higgins GS: CDK1 inhibition sensitizes normal cells to DNA damage
in a cell cycle dependent manner. Cell Cycle. 17:1513–1523.
2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Haider C, Grubinger M, Řezníčková E, Weiss
TS, Rotheneder H, Miklos W, Berger W, Jorda R, Zatloukal M, Gucky
T, et al: Novel inhibitors of cyclin-dependent kinases combat
hepatocellular carcinoma without inducing chemoresistance. Mol
Cancer Ther. 12:1947–1957. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou Z, Li Y, Hao H, Wang Y, Zhou Z, Wang
Z and Chu X: Screening Hub genes as prognostic biomarkers of
hepatocellular carcinoma by bioinformatics analysis. Cell
Transplant. 11(963689719893950)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Nakayama Y and Yamaguchi N: Role of cyclin
B1 levels in DNA damage and DNA damage-induced senescence. Int Rev
Cell Mol Biol. 305:303–337. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chai N, Xie HH, Yin JP, Sa KD, Guo Y, Wang
M, Liu J, Zhang XF, Zhang X, Yin H, et al: FOXM1 promotes
proliferation in human hepatocellular carcinoma cells by
transcriptional activation of CCNB1. Biochem Biophys Res Commun.
500:924–929. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Weng L, Du J, Zhou Q, Cheng B, Li J, Zhang
D and Ling C: Identification of cyclin B1 and Sec62 as biomarkers
for recurrence in patients with HBV-related hepatocellular
carcinoma after surgical resection. Mol Cancer.
11(39)2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chiu YT, Wong JK, Choi SW, Sze KM, Ho DW,
Chan LK, Lee JM, Man K, Cherny S, Yang W, et al: Novel pre-mRNA
splicing of intronically integrated HBV generates oncogenic chimera
in hepatocellular carcinoma. J Hepatol. 64:1256–1264.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jin Q, Dai Y, Wang Y, Zhang S and Liu G:
High kinesin family member 11 expression predicts poor prognosis in
patients with clear cell renal cell carcinoma. J Clin Pathol.
72:354–362. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chen J, Li S, Zhou S, Cao S, Lou Y, Shen
H, Yin J and Li G: Kinesin superfamily protein expression and its
association with progression and prognosis in hepatocellular
carcinoma. J Cancer Res Ther. 13:651–659. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li R, Jiang X, Zhang Y, Wang S, Chen X, Yu
X, Ma J and Huang X: Cyclin B2 overexpression in human
hepatocellular carcinoma is associated with poorprognosis. Arch Med
Res. 50:10–17. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li Y, Bai W and Zhang J: MiR-200c-5p
suppresses proliferation and metastasis of human hepatocellular
carcinoma (HCC) via suppressing MAD2L1. Biomed Pharmacother.
92:1038–1044. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yun MY, Kim SB, Park S, Han CJ, Han YH,
Yoon SH, Kim SH, Kim CM, Choi DW, Cho MH, et al: Mutation analysis
of p31comet gene, a negative regulator of Mad2, in human
hepatocellular carcinoma. ExperimeExp Mol Med. 39:508–513.
2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wong N, Yeo W, Wong WL, Wong NL, Chan KY,
Mo FK, Koh J, Chan SL, Chan AT, Lai PB, et al: TOP2A overexpression
in hepatocellular carcinoma correlates with early age onset,
shorter patients survival and chemoresistance. Int J Cancer.
124:644–652. 2009.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Xu B, Xu T, Liu H, Min Q, Wang S and Song
Q: MiR-490-5p suppresses cell proliferation and invasion by
targeting BUB1 in hepatocellular carcinoma cells. Pharmacology.
100:269–282. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ricke RM, Jeganathan KB and van Deursen
JM: Bub1 overexpression induces aneuploidy and tumor formation
through Aurora B kinase hyperactivation. J Cell Biol.
193:1049–1064. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chen QF, Xia JG, Li W, Shen LJ, Huang T
and Wu P: Examining the key genes and pathways in hepatocellular
carcinoma development from hepatitis B virus-positive cirrhosis.
