Introduction
Coronary artery obstruction presenting with acute
myocardial infarction is one of the leading causes of death
worldwide (1). Ischemia-reperfusion
(I/R) injury, a pervasive pathological disorder, occurs when the
blood flow to a tissue is blocked and reperfusion occurs when the
blockage is removed (2-4).
Acute myocardial I/R (AMI/R) injury is a complex phenomenon
involving atherosclerotic plaque rupture (5), which stops the oxygen and nutrient
supply, inducing cardiomyocyte death due to the reperfusion
process. Previous studies have demonstrated that mitochondrial
activities play a critical role in the early phase of AMI/R
(6,7).
Reactive oxygen species (ROS) from mitochondria
drive the acute damage in reperfusion (8), which directly causes mitochondrial ATP
disruption (9), calcium level
dysregulation (10) and leads to
cell apoptosis (8). The acute
oxidative stress injury and mitochondrial dysfunction are important
steps for AMI/R injury, which can induce cell or mitochondrial
membrane rupturing and myocardial infarction. The major source of
oxidative stress in I/R injury may be associated with complex I and
III of the electron transport chain in mitochondria (11). Therefore, alterations in the activity
of complex I and complex III in mitochondria may influence the
oxidative damage caused by ROS.
The ERK1/2 signaling pathway is one of the important
pathways that utilize proteins from the mitogen-activated protein
kinases family, which exert anti-apoptotic and cardioprotective
effects during AMI/R (12). However,
the extent of ERK phosphorylation is significantly raised in I/R
injury. Previous studies have demonstrated that ERK protects
against I/R injury through activating p90 ribosomal S6 kinase and
phosphorylation of Bcl-xl/Bcl-2-associated proteins to inhibit
cellular apoptosis (13,14). The ERK1/2 signaling pathway has been
identified as the pro-survival mediator against I/R injury. PD98059
is a highly selective inhibitor of ERK which binds to the inactive
form of the protein and prevents activation by its upstream
activators (15). PD98059 may act
against ERK signal transduction and therefore act as a powerful
tool to investigate the mechanisms of action behind I/R injury.
Homocysteine (Hcy), the sulfur-containing amino acid
form during methionine metabolism, which is associated with the
risk of I/R (16). Abnormally high
levels of Hcy in plasma have been indicated as a strong and
independent risk factor for cardiovascular disease (17). Recent studies have reported that Hcy
plays a critical role in I/R-induced oxidative stress, which causes
the activation of inflammatory pathways, impaired endothelial
function, atherogenesis, and cardiac remodeling (17). Furthermore, plasma Hcy expression
levels are also positively associated with blood pressure, a major
risk factor for cardiovascular disease (18,19).
However, the precise effects of Hcy and the ERK1/2
signaling pathway on mitochondrial dysfunction and oxidative stress
in AMI/R injury remains unclear. In the present study, AMI/R injury
animal models were established and treated with Hcy. Hcy was also
used in H9C2 cells that were subjected to hypoxia-reoxygenation
(H/R). Mitochondrial function and ROS production were
evaluated.
Materials and methods
Animal model of AMI/R
Male Sprague-Dawley rats (250±10 g) were purchased
from Vital River Laboratory Co. Ltd. The animal care and
experimental procedures were according to the Guide for the Care
and Use of Laboratory Animals published by the National Institutes
of Health. The study protocol was reviewed and approved by the
Ethics Committee of Cangzhou Central Hospital (approval no.
2018-029-01). Rats were maintained in environmentally controlled
rooms (25˚C) with 12 h light/dark cycles. Rats were randomly
divided into 4 experimental groups with 5 rats per group: Sham
operation control group, AMI/R group, AMI/R and Hcy group (AMI/R +
Hcy), and the AMI/R, Hcy and PD98059-treated group (AMI/R + Hcy +
PD98059). Rats were sacrificed 21 days after reperfusions. Blood
samples and heart tissue were collected to measure the cardiac
I/R-related protein expression.
The AMI/R rat model was established according to a
previous study (20). In brief,
ischemia was maintained for 30 min, at which time the slip knot was
released, initiating reperfusion for 3 h. After intraperitoneal
injection of 2% pentobarbital (30 mg/kg), the left anterior
descending coronary artery was ligated for 45 min before
reperfusion injury in the AMI/R group. To prevent infection, benzyl
penicillin sodium (400,000 U/kg) was injected once a day for 3
days. Successful AMI/R was confirmed by ST segment-characterized
electrocardiogram. The Hcy (1.6 mg/kg/day; Sigma-Aldrich; Merck
KGaA) was injected intravenously via the tail vein for 21 days
prior to the operation and up to 24 h after surgery. In the AMI/R +
Hcy + PD98059 group, ERK1/2 inhibitor PD98059 (10 mg/kg) was
administered throughout the entire period of Hcy treatment in the
AMI/R rats. All rats were euthanized following the AVMA Guidelines
on Euthanasia (21) after the
experiment. Intraperitoneal injections of a minimum of 200 mg/kg
sodium pentobarbital were used as the euthanasia solution.
Following the completion of the euthanasia procedure, death was
confirmed by ascertaining cardiac and respiratory arrest or noting
an animal's fixed and dilated pupils.
