Introduction
Bone homeostasis is maintained by the balance
between bone resorption by osteoclasts and bone formation by
osteoblasts. Osteoclasts are derived from the bone marrow-derived
mononuclear-macrophage (BMDM) cell lineage and osteoblasts are
derived from mesenchymal cells (1-3).
Osteoclasts are the only type of bone-resorbing cell that are
essential for bone development and remodelling, and their lack
leads to osteopetrosis, which is a disease manifested by an
increase in the non-proliferating bone mass (4). Conversely, increased numbers and
activity of osteoclasts under certain pathological conditions lead
to accelerated bone resorption and may lead to osteoporosis and
osteolytic diseases (5,6). To better understand the mechanism of
osteoclast-based diseases and develop relevant therapeutic methods,
the molecular basis of osteoclast differentiation and function, and
the regulatory mechanisms of osteoclast signalling must be
elucidated.
Numerous studies have confirmed that receptor
activator of NF-κB ligand (RANKL) is an important extracellular
regulatory molecule for osteoclast development that stimulates
osteoclast formation (5,7). Binding of RANKL to its receptor leads
to recruitment of tumour necrosis factor receptor-associated factor
6 and activation of NF-κB/mitogen-activated protein kinase (MAPK)
signalling, which is the most direct pathway for osteoclast
development (8). In addition,
calcineurin/mammalian target of rapamycin (mTOR)/nuclear factor of
activated T cells 2 (NFATC2) signalling has an important role in
RANKL-induced osteoclast development (9-11).
Calcineurin and mTOR are important negative regulators during bone
development, and their respective clinical target drugs cyclosporin
A (CsA) and rapamycin cause severe bone metabolic disease (12-14).
NFATC2 is a direct regulatory transcription factor for osteoclast
development that regulates osteoclastogenesis, as well as the
expression of osteolysis-associated molecules, including matrix
metallopeptidase (MMP)-9 and cathepsin K (5,10,15).
Activation of calcineurin/mTOR/NFATC2 is dependent on the
intracellular Ca2+ levels. The RANKL-induced calcium
influx-activated calcineurin-dependent NFATC2 pathway has been
reported to have an important role in osteoclast differentiation
(16); these authors observed a
continuous rather than a transient calcium influx, which was
critical for the continued activation of NFATC2. However, how RANKL
leads to activation and nuclear translocation of NFATC2 via
activation of the calcium influx has remained elusive. The
involvement in RANKL-induced osteoblast differentiation has been
reported for calcium influx mediated by transmembrane protein 64 by
Kim et al (17), the
Transient receptor potential cation channel subfamily V member 4
channel by Masuyama et al (18) and the store-operated Ca2+
entry (SOCE) membrane calcium channel ORAI1 by Hwang and Putney
(19); importantly, these calcium
influxes regulate osteoclast development through sustained
activation of NFATC2 and nuclear translocation. However, few
studies have investigated RANKL-induced osteoclast differentiation
via the endoplasmic reticulum (ER) sensor stromal interaction
molecule 1 (STIM1) of SOCE.
Combined immunodeficiencies (CID) are genetic
defects that result in defective T cell function with or without
intrinsic B cell abnormalities. The CID caused by the STIM1
gene mutation is called the Stormorken Syndrome. In the present
study, three STIM1 mutations were identified in patients with
Stormorken Syndrome; (the p.E136X recessive mutation, p.R429C
dominant mutation and p.R304W recessive mutation). The three
patients had different degrees of skeletal sclerosis (p.E136X and
p.R429C) and bone loss (p.R304W) disease. Osteoclast precursor
cells, which are BMDMs, were obtained from these patients and a
healthy individual, and subjected to in vitro experiments.
The osteoclast differentiation phenotype induced by
RANKL/macrophage colony-stimulating factor (M-CSF) was observed and
the signalling pathways involved in calcium influx via SOCE were
investigated.
