Introduction
Obesity, originating from a combination of genetic
and environmental factors, including lifestyle, culture, physiology
and behavior, has reached pandemic proportions in the
21st century (1,2). It presents substantial health
challenges and economic burden worldwide (1,2). Obesity
is capable of producing a variety of alterations in the body that
may predispose individuals to changes in cardiac morphology and
ventricular function (3,4), in a process known as cardiac
remodeling. Cardiac remodeling refers to the structural and
functional dysfunction caused by molecular and genetic changes in
cardiomyocytes under the influence of neurohumoral factors
(5). A number of potential
mechanisms have been hypothesized to underlie obesity-associated
cardiomyopathy, including inflammation, neurohumoral and metabolic
abnormalities (3). In addition,
oxidative stress has also been reported to serve a significant role
in myocardial abnormalities associated with obesity (6).
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2)
is a major regulator of redox signaling. Under physiological
conditions, kelch-like ECH-associated protein 1 (Keap1) binds to
and target Nrf2 for proteasomal degradation. However, during
oxidative stress, Keap1 becomes inactivated by oxidation of its
several cysteine residues or autophagic elimination in a
sequestosome-1-dependent manner (7).
Nrf2 is then released from this complex, where it enters the
nucleus to promote the expression of genes associated with
antioxidant activities. In the nucleus, Nrf2 can upregulate the
expression of a wide range of antioxidant genes, including
glutathione-S-transferase, heme oxygenase (HO-1), NADPH and quinone
1, by binding with the antioxidant response element (8). Although the Nrf2/Keap1 pathway is
physiologically important for the antioxidant defense against a
variety of cardiovascular diseases, its role in obesity-associated
cardiac remodeling remains to be elucidated.
Metformin (Met) is one of the most commonly
prescribed drugs for the treatment of type 2 diabetes (9) that has been previously demonstrated to
exhibit protective effects against cardiovascular disease. Met has
been reported to protect the myocardium against
isoproterenol-induced infarction (10,11),
attenuate cardiac remodeling in monosodium glutamate-induced
obesity in mouse models and inhibit isoproterenol- and pressure
overload-induced remodeling, in a manner that was either dependent
or independent of adenosine 5'-monophosphate (AMP)-activated
protein kinase (AMPK) activation (12-14).
The aim of the present study was to investigate the status of the
Nrf2/Keap1 signaling pathway underlying the protective effects of
Met against cardiac remodeling in mouse models of obesity.
Materials and methods
Experimental animals and groups
C57BL/6J male mice (age, 4-5 weeks, 15-17g, n=24)
were purchased from the Medical Experimental Animal Center of Henan
University of Science and Technology (Luoyang, China; License
number: SCXK 2018-0007). After adaptive feeding for 1 week, the
animals were randomly divided into the following three groups (n=8
per group): i) Control; ii) high-fat diet (HFD); and iii) HFD+Met
(300 mg/kg). All mice except for mice in the control group were fed
on a HFD for 24 weeks consecutively. The composition of HFD was 60%
fat, 20% carbohydrate and 20% protein, while the normal chow diet
consisted of 4.5% fat. In addition, mice in the HFD+Met group were
administered Met in drinking water daily from week 8 onwards
following the commencement of feeding on HFD. All mice were housed
in a temperature and humidity regulated room (temperature, 22±2˚C;
humidity, 50±5%) with controlled lighting (12-h light/dark cycle).
Water and food was freely available to the mice.
The study was approved by the Animal Care and Ethics
Committee of Henan University of Science and Technology (Luoyang,
China) and was performed in accordance with the National Institutes
of Health Guidelines for the Care and Use of Laboratory Animals
(15).
Metabolic measurements
On week 22 of the experiment, an oral glucose
tolerance test (OGTT; 0.2 g/kg) was performed. Blood samples (a
drop of blood) were collected through the tail artery at 5, 15, 30,
60 and 120 min after the administration of glucose by oral gavage,
following which plasma glucose concentrations were measured using a
blood glucose monitor (Roche Diagnostics). AUC was calculated
according to the approximate trapezoidal area formula (16), which could better reflect the changes
in the trend and time accumulation effect of blood glucose levels.
After OGTT, animals in each group continued to live in their
corresponding conditions as aforementioned until week 24. Body
weight, body length, waistline and food/water consumption were
monitored weekly, whilst Lee index (17) was calculated [(body
weight)1/3/body length] at the end
of the experiment.
Serum analysis and heart weight index
calculation
After feeding on HFD for 24 weeks, all mice were
anaesthetized by an intraperitoneal injection of sodium
pentobarbital (45 mg/kg), following which blood (~0.5 ml) was
collected from the orbital venous sinus. Fasting serum glucose
(cat. no. 133011, Zhongsheng North Control Biotechnology Co., Ltd.)
and insulin levels (cat. no. XY-E20353, Shanghai Biological
Technology Co., Ltd.) were then measured in the blood samples
collect in each group using corresponding commercial kits.
Homeostasis model assessment of insulin resistance (HOMA-IR) was
calculated for mice in each group according to protocols described
previously (18). Following
retroorbital exsanguination, all mice were immediately euthanized
by cervical dislocation. The mouse hearts were then removed
rapidly, washed in ice-cold 0.9% saline and weighed. Heart
weight/tibial length (HW/TL) and the left ventricular weight/tibial
length (LHW/TL) were calculated. ~1/3 of the left ventricular
samples were fixed in 4% paraformaldehyde at room temperature (RT)
for 24 h and then embedded in paraffin for subsequent morphological
analysis. The remaining left ventricle samples were flash-frozen in
liquid nitrogen and stored at -80˚C until further use.