Cell culture and the I/R model
H9C2 (2-1) cells (CRL-1446™) were obtained from the
American Type Culture Collection and cultured in DMEM containing
10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 2 mM glutamine,
100 U/ml of penicillin and 100 µg/ml of streptomycin and exposed to
H/R conditions as previously described (22). The H/R treatments consisted of 0.1%
O2 + 5% CO2 in serum-starvation FBS medium
for 4 h. After hypoxia, the cells were re-oxygenated by incubating
in 95% O2 + 5% CO2 environment. Then, H9C2
cells were randomly exposed to one of the following treatments:
Pretreated ERK inhibitor PD98059 (50 µM) for 30 min + Hcy (500 µM)
or Hcy (500 µM) only at 37˚C.
Western blotting
Rat normal or ischemic heart tissues were
homogenized in RIPA buffer (20 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1
mM EDTA, 1% Triton-X100, 1% sodium deoxycholate, 1 mM PMSF and 1
µg/ml leupeptin). The concentration of the protein was determined
using Lowry protein assay and equal amounts (20 µg) of protein were
separated by 10% SDS-PAGE. After transferring onto nitrocellulose
membranes, the membranes were blocked for 1 h with 5% non-fat milk
at 25˚C and then incubated with the following primary antibodies:
Phosphorylated (p)-p38 (1:1,000; Abcam; cat. no. ab4822), p38
(1:2,000; Abcam; cat. no. ab170099), VDAC1/porin (1:2,000; Abcam;
cat. no. ab14734), p-ERK (1:1,000; Abcam; cat. no. ab192591),
ERK1/2 (1:5,000; Abcam; cat. no. ab184699), peroxisome proliferator
activated receptor γ (PPARγ; 1:2,000; Abcam; cat. no. ab45036), p53
(1:5,000; Santa Cruz Biotechnology, Inc.; cat. no. sc-126),
cytochrome c (1:1,000; Abcam; cat. no. ab133504), cleaved caspase 3
(1:2,000; Santa Cruz Biotechnology, Inc.; cat. no. sc-56053) and
GAPDH (1:5,000; Santa Cruz Biotechnology, Inc.; cat. no. sc-32233)
at 4˚C overnight. The membrane was incubated with the corresponding
secondary antibody (goat anti-rabbit or goat anti-mouse; 1:10,000;
Abcam; cat. no. ab6702 or ab6708, respectively) at 25˚C and then
analyzed under a fluorescence microscope. The protein levels were
quantified using ImageJ 1.48 (National Institute of Health).
Determination of cellular and
mitochondrial ROS
The formation of ROS in H9C2 cells was measured
using the fluorescein dye, dichloro-dihydro-fluorescein diacetate
(DCF-DA). The cells were seeded into six-well plates
(5x105) and pre-incubated with 10 µM DCF-DA in the dark.
The fluorescence of DCF-DA was then detected at the excitation
wavelength of 488 nm and emission wavelength of 525 nm.
Mitochondrial ROS production was measured using the mitochondrial
superoxide indicator, MitoSOX™ (cat. no. LSM36008; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instruction. MitoSOX was added to the cells and incubated for 10
min at 37˚C after the various treatments. Subsequently, cells were
washed and detected using confocal microscopy imaging.
Measurement of heart function
Following 2 h after reperfusion, creatine kinase
(CK) and glutamic-oxaloacetic transaminase (GOT) levels were
determined using an automatic biochemistry analyzer (Toshiba model
750; Toshiba Corporation) following the manufacturer's
instructions. Left ventricular systolic pressure (LVSP), maximum
change rate of left ventricular pressure rise
(+dp/dtmax) and the maximum change rate of left
ventricular pressure fall (-dp/dtmax) were recorded
using echocardiography (Vivid E95; GE Healthcare) following the
manufacturer's protocol.
Statistical analysis
All data are presented as the mean ± SEM.
Statistical analyses were performed using one-way ANOVAs followed
by post-hoc Tukey's test for multiple comparisons in GraphPad Prism
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Hcy enhances ERK1/2 protein expression
and oxidative stress in rats with AMI/R injury
ERK1/2 expression was analyzed in a rat model of
AMI/R injury. The western blot analysis showed significant
differences between the AMI/R and AMI/R + Hcy groups, revealing
that the protein expression ratio of p-ERK1/2/total-ERK1/2 was
increased after Hcy treatment (Fig.
1A). To determine whether Hcy was capable of inducing oxidative
stress in AMI/R injury, AMI/R rats were treated with Hcy + PD98059
or Hcy alone, and the concentration of ROS was assessed by DCF-DA.
The results showed that the AMI/R + Hcy treatment resulted in a
significant enhancement in ROS generation as compared to the AMI/R
group (Fig. 1B). These results
demonstrated that the protein expression of ERK1/2 was enhanced and
that oxidative stress was induced after administration of Hcy in
rats with AMI/R injury.