Mol Med Rep. 18:4940–4950. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu X, Liao W, Yuan Q, Ou Y and Huang J:
TTK activates Akt and promotes proliferation and migration of
hepatocellular carcinoma cells. Oncotarget. 6:34309–34320.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Baffy G: Decoding multifocal
hepatocellular carcinoma: An opportune pursuit. Hepatobiliary Surg
Nutr. 4:206–210. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zaman GJR, de Roos JADM, Libouban MAA,
Prinsen MBW, de Man J, Buijsman RC and Uitdehaag JCM: TTK
inhibitors as a targeted therapy for CTNNB1 (β-catenin) mutant
cancers. Mol Cancer Ther. 16:2609–2617. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Liu W, Liang B, Liu H, Huang Y, Yin X,
Zhou F, Yu X, Feng Q, Li E, Zou Z and Wu L: Overexpression of
non-SMC condensin I complex subunit G serves as a promising
prognostic marker and therapeutic target for hepatocellular
carcinoma. Int J Mol Med. 40:731–738. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Liu K, Li Y, Yu B, Wang F, Mi T and Zhao
Y: Silencing non-SMC chromosome-associated polypeptide G inhibits
proliferation and induces apoptosis in hepatocellular carcinoma
cells. Can J Physiol Pharmacol. 96:1246–1254. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ju LL, Chen L, Li JH, Wang YF, Lu RJ, Bian
ZL and Shao JG: Effect of NDC80 in human hepatocellular carcinoma.
World J Gastroenterol. 23:3675–3683. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Liu B, Yao Z, Hu K, Huang H, Xu S, Wang Q,
Yang Y and Ren J: ShRNA-mediated silencing of the Ndc80 gene
suppress cell proliferation and affected hepatitis B virus-related
hepatocellular carcinoma. Clin Res Hepatol Gastroenterol.
40:297–303. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhang L, Huang Y, Ling J, Zhuo W, Yu Z,
Shao M, Luo Y and Zhu Y: Screening and function analysis of hub
genes and pathways in hepatocellular carcinoma via bioinformatics
approaches. Cancer Biomark. 22:511–521. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bao Z, Lu L, Liu X, Guo B, Zhai Y, Li Y,
Wang Y, Xie B, Ren Q, Cao P, et al: Association between the
functional polymorphism Ile31Phe in the AURKA gene and
susceptibility of hepatocellular carcinoma in chronic hepatitis B
virus carriers. Oncotarget. 8:54904–54912. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Li J, Gao JZ, Du JL, Huang ZX and Wei LX:
Increased CDC20 expression is associated with development and
progression of hepatocellular carcinoma. Int J Oncol. 45:1547–1555.
2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chen PF, Li QH, Zeng LR, Yang XY, Peng PL,
He JH and Fan B: A 4-gene prognostic signature predicting survival
in hepatocellular carcinoma. J Cell Biochem. 120:9117–9124.
2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wen DY, Lin P, Pang YY, Chen G, He Y, Dang
YW and Yang H: Expression of the long intergenic non-protein coding
RNA 665 (LINC00665) gene and the cell cycle in hepatocellular
carcinoma using the cancer genome atlas, the gene expression
omnibus, and quantitative real-time polymerase chain reaction. Med
Sci Monit. 24:2786–2808. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin SY, Pan HW, Liu SH, Jeng YM, Hu FC,
Peng SY, Lai PL and Hsu HC: ASPM is a novel marker for vascular
invasion, early recurrence, and poor prognosis of hepatocellular
carcinoma. Clin Cancer Res. 14:4814–4820. 2008.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Okuda K, Nakashima T, Sakamoto K, Ikari T,
Hidaka H, Kubo Y, Sakuma K, Motoike Y, Okuda H and Obata H:
Hepatocellular carcinoma arising in noncirrhotic and highly
cirrhotic livers: A comparative study of histopathology and
frequency of hepatitis B markers. Cancer. 49:450–455.
1982.PubMed/NCBI View Article : Google Scholar
|