Materials and methods
Subjects
BMDM samples were obtained from three male CID
patients (age, Patient 1, 4 years; Patient 2, 4 years; and Patient
3, 7 years) and one healthy male subject (age, 17 years. For the
healthy subject, bone marrow sample was obtained due to suspected
aplastic anemia, where part of the BMDM are used for in
vitro culture. The patient' final diagnosis was negative for
aplastic anemia. The experiments involving human subjects were
based on the Declaration of Helsinki and the European Declaration
of Human Rights, and informed consent was obtained from the parents
of the patients and the healthy donor. The study was approved by
the Ethical Review Committee of Yanjishan Hospital of Wannan
Medical College (Wuhu, China; no. 201627). Bone mineral density
(BMD) of the L1-4 lumbar vertebrae and left hip (femoral neck,
total hip joint) were measured by Hologic dual-energy X-ray
absorptiometry (Hologic). The BMD was provided as the bone mineral
content per unit area in mg/cm2. As shown in Table II, the Z value is provided by
Hologic, Inc. and is compared with the BMD value of the
corresponding age average in the Asian Children's Database
(20,21). Healthy was defined as 1>Z value
>-1; Mild low bone density as -1<Z; Low bone density as
-1.5<Z value<-1, Moderate loss for bone density as -2<Z
value <-1.5; and Severe loss for bone density as Z<-2.
 | Table IIBone mineral density
(mg/cm2) and Z value of stromal interaction molecule 1
mutation patients and a healthy subject. |
Table II
Bone mineral density
(mg/cm2) and Z value of stromal interaction molecule 1
mutation patients and a healthy subject.
| | Femoral neck | Wards triangle | Hip joint | Lumbar spine
L1-L4 |
|---|
| STIM1 status | BMD | Z-value | BMD | Z-value | BMD | Z-value | BMD | Z-value |
|---|
| WT | 911.3 | 0.12 | 751.2 | 0.32 | 1019.3 | 0.41 | 1124.7 | 0.57 |
| p.E136X | 1125.7 | 1.51 | 895.8 | 1.35 | 1158.4 | 1.6 | 1354.6 | 1.52 |
| p.R429C | 1185.4 | 1.59 | 862.3 | 1.3 | 1162.3 | 1.61 | 1248.6 | 1.4 |
| p.R304W | 703.6 | -1.17 | 451.9 | -1.84 | 812.3 | -1.23 | 815.5 | -1.45 |
Genomic sequencing
Genomic DNA was isolated from skin fibroblasts of
CID patients and the healthy control using DNAzol Reagent (Thermo
Fisher Scientific, Inc.) according to manufacturer's protocol.
High-fidelity KOD DNA polymerase (Biosciences) was used to
PCR-amplify the STIM1 exon sequence. The primer sequences were as
follows: Forward, 5'-CTTAACTGTTCGGGGACACA-3' and reverse,
5'-ACAGCGGAAGAGTGACACTG-3'. The amplicons were purified with the
QIAquick Gel Extraction kit (Qiagen GmbH) and directly sent for
sequencing (Genewiz, Inc.). If a mutation was detected, the PCR and
sequencing reactions were repeated a further three times using at
least two separate genomic DNA preparations. Sequence alignments
were performed using T-Coffee software (Swiss Institute of
Bioinformatics) (22); DNA
sequencing traces were presented using PeakTrace Basecaller
software (version 6.46; Nucleics Pty., Ltd.). Single-nucleotide
polymorphism (SNP) searches were performed in the dbSNP database
(www.ncbi.nlm.nih.gov/SNP) of the National Council
for Biotechnology Information.
Acquisition of BMDMs
A total of 2 ml bone marrow samples was collected in
a sterile polyacrylamide centrifuge tube using a non-invasive
Sprotte spinal needle (Pajunk) and centrifuged to separate the
cells and debris (2,000 x g, 4˚C, 5 min). The supernatant was
discarded and the pellet was re-suspended in red blood cell lysis
buffer, followed by incubation at 4˚C for 10 min. The cells were
centrifuged, re-suspended and counted, and the concentration was
adjusted to 2x106 cells/ml. The cells were cultured in
37˚C in a humidified atmosphere with 5% CO2, and grown
in complete Dulbecco's modified Eagle's medium (DMEM, Gibco; Thermo
Fisher Scientific, Inc.) containing 10% fetal bovine serum (FBS,
Gibco; Thermo Fisher Scientific, Inc.) and 5 ng/ml M-CSF to ensure
survival of the BMDMs. The medium was replaced every 2-3 days and
mature BMDMs were harvested after 7 days. Mature BMDMs were stored
in complete DMEM containing 1% DMSO and 10% FBS at -20˚C for use in
subsequent experiments. pGMLV-SV40T lentivirus (Genomeditech Co.