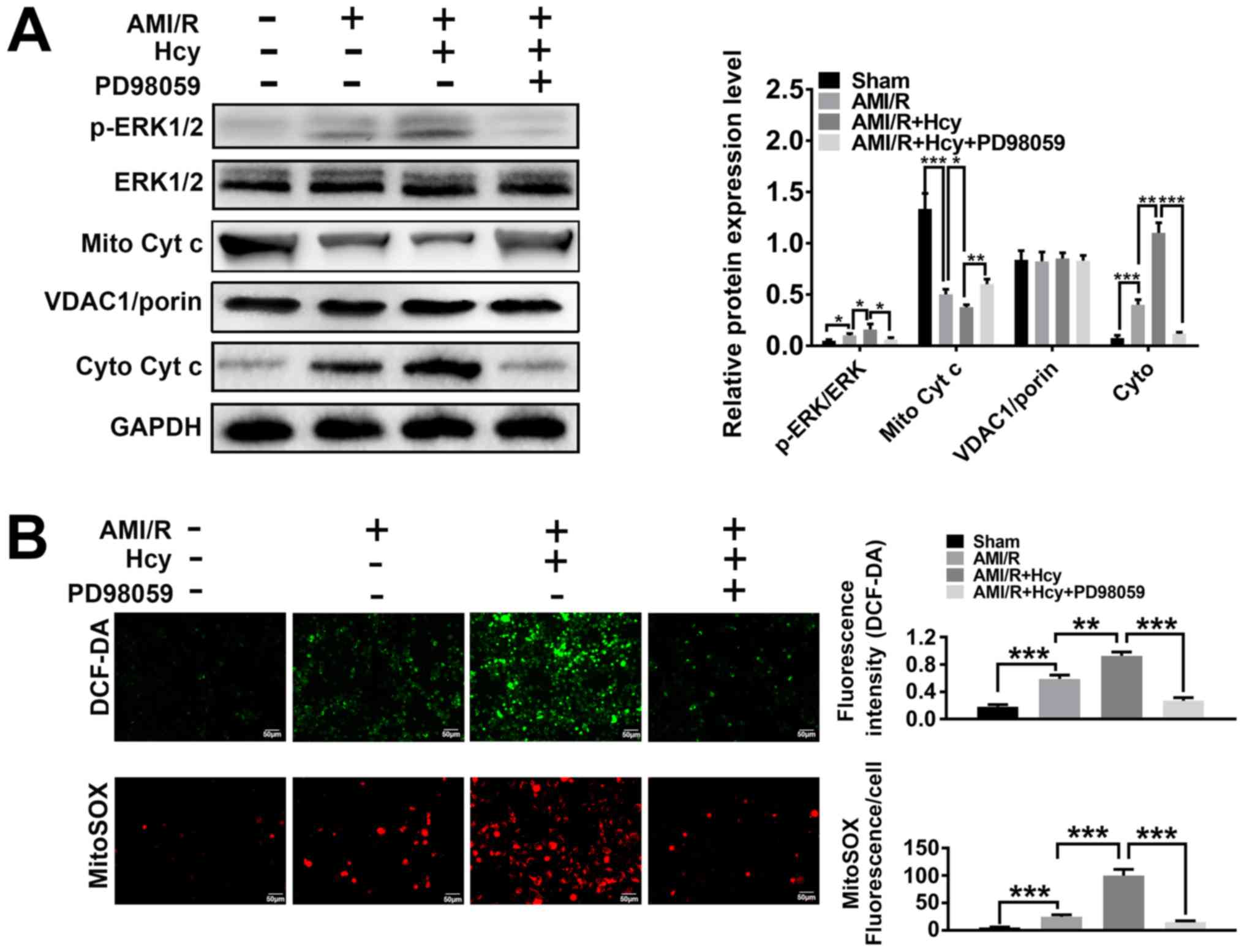 | Figure 1ERK1/2 inhibitor PD98059 reverses the
effects of Hcy on ERK 1/2 expression, the cytoplasmic translocation
of Cyt c and cellular and mitochondrial ROS production in rats with
AMI/R injury. (A) Western blotting for ERK and Cyt c after Hcy
treatment. Bar graphs show semi-quantitative analysis of the levels
of ERK and Cyt c, as determined by band density analysis (n=5). (B)
Reactive oxygen species production in the cell matrix and
mitochondria after Hcy treatment in rats with AMI/R. Representative
images of the green DCF-DA or red MitoSOX staining (n=5).
*P<0.05, **P<0.01,
***P<0.005. AMI/R, acute myocardial
ischemia/reperfusion; Cyto, cytoplasmic; Cyt c, cytochrome c;
DCF-DA, dichloro-dihydro-fluorescein diacetate; Hcy,
DL-homocysteine; Mito, mitochondrial; p, phosphorylated. |
Hcy induces cytochrome c translocation
and mitochondrial dysfunction in rats with AMI/R injury
To explore the effects of Hcy on AMI/R-injury
induced mitochondria-mediated apoptosis, the protein expression
levels of cytochrome c were investigated by western blotting.
Significantly decreased expression levels of mitochondrial
cytochrome c were detected in the AMI/R + Hcy group as compared to
the AMI/R group (Fig. 1A). By
contrast, compared with the AMI/R group, a significant increase in
the protein expression levels of cytochrome c in the cytosolic
fraction was observed in the AMI/R + Hcy group (Fig. 1A). To determine whether Hcy induced
mitochondrial dysfunction in the AMI/R injury rats, the
mitochondrial oxidative damage was evaluated. Immunofluorescence
staining showed that the number of MitoSOX-positive cells was
significantly higher in the AMI/R + Hcy group compared with the
AMI/R group (Fig. 1B). The data
revealed that Hcy significantly enhanced the release of
mitochondrial cytochrome c into the cytosol and increased ROS
generation from mitochondria in AMI/R injury rats.