Histological examination
Interstitial fibrosis of the left ventricle was
determined using Masson's trichrome staining (19). The sections were deparaffinized using
xylene and washed with various levels of ethanol, dried and stained
for 10 min at RT with Masson composite solution. A total of 2
samples from each group were taken for preliminary morphology and
molecular biological experiments, with the remaining 6 samples in
each group for subsequent experiments. There were 6 heart tissues
in each group, 3 sections of each tissue and 10 randomized visual
fields in light microscope (Olympus; magnification, x200) were
selected for statistics. To assess the degree of fibrosis, the
images were quantitatively analyzed by morphometry using the
Image-Pro Plus software (version 1.61; Media Cybernetics,
Inc.).
Reverse transcription quantitative-PCR
(RT-qPCR)
Total RNA was extracted from the left ventricular
tissue (20) using the
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) on ice and reverse-transcribed using a high-capacity
complementary DNA reverse transcription kit (cat. no. 4368814;
Applied Biosystems) according to the manufacturer's protocol. The
temperature protocol was as follows: Denaturation at 70˚C for 10
min, then extension at 37˚C for 60 min and a final inactivation at
94˚C for 10 min. qPCR was performed using SYBR™ Green PCR Master
Mix kit (cat. no. CW2623; CWBio) according to manufacturer's
protocol. mRNA levels of cardiac fibrosis markers, including
transforming growth factor-β1 (TGF-β1), collagen I (Col I) and
collagen III (Col III), in addition those of genes associated with
oxidative stress, including keap1, Nrf2 and HO-1, were quantified.
Primer sequences are shown in Table
SI. qPCR was performed in a Step One Plus Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) for 40 cycles,
specifically, pre-denaturation at 94˚C for 20 sec, and then
entering the cycle of 94˚C for 15 sec, and 60˚C for 1 min. The
2-ΔΔCq method was used to quantify
the genes (20), and GAPDH
gene was used as the internal control.
Western blot analysis
Total proteins were extracted from the left
ventricle using ice-cold RIPA buffer (cat. no. CW2333; CWBio).
Denatured protein samples were quantified using the BCA method, and
30 µg protein was subjected to 10% SDS-PAGE analysis. After
electrophoresis, proteins were transferred to PVDF membranes (EMD
Millipore), which were then blocked at RT for 1 h with 5% non-fat
dry milk in TBS supplemented with 0.1% Tween-20 (TBST) and
subsequently incubated with the respective primary antibodies
diluted in 5% non-fat dry milk at 4˚C overnight. Dilutions used for
each primary antibody was 1:500 for Nrf2, 1:500 for Keap-1, 1:500
for HO-1 and 1:8,000 for GAPDH. Rabbit and mouse polyclonal
antibodies specific for Nrf2 (cat. no. 16396-1-AP), Keap1 (cat. no.
10503-2-AP), HO-1 (cat. no. 10701-1-AP) were purchased from
Proteintech; and GAPDH (cat. no. BM3896) were purchased from Boster
Biological Technology. After washing with TBST three times,
membranes were incubated with anti-rabbit (cat. no. BA1054; Boster
Biological Technology) or anti-mouse (cat. no. BA1050; Boster
Biological Technology) horseradish peroxidase conjugated secondary
antibodies (1:5,000) for 1 h at RT. Protein bands were visualized
using chemiluminescence using the ECL chemiluminescent substrate
(cat. no. BL523A; Biosharp). GAPDH was used as the loading control
for total protein. Quantification of bands was performed using the
ImageJ Software 1.50 (National Institutes of Health).
Determination of nuclear Nrf2 using
the immunofluorescence staining method
Paraffin-embedded samples were cut into 5-µm thick
sections, which were deparaffinized with xylene followed by
rehydration using a descending alcohol series, subjected to antigen
retrieval in EDTA buffer (pH 8.0) using a microwave (at medium
strength for 10-15 min) and then placed in 3% BSA (cat. no.
9048-46-8; Sigma-Aldrich; Merck KGaA) to block non-specific
staining for 30 min at room temperature. Sections were then
incubated with anti-Nrf2 antibodies (1:100; cat. no. 16396-1-AP;
Proteintech) at 4˚C overnight, followed by incubation with the
fluorescent-labeled secondary antibody (1:300; cat. no. BA1032;
Boster Biological Technology) in darkness and room temperature for
50 min. After counterstaining with 1 µg/ml DAPI for 5 min at RT,
the sections were dehydrated and viewed under a fluorescence
microscope (x400; Nikon Corporation).
Statistical analysis
In all experiments, values were expressed as the
mean ± SEM (n=8). Differences between groups were evaluated using
the Student-Newman-Keuls test after One-Way ANOVA (GraphPad Prism
5.0; GraphPad Software, Inc.). P<0.05 was considered to indicate
a statistically significant difference.
Results
Met treatment alleviates obesity and
glucose metabolic disorder in mice
The effects of Met on the metabolic parameters of
obese mice were first examined. Met treatment significantly reduced
the body weight, waist circumference (Fig. 1A and B) and Lee index (Fig. 1C) in obese mice, whilst significantly
reducing the fasting blood glucose (Fig.
1D) and serum insulin levels (Fig.