Ltd.) was used for immortalization of the BMDM cells. The
immortalized cell line was generated by transfecting primary BMDM
cells with a vector expressing the oncogene pSV40Tag for 72 h, in
accordance with a protocol established previously (23). Its characteristics are maintained in
the media aforementioned for >30 generations where no cell
differentiation or lack of proliferation were observed.
Plasmids and antibodies
Plasmid vectors encoding wild type STIM1
(MIGR-STIM1-IRES-RFP, ca.STIM1 thereafter), and NFATC2 small
interfering (si)RNA were constructed by Shanghai GenePharma Co.,
Ltd. The species reactivity, conjugation, manufacturer, product
catalog numbers and dilutions of all antibodies used are shown in
Table I.
| Table IInformation of all antibodies used in
the present study. |
Table I
Information of all antibodies used in
the present study.
| Name | Host | Species
reactivity | Conjugation | Supplier | Cat. no. | Dilution used |
|---|
| STIM1 | Rabbit | Human | Unconjugated | Cell Signaling
Technology, Inc. | 5668 | 1:20 (FC) |
| CD11b | Mouse | Human | Unconjugated | Abcam | Ab34216 | 1:50 (FC) |
| TRAP | Rabbit | Human | Unconjugated | Abcam | ab2391 | 1:50 (FC) |
| Ki67 | Rabbit | Human | Alexa Fluor
488 | Cell Signaling
Technology, Inc. | 11882 | 1:50 (FC) |
| p-mTOR | Rabbit | Human | Unconjugated | Cell Signaling
Technology, Inc. | 5536 | 1:20 (FC) |
| p-AKT (T308) | Rabbit | Human | Unconjugated | Cell Signaling
Technology, Inc. | 13038 | 1:20 (FC) |
| p-AKT (S473) | Rabbit | Human | Unconjugated | Cell Signaling
Technology, Inc. | 4060 | 1:20 (FC) |
| p-S6
(S235/236) | Rabbit | Human | Unconjugated | Cell Signaling
Technology, Inc. | 4858 | 1:20 (FC) |
| p-NFATC2
(Ser54) | Rabbit | Human | Unconjugated | eBioscience; Thermo
Fisher Scientific, Inc. | 44-944G | 1:20 (FC) |
| NFATC2 | Mouse | Human | Unconjugated | R&D
Systems | MAB6499 | 1:20 (FC) |
| MMP9 | Rabbit | Human | Unconjugated | Cell Signaling
Technology, Inc. | 13667 | 1:50 (FC) |
| Cathepsin K | Rabbit | Human | Unconjugated | eBioscience; Thermo
Fisher Scientific, Inc. | PA5-14270 | 1:50 (FC) |
| Secondary
antibody | Goat | Rabbit | Alexa Fluor
647 | Thermo Fisher
Scientific, Inc. | A32733 | 1:100 (FC) |
| Secondary
antibody | Goat | Mouse | Alexa Fluor
488 | Thermo Fisher
Scientific, Inc. | A28175 | 1:100 (FC) |
|
(FcgR)II/FcgRIII | Mouse | Human | Unconjugated | eBioscience; Thermo
Fisher Scientific, Inc. | MFCR00-4 | 1:100 (FC) |
Flow cytometry
Since the three naturally-mutant strains are rare
(24) and the number of cells
obtained is insufficient to detect protein expression using western
blot analysis, the expression of all proteins was detected by flow
cytometry. BMDMs were treated with rapamycin (0.1 nM; AdooQ
Bioscience) and CsA (7 nM; AdooQ) or lentiviral concentrate. After
3 days of stimulation with recombinant human M-CSF (20 ng/ml; Kexin
Biomedical Technology), the BMDMs were induced using 100 ng/ml
RANKL (ACROBiosystems) for 3 days. The cells were treated with
intracellular immobilization buffer (Thermo Fisher Scientific,
Inc.) and blocked with an anti-Fc region of immunoglobulin receptor
(FcgR)II/FcgRIII antibody for 2 h. The permeabilized cells were
fixed by the slow addition of ice-cold 100% methanol to the
pre-chilled cells under gentle vortexing, to a final concentration
of 90% methanol. The cells were incubated with anti-STIM1, CD11b,
TRAP, anti-Ki67, p-mTOR (S2448), phosphorylated (p)-AKT (T308),
p-AKT (S473), p-S6 (S235/236), p-NFATC2, anti-MMP9 or
anti-Cathepsin K primary antibodies overnight at 4˚C. A
fluorescently labelled secondary antibody was coupled to the
primary antibody for 2 h at room temperature in the dark. All flow
cytometry experiments were also performed with homotyped
non-specific negative control antibodies. The samples were analysed
using the BD FACSCanto™ II flow cytometer (BD
Biosciences).