Hcy causes cardiac dysfunction in rats
with AMI/R injury
Using cardiac dysfunction analysis, the current
study determined the effects of Hcy on AMI/R injury-induced cardiac
dysfunction. Compared with the AMI/R group, Hcy significantly
decreased the LVSP and +dp/dtmax. However, the
-dp/dtmax has been enhanced by Hcy (Fig. 2). Furthermore, the activity of CK and
GOT was significantly increased by Hcy in AMI/R injury rats
(Fig. 2).
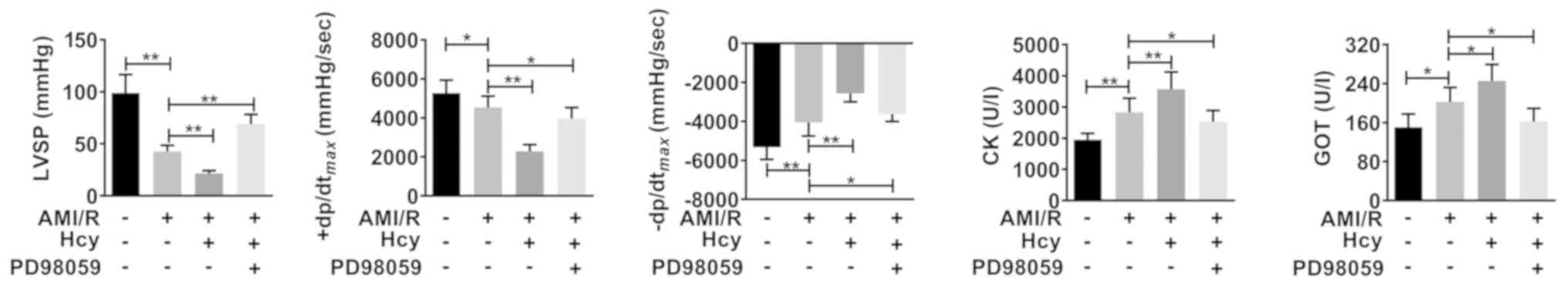 | Figure 2ERK1/2 inhibitor PD98059 reverses the
effects of Hcy on inducing cardiac dysfunction in rats with AMI/R
injury. PD98059 ameliorated the changes in LVSP,
+dp/dtmax and -dp/dtmax in the rats with
AMI/R injury. Bar graphs show semi-quantitative analysis of the
levels of CK and GOT as determined by an automatic biochemistry
analyzer, as well as the LVSP, +dp/dtmax and
-dp/dtmax, as determined by echocardiography. n=5;
*P<0.05 and **P<0.01. AMI/R, acute
myocardial I/R; CK, creatine kinase; GOT, glutamic-oxaloacetic
transaminase; LVSP, left ventricular systolic pressure; Hcy,
D,L-homocysteine; +dp/dtmax, the maximum change rate of
left ventricular pressure rise; -dp/dtmax, the maximum
change rate of left ventricular pressure fall. |
ERK1/2 inhibitor reverses the effects
of Hcy on mitochondrial dysfunction and oxidative stress in rats
with AMI/R injury
To assess whether ERK1/2 was involved in the effects
of Hcy on inducing mitochondrial dysfunction and oxidative stress,
cellular ROS production and mitochondrial function was evaluated in
rats with AMI/R injury. A significant increase in mitochondrial
cytochrome c expression levels was observed after PD98059 treatment
as compared to the AMI/R injury rats following Hcy treatment
(Fig. 1A). These data suggested that
PD98059 treatment caused a suppressive effect on cellular and
mitochondrial ROS production. Furthermore, PD98059 significantly
inhibited the release of mitochondrial cytochrome c into the
cytosol in the AMI/R + Hcy rats after Hcy treatment. As shown in
Fig. 1B, PD98059 significantly
inhibited ROS generation after Hcy treatment in AMI/R injury rats.
Consistent with this observation, mitochondrial ROS production was
also attenuated in the PD98059-treated animals as compared with the
AMI/R + Hcy group (Fig. 1B).
ERK1/2 inhibitor reverses the effects
of Hcy on inducing cardiac dysfunction in rats with AMI/R
injury
Whether ERK1/2 was involved in the effects of Hcy on
inducing cardiac dysfunction in rats with AMI/R injury was further
evaluated. After ERK1/2 inhibitor PD98059 treatment, the effects of
Hcy on LVSP, +dp/dtmax and -dp/dtmax were
significantly reversed. In addition, the activity of both CK and
GOT was significantly decreased by PD98059 treatment in the AMI/R
injury rats following Hcy treatment (Fig. 2).