1E) in addition to ameliorating insulin resistance (Fig. 1F) when compared with untreated mice
in the HFD group. Met also significantly improved oral glucose
tolerance compared with that in the HFD only group (Fig. 1G). These results suggest that Met
treatment alleviated metabolic disorder and obesity syndrome in
mice with HFD-induced obesity.
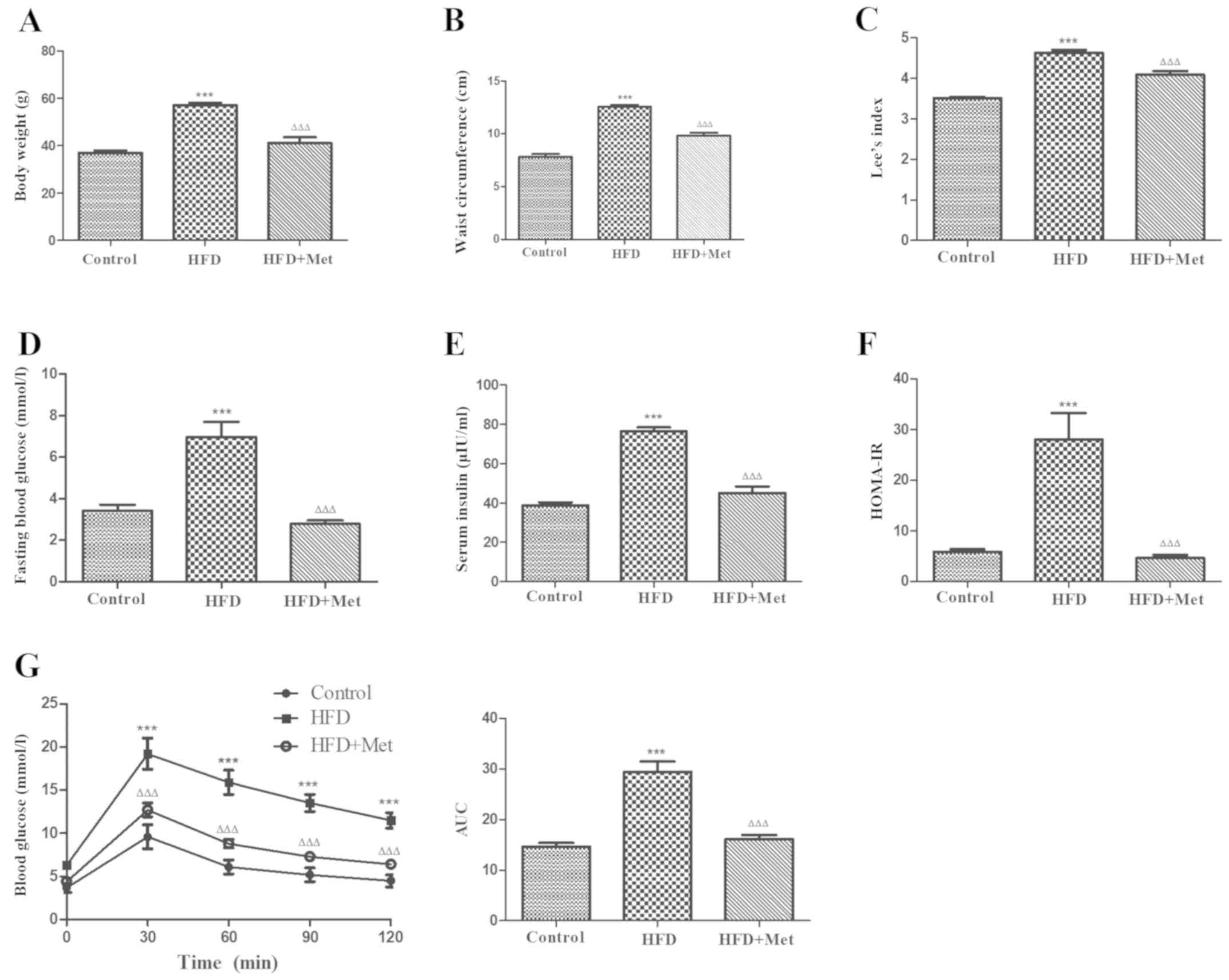 | Figure 1Met ameliorates obesity and metabolic
disorders in HFD-induced obese mice after 24 weeks. (A) Met
decreased the body weight, (B) waist circumference and (C) the Lee
index in mice from the HFD+Met group compared with those in the HFD
group. (D) Between the two HFD groups of mice, hyperglycemia, (E)
hyperinsulinaemia and (F) HOMA-IR were significantly improved after
Met treatment compared with those in untreated mice. (G) The effect
of Met on impaired glucose tolerance, as measured by the oral
glucose tolerance test. Right: AUC. Data are expressed as mean ±
SEM, n=8 per group. ***P<0.001
vs. control; ΔΔΔP<0.001 vs. HFD. AUC, area under the
curve; HFD, high fat diet; HOMA-IR, homeostasis model assessment of
insulin resistance; OGTT, oral glucose tolerance test; Met,
metformin. |
Met prevents cardiac remodeling in
HFD-induced obese mice
To investigate the effects of Met on cardiac
remodeling associated with obesity, heart weight index and degree
of cardiac fibrosis were evaluated in obese mice. HW (Fig. 2A and B) and LHW indices (Fig. 2C and D) were both found to be significantly
increased in the HFD group compared with those in the control
group. Masson's staining revealed that the degree of cardiac
fibrosis was significantly increased in the HFD group compared with
that in the control group (Fig. 2E),
which was also indicated by the RT-qPCR results (Fig. 2F), suggesting the development of
cardiac remodeling in obese mice. Met treatment significantly
reversed this cardiac remodeling, as indicated by significant
reductions in the HW index and levels of cardiac fibrosis compared
with those in the HFD group (Fig.