Carboxyfluorescein succinimidyl ester
(CFSE) labelling of live cells
BMDMs (1x105/ml) subjected to different
treatments were loaded with 10 µM CFSE (eBioscience; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
cells were evaluated by flow cytometry using an excitation
wavelength of 494 nm and emission wavelength of 521 nm.
Lentiviral transfection
The siRNA for NFATC2, scrambled siRNA, STIM1
overexpression plasmids and control plasmids were purchased from
Genomeditech Co. Ltd. For each transfection reaction, 4 µg/ml SiRNA
or 4 µg/ml plasmids and 10 µg/ml of the PPACK packaging plasmid
(System Biosciences) were co-transfected into 293T cells (American
Type Culture Collection) using Lipofectamine® 3000
(Thermo Fisher Scientific, Inc.) according to manufacturer's
protocol. The 293T cells were cultured in DMEM supplemented with
10% FBS and 2 mM L-glutamine in 37˚C in a humidified atmosphere
with 5% CO2. After 2 days, the supernatant was collected
and filtered by centrifugation in Amicon Ultra-15-centrifugal
filter tubes according to the manufacturer's protocol. (Merck
Millipore; Merck KGaA). The viral titres were determined by
gradient dilution. The resulting concentrated lentiviral fluid was
used for direct infection of BMDMs. The BMDM cell suspension was
seeded into a 6-well plate and cultured overnight under the same
conditions; the corresponding lentiviral solution (multiplicity of
infection, 20) was added to the cells, followed by culture for 72
h. Subsequently, the cells were harvested, and the NFATC2-knockdown
and STIM1-overexpressing cells were subjected to flow cytometric
assays.
Determination of intracellular
Ca2+ concentration
A total of 5x103 BMDMs were seeded onto
poly-L-lysine-coated 96-well plates for attachment. The cells were
first loaded with 1 mM Fura-2-AM (AAT Bioquest) and re-suspended
using Ca2+-free saline. Fluorescence intensity
measurements were performed using an Epoch full wavelength
microplate reader (Omega Bio-Tek, Inc.). After 250 sec, 30 nM
thapsigargin (Abcam) was added to induce Ca2+ release
from the ER, and an equal volume of 40 mM Ca2+
physiological saline solution was added at 500 sec to induce SOCE.
Fura-2 fluorescence was excited at 340 and 380 nm, with their
emission measured at 510 nm and plotted as the 340/380 nm ratio to
represent the relative intracellular Ca2+ levels.
Detection of calcineurin activity
After collection of the 5x104 BMDMs
treated with M-CSF/RANKL in the presence or absence of CsA, the
cells were incubated with DMSO for 30 min and then stimulated with
10 nM caerulein for 15 min. The cells were lysed and the
calcineurin activity was determined using the Calcineurin Cell
Activity Assay kit (Abcam) according to the manufacturer's
protocol.
Detection of the cell proliferation
activity
A total of 5x103 BMDMs were seeded into
96-well plates and treated with CsA, Rap, NFATC2 siRNA and ca.STIM1
or negative controls. The cells were then incubated with 10 µl Cell
Counting Kit-8 stain (Dojindo Molecular Technologies, Inc.) for 2 h
according to the manufacturer's protocol, and the absorbance at 450
nm was detected by a microplate reader (Omega Bio-Tek, Inc.).
Statistical analysis
The data were analysed using one-way analysis of
variance and paired Student's t-test. The statistical analyses and
graphical representation of the data were performed using GraphPad
Prism 6.0 (GraphPad Inc.) and SPSS version 19.0 (IBM Corp.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Defects in STIM1 cause a disorder in
SOCE in BMDMs
Genomic DNA sequencing of patient 1 revealed a
380_381insA mutation resulting in insertion of an adenine between
positions 380 and 381 in the exon 3 sequence of STIM1 (Fig. 1A). This mutation caused a change in 8
amino acids (RSIQLDRG) and led to a nonsense mutation and premature
termination of the STIM1 translation codon (p.E136X; Fig. 1D). Patient 2 developed a C→T
substitution mutation (c.1285C>T) at position 1,285 of exon 11
of STIM1 (Fig. 1B), which was a
missense mutation that resulted in a single amino acid change in
the STIM1 sequence (p.R429C; Fig.