ERK1/2 inhibitor reverses the effects
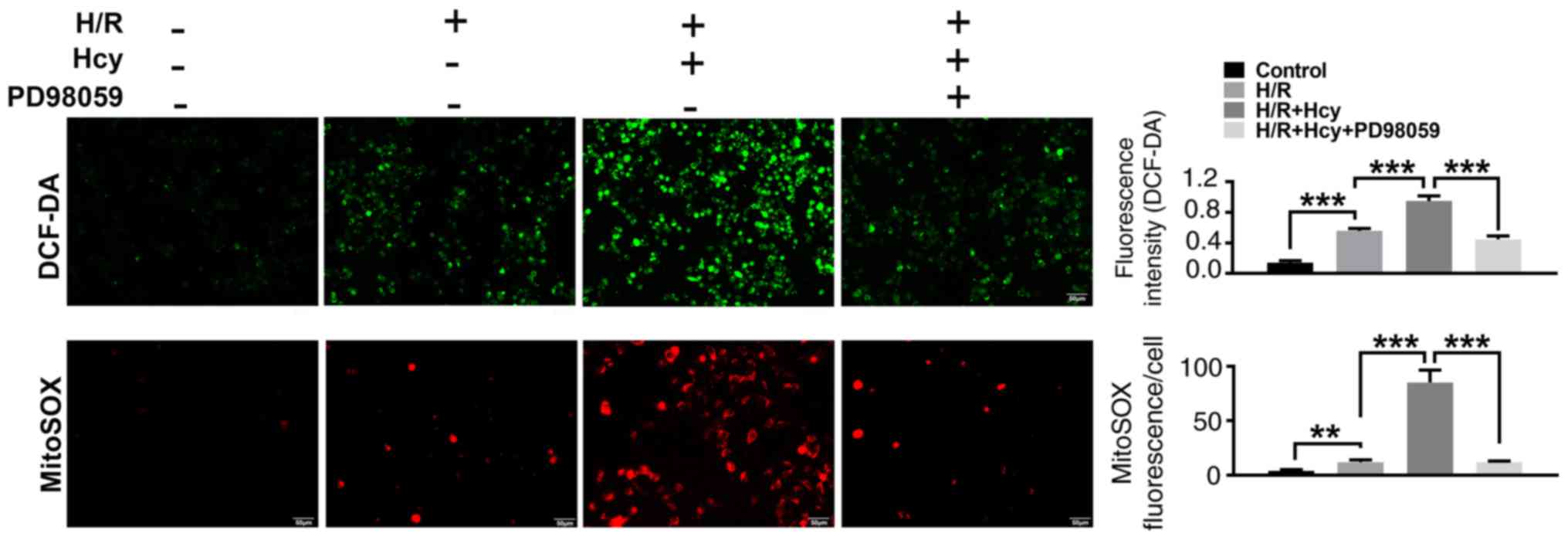
of Hcy on mitochondrial dysfunction and cell injury in H/R H9C2
cells
To verify the beneficial effects of ERK1/2 inhibitor
on Hcy-induced cardiac dysfunction, H9C2 cells were exposed to H/R
conditions and treated with or without Hcy or PD98059. After
co-treatment with Hcy and H/R, the p-ERK1/2/ERK1/2 ratio was
significantly increased compared to the H/R group (Fig. 3A). Subsequently, the rate of
apoptosis was determined by western blotting and annexin-V staining
assay. Compared with the H/R group, the proportion of annexin-V
positive cells and the expression of p53 were significantly
increased (Fig. 3A and B); however, the levels of p-p38/total p38
and PPARγ were significantly decreased in the H/R + Hcy group
(Fig. 3A). Pretreatment with PD98059
could reverse the Hcy-induced increase in the levels of apoptosis
in H9C2 cells (Fig. 3A and B). Similar results were observed using
DCF-DA fluorescence. The H/R + Hcy + PD98059 group demonstrated
significantly reduced oxidative stress compared with that in the
H/R + Hcy group (Fig. 4). The data
revealed that the ERK1/2 inhibitor could prevent the Hcy-induced
apoptosis and oxidative stress.