2).
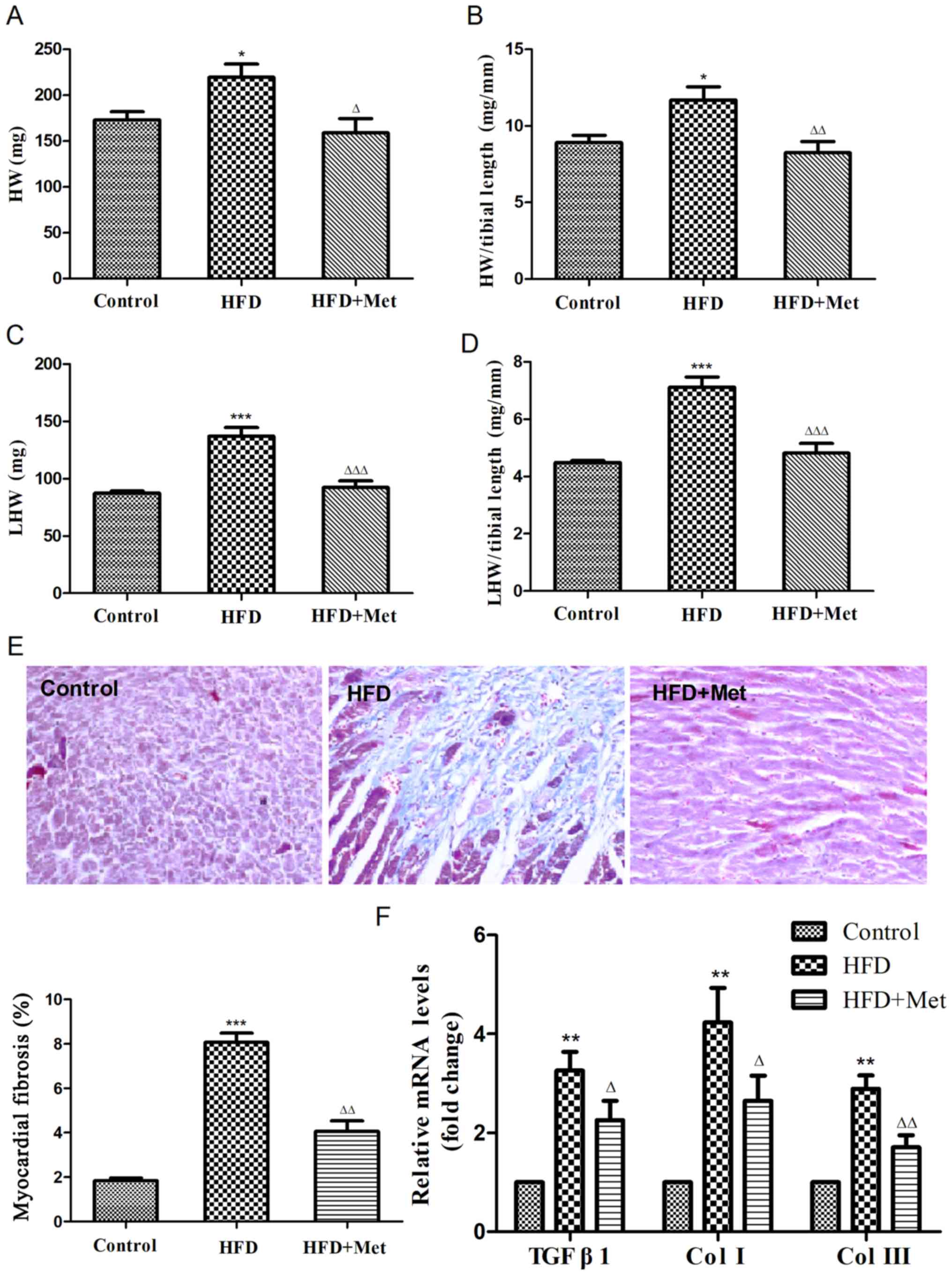 | Figure 2Met reduces the heart weight index and
ameliorates myocardial fibrosis in mice fed on HFD after 24 weeks.
(A) Heart weight, (B) heart weight index, (C) left ventricular
weight and (D) left ventricular weight index were calculated. (E)
Representative figures of myocardial interstitial fibrosis using
Masson's trichrome staining (magnification, x200) and the
quantified results of Masson's staining. (F) Reverse
transcription-quantitative PCR analysis of TGFB1,
COL1A1 and COL3A1 expression. Data are expressed as
mean ± SEM, n=6 per group. *P<0.05,
**P<0.01 and
***P<0.001 vs. Control;
ΔP<0.05, ΔΔP<0.01 and
ΔΔΔP<0.001 vs. HFD. COL, collagen gene; HFD,
high fat diet; HW, heart weight; LHW, left ventricular weight; Met,
metformin; TGFB1, transforming growth factor β1 gene. |
Met enhances the endogenous
antioxidant system in the heart
To elucidate the mechanism underlying the protective
effects of Met against cardiac remodeling, the expression of genes
and proteins associated with Nrf2/Keap1 signaling were analyzed
using RT-qPCR and western blotting. Gene and protein expression
levels of Nrf2 exhibited slight increases in the heart tissues of
mice in the HFD group compared with those in the control group
(Fig. 3A, D and E),
whilst an opposite trend was observed in Keap1 expression (Fig. 3B, D
and F). Expression of HO-1 mRNA, a
downstream target gene of Nrf2, was found to be slightly reduced in
the heart tissue of mice in the HFD group compared with that in the
control group (Fig. 3C). Significant
reductions were observed in the protein levels of HO-1 in the heart
tissues of mice in the HFD group compared with mice in the control
group (Fig. 3D and G). It was found that whilst Met treatment
significantly reduced Keap1 protein expression (Fig. 3D and F), no effect was observed on KEAP1
gene expression between the HFD and HFD+Met groups (Fig. 3B). At the same time, Met treatment
significantly upregulated Nrf2 and HO-1 expression on both mRNA and
protein levels in heart tissues compared with those in untreated
mice in the HFD group (Fig. 3A,
C, E
and G).