1D). Patient 3 developed a C→T substitution mutation
(c.910C>T) at position 910 of exon 6 of STIM1 (Fig. 1C), which resulted in a missense
mutation in a single amino acid position in the STIM1 sequence
(p.R304W; Fig. 1D). The p.E136X and
p.R429C mutations resulted in loss of STIM1 expression, while
p.R304W mutations had no effect on STIM1 expression (Fig. 1E). Studies have indicated that
p.E136X and p.R429C mutations result in truncation of STIM1 during
its expression and conformational changes, causing loss of function
of SOCE (25,26), while p.R304W mutations cause
conformational changes in STIM1 leading to constitutive activation
of SOCE (27). The STIM1-wild-type
(STIM1wt) BMDMs produced transient Ca2+
release from the ER in response to thapsigargin (TG), a Sarco ER
calcium adenosine triphosphatase pump blocker, followed by an
increase in the Ca2+ concentration in the medium,
leading to sustained extracellular Ca2+ influx which is
SOCE (Fig. 1F). This normal SOCE was
abolished in BMDMs with STIM1p.E136X and
STIM1p.R429C mutations, which responded to the
TG-induced Ca2+ release from the ER, but extracellular
Ca2+ did not enter the cell even after ER storage
depletion (Fig. 1F). For BMDMs with
STIM1p.R304W mutations, the intracellular
Ca2+ concentration varies with the extracellular
Ca2+, suggesting that their SOCE channel is continuously
open (Fig. 1F). More importantly,
three STIM1-mutant patients had a disorder of bone metabolism,
indicating that the BMD of patients with p.R304W was too low, while
the BMD of patients with p.E136X and p.R429C was too high (Table II). These results suggest that
mutations in STIM1 affect the function of SOCE in BMDMs, which may
be associated with bone metabolic disorders in patients carrying
these mutations.
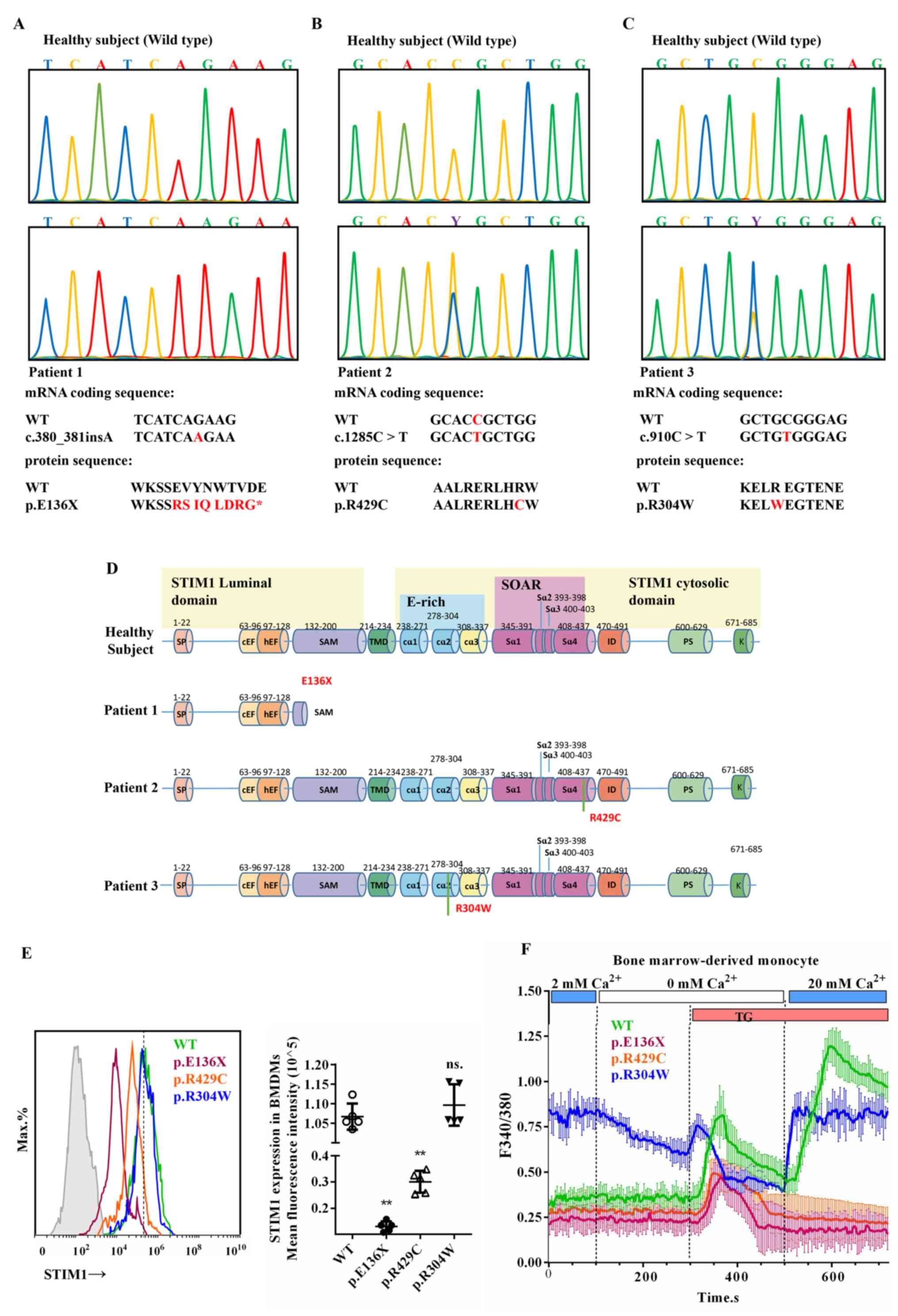 | Figure 1Missense or nonsense mutations in
STIM1 cause disorder of the SOCE in BMDMs. (A-C) Sequence analysis
of genomic STIM1 DNA obtained from (A) subject 1, (B) subject 2,
(C) subject 3 and comparison with the corresponding sequence of the