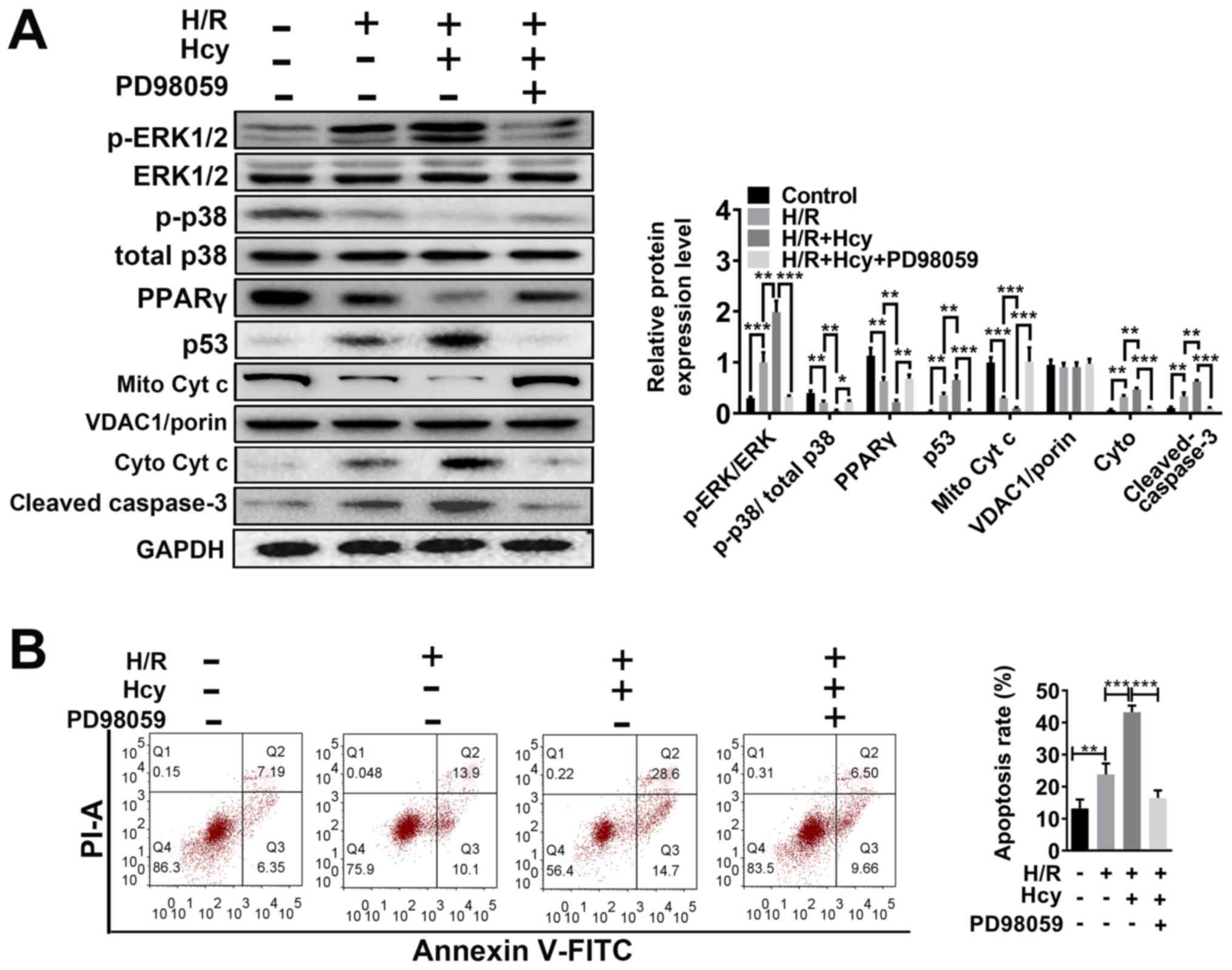 | Figure 3ERK1/2 inhibitor PD98059 reverses the
effects of Hcy on inducing Cyt c cytoplasmic translocation and
apoptosis in H/R H9C2 cells. (A) Western blotting was performed to
determine the expression levels of ERK and apoptosis-related
proteins. Bar graphs show semiquantitative analysis of the levels
of p/total ERK, p-p38, PPARγ, p53, cleaved caspase 3 and Cyt c, as
determined by band density analysis. (B) The apoptosis rate was
evaluated using Annexin V-FITC and PI staining, using an imaging
flow cytometer. *P<0.05, **P<0.01,
***P<0.005. Cyto, cytoplasmic; Cyt c, cytochrome c;
Hcy, DL-homocysteine; H/R, hypoxia-reoxygenation; Mito,
mitochondrial; p, phosphorylated; PI; propidium iodide; PPARγ,
peroxisome proliferator activated receptor γ. |
To confirm the findings in vitro, the effects
of ERK1/2 inhibitor on Hcy-stimulated mitochondrial dysfunction
were also examined in H9C2 cells. As shown in Fig. 3A, the mitochondrial cytochrome c
levels were significantly increased in the H/R + Hcy + PD98059
group compared with the H/R + Hcy group. Consistent with the in
vivo data, the mitochondrial ROS production was suppressed
following PD98059 treatment, compared with the AMI/R + Hcy group
(Fig. 4). Taken together, these
results demonstrated that the ERK1/2 inhibitor decreased ROS
generation and suppressed cell apoptosis, thereby exerting a
protective role in Hcy-induced cardiac dysfunction in H9C2
cells.
Discussion
A number of studies have shown that coronary heart
disease is a major cause of death and disability worldwide
(23). Coronary heart disease is
usually associated with the detrimental effects of AMI/R (5). I/R not only appears in different organs
but is also involved in various pathological processes, such as
heart failure. Previous studies have shown that the apoptosis of
cardiomyocytes is the most important pathogenic mechanisms behind
AMI/R injury (24,25). Reperfusion injury after ischemia is
characterized by myocardial stunning, myocyte death and
microvascular dysfunction. The mechanisms of action behind AMI/R
remain complex. Recent advances have indicated that oxidative
stress, mitochondrial membrane depolarization, calcium overloading
and inflammation are all involved. There are numerous kinases and
signaling pathway involved in I/R-induced cell apoptosis.