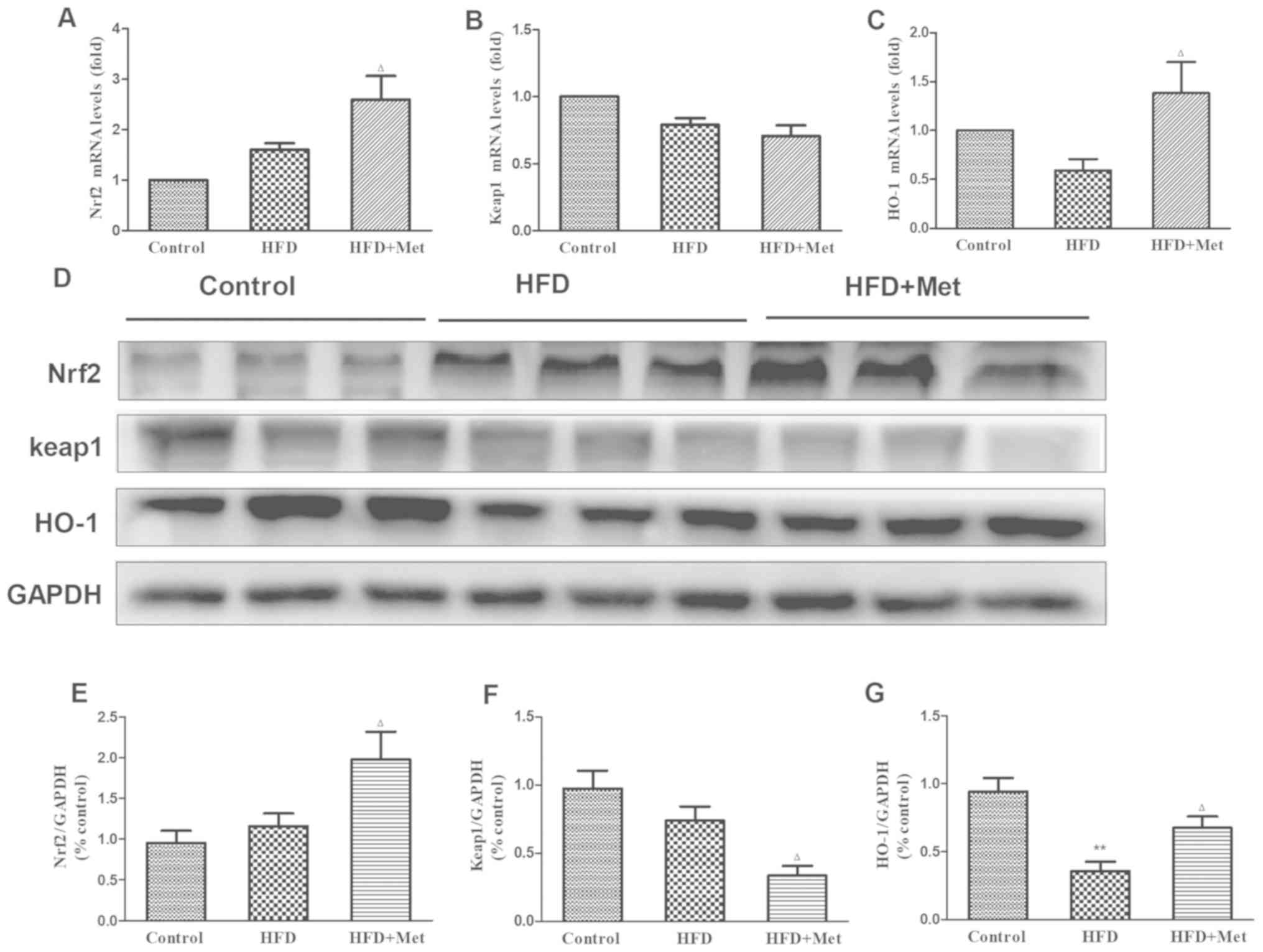 | Figure 3Effect of Met on the expression level
of genes and proteins associated with the Nrf2/Keap1 signaling
pathway in the cardiac tissue of the three experimental groups. (A)
mRNA levels of NFE2L2, (B) KEAP1 and (C)
HMOX-1. (D) Representative western blot images of proteins
associated with the Nrf2/Keap1 signaling pathway. (E) Densitometry
analysis of Nrf2, (F) Keap1 and (G) HO-1 expression. The relative
densitometry is expressed as the ratio of Nrf2, Keap1 or HO-1 to
GAPDH. Data are presented as mean ± SEM; n=6 per group.