healthy subject. (D) Linear patterns of the normal and mutated
STIM1 proteins. (E) STIM1 expression in BMDMs from patients with
the mutations STIM1p.E136X, STIM1p.r429c and
STIM1p.r304w, and the healthy subject. (F) The 340/380
nm ratio in Fura-2-loaded BMDMs from patients carrying the
mutations STIM1p.E136X, STIM1p.r429c and
STIM1p.r304w, and the healthy subject. TG was used to
empty the intracellular Ca2+ stores in the absence of
extracellular Ca2+ (0 mM Ca2+) and SOCE was
measured by addition of 20 mM extracellular Ca2+. Values
are expressed as the mean ± standard deviation (n=6).
**P<0.01 vs. WT BMDMs; ns, no significance vs. WT
BMDMs. WT, wild-type; SOCE, store-operated Ca2+ entry;
STIM1, stromal interaction molecule 1; BMDMs, bone marrow-derived
mononuclear macrophages; TG, thapsigargin. SOAR, STIM1-Orai1
activation region. |
STIM1 deficiency causes BMDM
proliferation and differentiation disorders in response to
RANKL/M-CSF
RANKL/M-CSF treatment induced calcium influx in BMDM
cells but did not alter STIM1 expression in BMDMs (Fig. 2A and B), suggestive of SOCE activation. STIM1
mutations cause disorder of RANKL/M-CSF-induced calcium influx in
BMDMs, including elevated basic calcium levels in the
STIM1p.R304W BMDMs, no sensitive calcium influx in
STIM1p.R429C BMDMs and no response in
STIM1p.E136X BMDMs (Fig.
2B). The formation of osteoclasts is the result of the
interaction between M-CSF and RANKL (1). Under the combined induction by
RANKL/M-CSF, STIM1wt BMDMs (Cd11b+)
differentiated into osteoclasts (TRAP+) and fused,
whereas the STIM1p.R429C and STIM1p.E136X
BMDMs exhibited significant reductions in RANKL/M-CSF-induced
osteoclast differentiation, and the STIM1R304W BMDMs
exhibited a significant increase in osteoclastogenesis (Fig. 2C). Similarity, the cell proliferation
activity of the normal BMDMs increased in a time-dependent manner,
whereas the STIM1p.R429C and STIM1p.E136X
BMDMs exhibited a significant decrease and STIM1p.R304W
BMDMs exhibited a significant increase in proliferation in response
to RANKL/M-CSF treatment (Fig. 2D).