Activation of pro-survival kinases, such as the PI3K-Akt and
ERK1/2, have been shown to be critical in AMI/R-induced
cardioprotection (26). Hcy plays a
critical role in the metabolism of sulfur amino acids and is
associated with cardiovascular vascular disorders (27). The auto-oxidation process of Hcy is
highly reactive at the physiological pH and leads to the production
of superoxide and hydrogen peroxide (28). This phenomenon indicates that ROS
production from the auto-oxidation of Hcy remains one of the
mechanisms of action contributing to Hcy-induced cell injury. A
previous study reported that increasing Hcy expression levels in
plasma may enhance smooth muscle cell proliferation and collagen
production, resulting in vascular disease (29). However, the effects and mechanisms of
action behind Hcy induced cellular injury in AMI/R have not yet
been fully elicited. Considering that ERK1/2 pathway activation,
oxidative stress and mitochondrial dysfunction all play a critical
role in the process of AMI/R injury, the present study analyzed the
functional relevance of these factors in Hcy-induced cell injury in
AMI/R. The results of the present study showed that after Hcy
treatment in AMI/R rats, ERK1/2 prosphorylation and oxidative
stress were significantly elevated. Hcy also enhanced the release
of mitochondrial cytochrome c into the cytosol and increased the
ROS generation from mitochondria in AMI/R rats. These results are
in accordance with previous research which indicated that the role
of Hcy in endothelial dysfunction is mediated by oxidative stress
and inflammation (30). Furthermore,
the LVSP, +dp/dtmax and -dp/dtmax, as well as
the activity of CK and GOT were all significantly increased by Hcy
during AMI/R injury. These data are consistent with previous
studies which reported that Hcy may be involved in cardiovascular
diseases through a number of mechanisms and that Hcy may alter
arterial structure and function (31,32).
As the ERK1/2 signaling pathway is known to regulate
the inflammatory processes in cardiovascular disease, the ERK1/2
signaling pathway may become a new therapeutic target for heart
failure (33,34). To further explore the significance of
the ERK1/2 signaling pathway in cardioprotection during Hcy
treatment in AMI/R, the ERK inhibitor, PD98059, was used to
investigate the role of the ERK1/2 signaling pathway in Hcy-induced
cell injury. The present results indicated that the ERK1/2
inhibitor not only protected I/R injury rats from Hcy-induced
mitochondrial dysfunction and oxidative stress but also improved
the myocardial function following Hcy-induced cardiac dysfunction.
Furthermore, in the cell model, the inhibition of ERK1/2 also
decreased ROS generation and apoptosis, thereby suggesting a
protective effect against Hcy-induced cardiac dysfunction in H9C2
cells.
In conclusion, the present study demonstrated that
the protective effect of the ERK1/2 inhibitor could reverse the
Hcy-induced cellular injury. ERK1/2 inhibitors may be a new
therapeutic method to treat Hcy-induced cardiac dysfunction in
AMI/R.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW, HN and JZ performed the experiments and wrote
the manuscript, collected and analyzed the experimental data,
revised the manuscript, designed the experiments and approved the
final version manuscript.
Ethics approval and consent to
participate
The study protocol was reviewed and approved by the
Ethics Committee of Cangzhou Central Hospital (no.
2018-029-01).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thygesen K, Alpert JS, Jaffe AS, Simoons
ML, Chaitman BR and White HD: Joint ESC/ACCF/AHA/WHF Task Force for
Universal Definition of Myocardial Infarction; Authors/Task Force
Members Chairpersons, Thygesen K, Alpert JS, et al. Third
universal definition of myocardial infarction. J Am Coll Cardiol.
60:1581–1598. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med.
17(1391)2011.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Heusch G, Libby P, Gersh B, Yellon D, Böhm
M, Lopaschuk G and Opie L: Cardiovascular in coronary artery
disease and heart failure. Lancet. 383:1933–1943. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ganote C, Worstell J and Kaltenbach J:
Oxygen-induced enzyme release after irreversible myocardial injury.
Effects of cyanide in perfused rat hearts. Am J Pathol. 84:327–350.
1976.PubMed/NCBI
|
|
7
|
Zweier JL, Flaherty JT and Weisfeldt ML:
Direct measurement of free radical generation following reperfusion
of ischemic myocardium. Proc Natl Acad Sci USA. 84:1404–1407.
1987.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Loor G, Kondapalli J, Iwase H, Chandel NS,
Waypa GB, Guzy RD, Vanden Hoek TL and Schumacker PT: Mitochondrial
oxidant stress triggers cell death in simulated
ischemia-reperfusion. Biochim Biophys Acta. 1813:1382–1394.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Di Lisa F and Bernardi P: Modulation of
mitochondrial permeability transition in ischemia-reperfusion
injury of the heart. Advantages and limitations. Curr Med Chem.
22:2480–2487. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Paradies G, Petrosillo G, Pistolese M and
Ruggiero FM: Reactive oxygen species affect mitochondrial electron
transport complex I activity through oxidative cardiolipin damage.
Gene. 286:135–141. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Abe Ji, Baines CP and Berk BC: Role of
mitogen-activated protein kinases in ischemia and reperfusion
injury: The good and the bad. Circ Res. 86:607–609. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Das A, Salloum FN, Xi L, Rao YJ and
Kukreja RC: ERK phosphorylation mediates sildenafil-induced
myocardial protection against ischemia-reperfusion injury in mice.
Am J Physiol Heart Circ Physiol. 296:H1236–H1243. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee
JC, Feuerstein GZ, Thomas H, Maleeff B and Ohlstein EH: Inhibition
of extracellular signal-regulated kinase enhances
ischemia/reoxygenation-induced apoptosis in cultured cardiac
myocytes and exaggerates reperfusion injury in isolated perfused
heart. Circ Res. 86:692–699. 2000.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Di Cristo G, Berardi N, Cancedda L,
Pizzorusso T, Putignano E, Ratto GM and Maffei L: Requirement of
ERK activation for visual cortical plasticity. Science.