**P<0.01 vs. Control;
ΔP<0.05 vs. HFD. HFD, high fat diet; HO-1, heme
oxygenase 1; Keap1, kelch-like ECH-associated protein 1;
HMOX-1, HO-1 gene; Met, metformin; Nrf2, nuclear factor
(erythroid-derived 2)-like 2; NFE2L2, Nrf2 gene. |
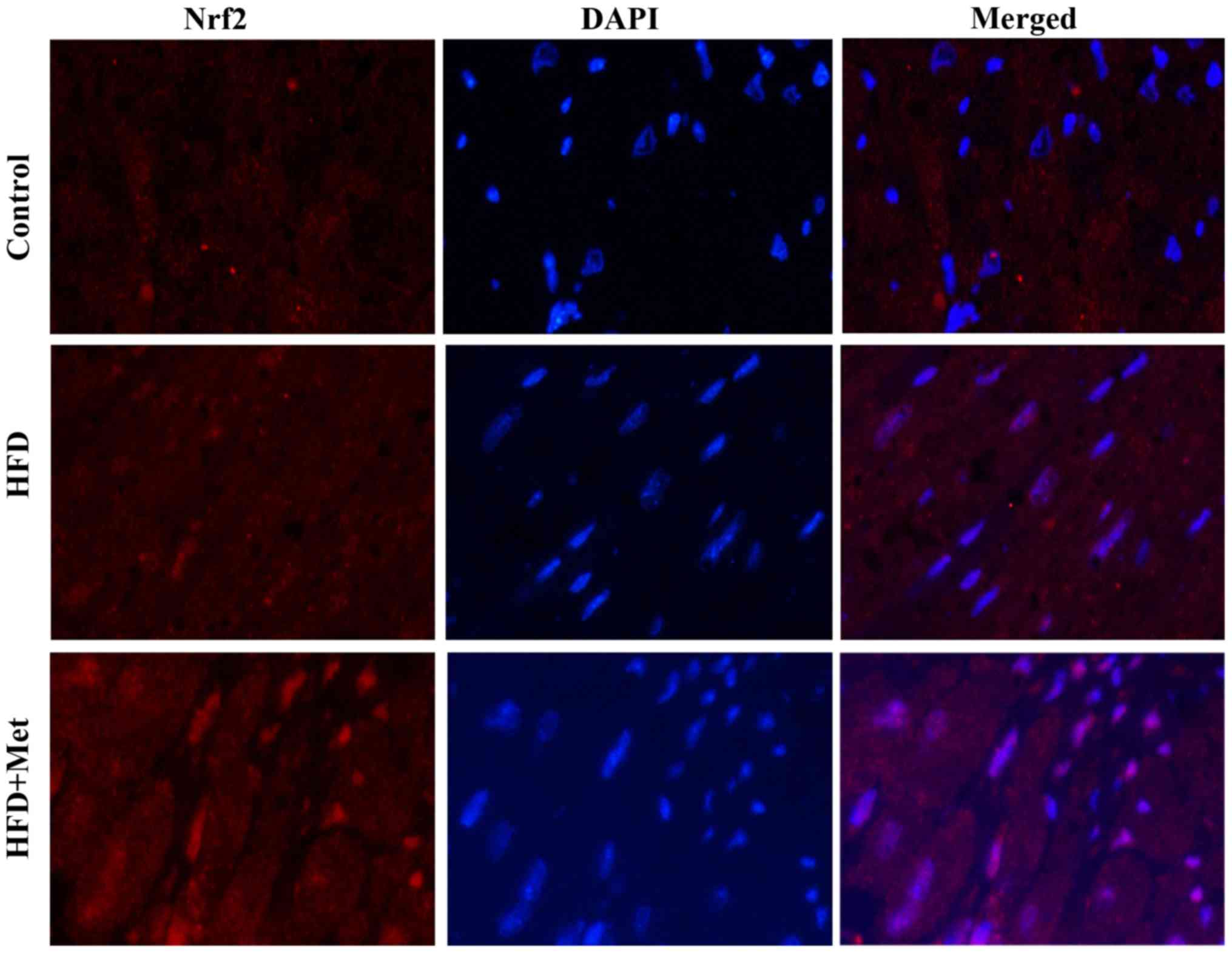
Met promotes Nrf2 translocation into
the nucleus in the heart tissue
Fig. 4 shows
representative immunofluorescence staining images of Nrf2
expression in the heart tissues isolated from mice from the three
experimental groups. Nrf2 staining showed slight increases in the
heart tissue of mice from the HFD group compared with that in
control mice. By contrast, Met induced a marked increase in nuclear
Nrf2 staining, suggesting that Met treatment may activate Nrf2
signaling by inducing the nuclear translocation of Nrf2.
Discussion
The results of the present study indicated that
long-term Met treatment provided protection against
obesity-associated cardiac remodeling in HFD-induced obese mice.
This may be mediated though the reduction of metabolic disorder and
enhancement of the Nrf2/Keap1 signaling pathway of the endogenous
antioxidant system. These findings suggest the potential of Met as
a therapeutic agent for patients of obesity at risk of cardiac
remodeling.