CSFE is a nuclear fluorescent dye that indirectly represents the
number of cell divisions. Ki67 is a cell cycle entry marker
molecule that is expressed on all cells in the cell cycle except
for those in G0 phase and indicates the degree of cell
proliferation. The cell division ability and the number of cells
entering the cell cycle were significantly higher in
STIM1wt BMDMs compared with that in
STIM1p.R429C and STIM1p.E136X BMDMs, and
significantly lower compared with that in STIM1p.R304W
BMDMs (Fig. 2E and F), under the combined induction of
RANKL/M-CSF. Cathepsin K and MMP9 process a variety of biologically
active molecules, participate in bone resorption of osteoclasts and
indirectly reflect the bone resorption ability of osteoclasts
(28,29). Cathepsin K and MMP9 process a variety
of biologically active molecules, participate in bone resorption of
osteoclasts and indirectly reflect the osteoclast ability of
osteoclasts (30). Following
stimulation by RANKL/M-CSF, the upregulation of Cathepsin K and
MMP9 expression in STIM1p.R304W cells was more
pronounced compared with that in the WT cells than
STIM1p.R429C and STIM1p.E136X cells.
(Fig. 2G and H). These results indicate that STIM1 is
involved in RANKL/M-CSF-induced osteoclast proliferation,
differentiation and bone resorption.
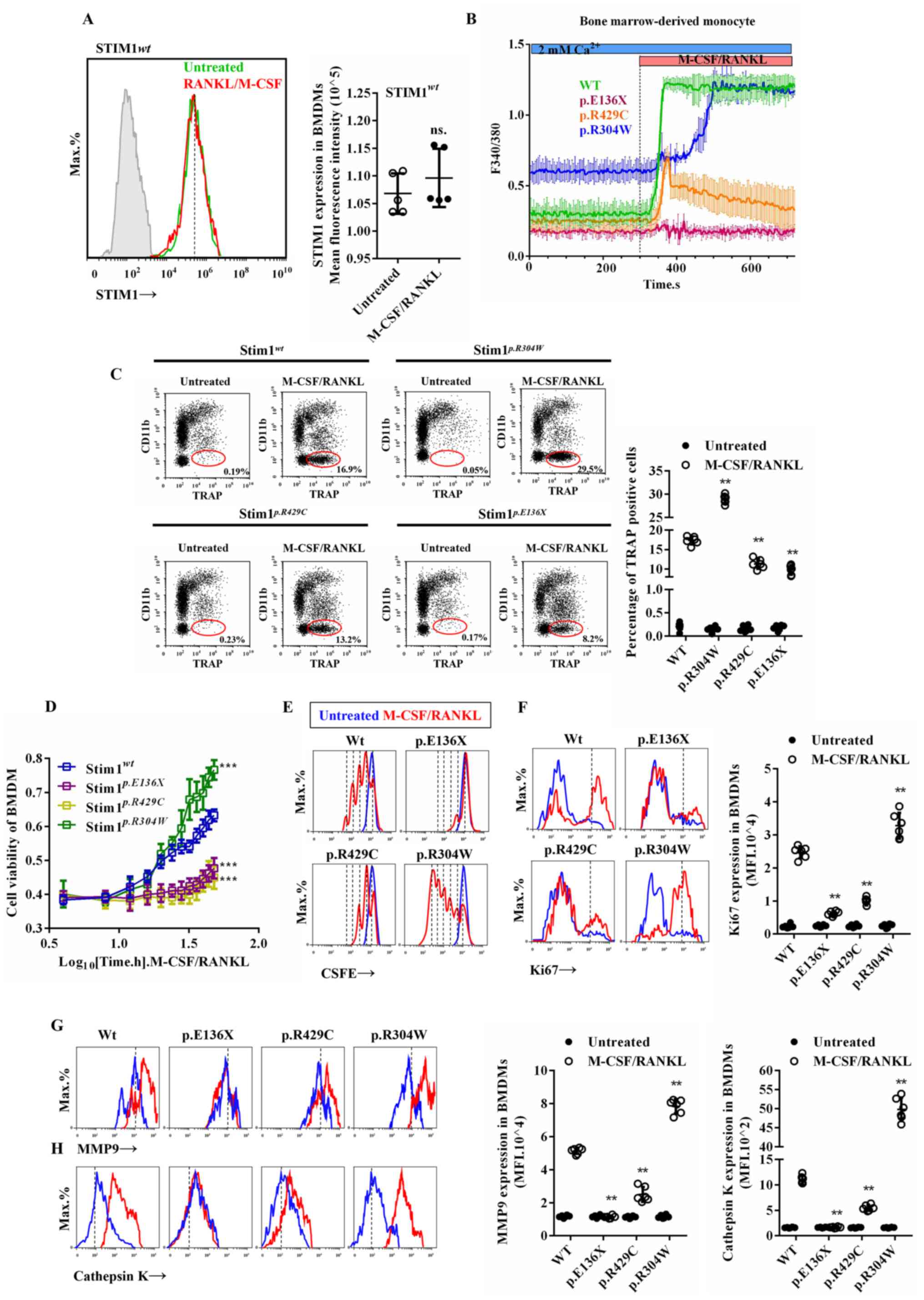 | Figure 2STIM1 deficiency affects BMDM
proliferation and differentiation. (A) Expression of STIM1 in BMDMs
of a healthy subject induced by M-CSF/RANKL. (B) Calcium signals in
response to M-CSF/RANKL in Fura-2-loaded BMDMs from patients with
the mutations STIM1p.E136X,
STIM1p.r429c and STIM1p.r304w, as
well as the healthy subject. (C) The proportion of
Cd11b+ and TRAP+ cells detected by flow
cytometry prior to and after treatment of STIM1wt or
STIM1mut BMDMs with M-CSF/RANKL. (D) Cell proliferation
of BMDMs at different time-points under M-CSF/RANKL treatment. (E
and F) Intensity of (E) the CSFE fluorescence probe and (F) ki67