292:2337–2340. 2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wald DS, Law M and Morris JK: Homocysteine
and cardiovascular disease: Evidence on causality from a
meta-analysis. BMJ. 325(1202)2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ganguly P and Alam SF: Role of
homocysteine in the development of cardiovascular disease. Nutri J.
14(6)2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Malinow M, Levenson J, Giral P, Nieto F,
Razavian M, Segond P and Simon A: Role of blood pressure, uric
acid, and hemorheological parameters on plasma homocyst (e)ine
concentration. Atherosclerosis. 114:175–183. 1995.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hoogeveen EK, Kostense PJ, Beks PJ,
Mackaay AJ, Jakobs C, Bouter LM, Heine RJ and Stehouwer CD:
Hyperhomocysteinemia is associated with an increased risk of
cardiovascular disease, especially in non-insulin-dependent
diabetes mellitus: A population-based study. Arterioscler Thromb
Vasc Biol. 18:133–138. 1998.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Shao L, Wu D, Zhang P, Li W, Wang J, Su G,
Liao Y, Wang Z and Liu K: The significance of microthrombosis and
fgl2 in no-reflow phenomenon of rats with acute myocardial
ischemia/reperfusion. Clin Appl Thromb Hemost. 19:19–28.
2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Leary S, Underwood W, Anthony R, Cartner
S, Corey D, Grandin T, Greenacre C, Gwaltney-Brant S, McCrackin MA,
Meyer R, et al: AVMA guidelines for the euthanasia of animals: 2013
edition.
|
|
22
|
Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan
W, Guo C, Zhou D, Wang Y, Xi M and Wen A: Cardioprotective effect
of Danshensu against myocardial ischemia/reperfusion injury and
inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2
phosphorylation. Eur J Pharmacol. 699:219–226. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Feigin VL, Forouzanfar MH, Krishnamurthi
R, Mensah GA, Connor M, Bennett DA, Moran AE, Sacco RL, Anderson L,
Truelsen T, et al: Global and regional burden of stroke during
1990-2010: Findings from the Global Burden of Disease Study 2010.
Lancet. 383:245–255. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Scarabelli TM, Stephanou A, Pasini E,
Comini L, Raddino R, Knight RA and Latchman DS: Different signaling
pathways induce apoptosis in endothelial cells and cardiac myocytes
during ischemia/reperfusion injury. Circ Res. 90:745–748.
2002.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Xu Q, Li X, Lu Y, Shen L, Zhang J, Cao S,
Huang X, Bin J and Liao Y: Pharmacological modulation of autophagy
to protect cardiomyocytes according to the time windows of
ischaemia/reperfusion. Br J Pharmacol. 172:3072–3085.
2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kim SJ, Yoo KY, Jeong CW, Kim WM, Lee HK,
Bae HB, Kwak SH, Li M and Lee J: Urinary trypsin inhibitors afford
cardioprotective effects through activation of PI3K-Akt and ERK
signal transduction and inhibition of p38 MAPK and JNK. Cardiology.
114:264–270. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hassan A, Hunt BJ, O'sullivan M, Bell R,
D'souza R, Jeffery S, Bamford JM and Markus HS: Homocysteine is a
risk factor for cerebral small vessel disease, acting via
endothelial dysfunction. Brain. 127:212–219. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Misra HP: Generation of superoxide free
radical during the autoxidation of thiols. J Biol Chem.
249:2151–2155. 1974.PubMed/NCBI
|
|
29
|
Majors A, Ehrhart LA and Pezacka EH:
Homocysteine as a risk factor for vascular disease. Enhanced
collagen production and accumulation by smooth muscle cells.
Arterioscler Thromb Vasc Biol. 17:2074–2081. 1997.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Basu A, Jenkins AJ, Stoner JA, Thorpe SR,
Klein RL, Lopes-Virella MF, Garvey WT and Lyons TJ: DCCT/EDIC
Research Group. Plasma total homocysteine and carotid intima-media
thickness in type 1 diabetes: A prospective study. Atherosclerosis.
236:188–195. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang S, Bai YY, Luo LM, Xiao WK, Wu HM
and Ye P: Association between serum homocysteine and arterial
stiffness in elderly: A community-based study. J Geriatr Cardiol.
11:32–38. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Balakumar P, Singh AP and Singh M: Rodent
models of heart failure. J Pharmacol Toxicol Methods. 56:1–10.
2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Purcell NH, Wilkins BJ, York A,
Saba-El-Leil MK, Meloche S, Robbins J and Molkentin JD: Genetic
inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and
heart failure but has no effect on hypertrophy in vivo. Proc Natl
Acad Sci USA. 104:14074–14079. 2007.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bueno OF, De Windt LJ, Tymitz KM, Witt SA,
Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, et
al: The MEK1-ERK1/2 signaling pathway promotes compensated cardiac
hypertrophy in transgenic mice. EMBO J. 19:6341–6350.
2000.PubMed/NCBI View Article : Google Scholar
|