It is estimated that there are 671 million
individuals with obesity in the world, with 62% occurring in
developing countries (21). Morbid
obesity has been associated with insulin resistance, diabetes
mellitus and organ damage, including atherosclerosis and left
ventricular hypertrophy (LVH). LVH is known has been previously
found to be independently associated with adiposity (22). HFD is associated with the
manifestation of obesity, metabolic disturbances, cardiac
hypertrophy and interstitial fibrosis (23). Results of the present study indicated
that mice fed on HFD for 24 weeks exhibited apparent metabolic
syndrome, with symptoms including increases in body weight, waist
circumference, Lee index, fasting blood glucose, serum insulin
levels and in the HOMA-IR index. All of the aforementioned
pathological changes could be mitigated by long-term Met treatment,
which also improved glucose tolerance. In addition to the reported
beneficial effects of Met on the metabolism, interest has also been
garnered in the effects of Met on cardiovascular diseases (13,24,25),
cancer (26) and aging (27). However, the underlying mechanism of
Met action remain elusive. Although Met-induced activation of the
energy-sensor AMPK has been well documented (28), AMPK-independent mechanisms have also
been reported, including suppression of TGF-β1 expression (29), inhibition of reactive oxygen species
generation by blocking the NADPH oxidase pathway (30) and the activation of endothelial
nitric oxide synthase (31).
Pre-treatment with Met has been previously demonstrated to activate
the Nrf2 antioxidant signaling pathways in the hippocampus of rats
with global cerebral ischemia (32).
In the present study, the HW and the LHW indices were found to be
significantly increased in mice from the HFD group compared with
the control group. Masson's staining of the myocardial tissue
confirmed the existence of myocardial fibrosis in mice from the HFD
group. In addition, gene expression levels of TGF-β1, Col I and Col
III in the myocardial tissue, markers of cardiac remodeling, were
revealed to be markedly elevated in mice from the HFD group. All of
these aforementioned observations support the notion that cardiac
remodeling occurred in mice from the HFD group in the present
study. Long-term treatment with Met significantly ameliorated
cardiac remodeling, which was demonstrated by reduced HW index, LHW
index and myocardial fibrosis. Amelioration of cardiac remodeling
was also indicated by the observed reductions in TGF-β1, Col I and
Col III gene expression in myocardial tissues in HFD mice treated
with Met.
The Nrf2/Keap1 pathway is physiologically important
for defense against oxidative stress (33). The present study suggested that Nrf2
in the heart may serve as a compensatory mechanism in response to
oxidative stress in mice fed with HFD. Met reduced Keap1 protein
levels, resulting in the activation of Nrf2 and subsequent
translocation into the nucleus, leading to the upregulation of
downstream antioxidative enzymes such as HO-1. Together, these data
suggested that Met has protective effects against
obesity-associated cardiac remodeling, which may potentially be due
to its effect on alleviating metabolic disorders and enhancing
endogenous antioxidant function. However, further studies are
required to elucidate the direct targets of Met in the regulation
of Nrf2/Keap1 signaling.
In summary, metabolic disorders and adverse cardiac
remodeling were found to be evident in mice with HFD-induced
obesity in the present study. Met exerted potent protective effects
against the development of metabolic disorders and cardiac
remodeling, which were associated with its effect on enhancing
endogenous antioxidant activities by activating the Nrf2/Keap1
signaling pathway. The present study suggested that Met serve as an
effective treatment option for obesity-associated cardiac
remodeling, where the Nrf2/Keap1 pathway may be another potential
therapeutic target.
Supplementary Material
Sequences of primers.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Henan Key
Scientific Research Projects (grant no. 19A350003).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD designed the study and wrote the manuscript. MZ,
HL, GL and YL performed the experiments. SF helped to analyze the
data and revise the manuscript. All of the authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Animal Care and Ethics
Committee of Henan University of Science and Technology (Luoyang,
China; grant no. 19A350003) and was in accordance with the National
Institutes of Health Guidelines for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang YC, McPherson K, Marsh T, Gortmaker
SL and Brown M: Health and economic burden of the projected obesity
trends in the USA and the UK. Lancet. 378:815–825. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Scheen AJ and Van Gaal LF: Combating the
dual burden: Therapeutic targeting of common pathways in obesity
and type 2 diabetes. Lancet Diabetes Endocrinol. 2:911–922.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tiwari S and Ndisang JF: The role of
obesity in cardiomyopathy and nephropathy. Curr Pharm Des.
20:1409–1417. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kenchaiah S, Evans JC, Levy D, Wilson PW,
Benjamin EJ, Larson MG, Kannel WB and Vasan RS: Obesity and the
risk of heart failure. New Engl J Med. 347:305–313. 2002.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling-concepts and clinical implications: A consensus paper
from an international forum on cardiac remodeling. Behalf of an
international forum on cardiac remodeling. J Am Coll Cardiol.
35:569–582. 2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rhee SG and Bae SH: The antioxidant
function of sestrins is mediated by promotion of autophagic
degradation of Keap1 and Nrf2 activation and by inhibition of
mTORC1. Free Radic Biol Med. 88:205–211. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bae SH, Sung SH, Oh SY, Lim JM, Lee SK,
Park YN, Lee HE, Kang D and Rhee SG: Sestrins activate Nrf2 by
promoting p62-dependent autophagic degradation of Keap1 and prevent
oxidative liver damage. Cell Metab. 17:73–84. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Suzuki T, Motohashi H and Yamamoto M:
Toward clinical application of the Keap1-Nrf2 pathway. Trends
Pharmacol Sci. 34:340–346. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Foretz M, Guigas B, Bertrand L, Pollak M
and Viollet B: Metformin: From mechanisms of action to therapies.
Cell Metab. 20:953–966. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Calvert JW, Gundewar S, Jha S, Greer JJ,
Bestermann WH, Tian R and Lefer DJ: Acute metformin therapy confers
cardioprotection against myocardial infarction via
AMPK-eNOS-mediated signaling. Diabetes. 57:696–705. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Soraya H, Khorrami A and Garjani A,
Maleki-Dizaji N and Garjani A: Acute treatment with metformin
improves cardiac function following isoproterenol induced
myocardial infarction in rats. Pharmacol Rep. 64:1476–1484.
2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Burlá AK, Lobato NS, Fortes ZB, Oigman W
and Neves MF: Cardiac fibrosis and vascular remodeling are
attenuated by metformin in obese rats. Int J Cardiol. 165:483–487.
2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Fu YN, Xiao H, Ma XW, Jiang SY, Xu M and
Zhang YY: Metformin attenuates pressure overload-induced cardiac
hypertrophy via AMPK activation. Acta Pharmacol Sin. 32:879–887.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cha HN, Choi JH, Kim YW, Kim JY, Ahn MW
and Park SY: Metformin inhibits isoproterenol-induced cardiac
hypertrophy in mice. Korean J Physiol Pharmacol. 14:377–384.
2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: Special Report: The 1996 guide for the care and use
of laboratory animals. ILAR J. 38:41–48. 1997.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Canis R, Demirkok SS, Osar Z, Balci H and
Can G: Effects of inhaled budesonide on insulin sensitivity in
nondiabetic patients with asthma and chronic obstructive pulmonary
disease. Adv Ther. 24:560–570. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Han Y, Wu JZ, Shen JZ, Chen L, He T, Jin
MW and Liu H: Pentamethylquercetin induces adipose browning and
exerts beneficial effects in 3T3-L1 adipocytes and high-fat
diet-fed mice. Sci Rep. 7(1123)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Matthews DR, Hosker JP, Rudenski AS,
Naylor BA, Treacher DF and Turner RC: Homeostasis model assessment:
Insulin resistance and beta-cell function from fasting plasma
glucose and insulin concentrations in man. Diabetologia.
28:412–419. 1985.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Simon J, Nemeth E, Nemes A, Husveth-Toth
M, Radovits T, Foldes G, Kiss L, Bagyura Z, Skopal J, Merkely B and
Gara E: Circulating relaxin-1 level is a surrogate marker of
myocardial fibrosis in HFrEF. Front Physiol. 10(690)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) methods. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ng M, Fleming T, Robinson M, Thomson B,
Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF,
et al: Global, regional, and national prevalence of overweight and
obesity in children and adults during 1980-2013: A systematic
analysis for the Global Burden of Disease Study 2013. Lancet.
384:766–781. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Brady TM: The role of obesity in the
development of left ventricular hypertrophy among children and
adolescents. Curr Hypertens Rep. 18(3)2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang Y, Fang X, Dai M, Cao Q, Tan T, He
W, Huang Y, Chu L and Bao M: Cardiac-specific down-regulation of
carnitine palmitoyltransferase-1b (CPT-1b) prevents cardiac
remodeling in obese mice. Obesity (Silver Spring). 24:2533–2543.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sasaki H, Asanuma H, Fujita M, Takahama H,
Wakeno M, Ito S, Ogai A, Asakura M, Kim J, Minamino T, et al:
Metformin prevents progression of heart failure in dogs: Role of
AMP-activated protein kinase. Circulation. 119:2568–2577.
2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Xie Z, Lau K, Eby B, Lozano P, He C,
Pennington B, Li H, Rathi S, Dong Y, Tian R, et al: Improvement of
cardiac functions by chronic metformin treatment is associated with
enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes.
60:1770–1778. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Daugan M, Dufaÿ Wojcicki A, d Hayer B and
Boudy V: Metformin: An anti-diabetic drug to fight cancer.
Pharmacol Res. 113:675–685. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Barzilai N, Crandall JP, Kritchevsky SB
and Espeland MA: Metformin as a tool to target aging. Cell Metab.
23:1060–1065. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Luo T, Nocon A, Fry J, Sherban A, Rui X,
Jiang B, Xu XJ, Han J, Yan Y, Yang Q, et al: AMPK Activation by
metformin suppresses abnormal extracellular matrix remodeling in
adipose tissue and ameliorates insulin resistance in obesity.
Diabetes. 65:2295–2310. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M,
Shen Q, Zhu Y and Zhang Y: Metformin attenuates cardiac fibrosis by
inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc Res.
87:504–513. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Batchuluun B, Inoguchi T, Sonoda N, Sasaki
S, Inoue T, Fujimura Y, Miura D and Takayanagi R: Metformin and
liraglutide ameliorate high glucose-induced oxidative stress via
inhibition of PKC-NAD(P)H oxidase pathway in human aortic
endothelial cells. Atherosclerosis. 232:156–164. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kawabata H and Ishikawa K:
Cardioprotection by metformin is abolished by a nitric oxide
synthase inhibitor in ischemic rabbit hearts. Hypertens Res.
26:107–110. 2003.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ashabi G, Khalaj L, Khodagholi F,
Goudarzvand M and Sarkaki A: Pre-treatment with metformin activates
Nrf2 antioxidant pathways and inhibits inflammatory responses
through induction of AMPK after transient global cerebral ischemia.
Metab Brain Dis. 30:747–754. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li J, Ichikawa T, Jin Y, Hofseth LJ,
Nagarkatti P, Nagarkatti M, Windust A and Cui T: An essential role
of Nrf2 in American ginseng-mediated anti-oxidative actions in
cardiomyocytes. J Ethnopharmacol. 130:222–230. 2010.PubMed/NCBI View Article : Google Scholar
|