expression in BMDMs treated with M-CSF/RANKL. (G and H) Expression
levels of (G) cathepsin K and (H) MMP9 following induction with
RANKL/M-CSF. Values are expressed as the mean ± standard deviation
(n=6). **P<0.01 and ***P<0.001 vs. WT
BMDMs or untreated BMDMs. BMDMs, bone marrow-derived mononuclear
macrophages; STIM1, stromal interaction molecule 1; wt, wild-type;
mut, mutant; MMP9, matrix metallopeptidase 9; TRAP,
tartrate-resistant acid phosphatase; RANKL, receptor activator of
NF-κB ligand; M-CSF, macrophage colony-stimulating factor; CSFE,
carboxyfluorescein succinimidyl ester. |
STIM1 defects lead to abnormal
osteoclastogenesis signalling in BMDMs
Calcineurin is a target protein of intracellular
Ca2+ that enters by SOCE and exerts a wide range of
physiological activities (31).
RANKL/M-CSF induced a rapid increase in calcineurin activity in
normal BMDMs, a comparatively greater increase in
STIM1p.R304W BMDMs, a weaker increase in
STIM1p.R429C and no response in
STIM1p.E136X BMDMs (Fig. 3A). In the BMDMs from all subjects,
calcineurin activity was inhibited by CsA (Fig. 3A), an immunosuppressant that targets
the Ca2+- and calmodulin-regulated phosphatase
calcineurin and inhibits the activation of the Akt/mTOR signaling
pathway (29). The Akt/mTOR pathway
is essential for osteoclast development. Next, the effect of SOCE
on RANKL/M-CSF-induced Akt/mTOR activity was explored. Due to the
limited amount of precious cell samples, flow cytometry was used to
detect the phosphorylation levels of intracellular protein kinases.
The mTORS2448, AktS473 and AktT308
phosphorylation levels were significantly increased in the
STIM1wt BMDMs following induction by RANKL/M-CSF
(Fig. 3B-D). The Akt/mTOR activation
levels were significantly higher in the STIM1p.R304W
BMDMs and lower in the STIM1p.R429C BMDMs than those in
the STIM1wt BMDMs, while the Akt/mTOR signal of
STIM1p.E136X BMDMs did not respond to
RANKL/M-CSF-induced activation (Fig.
3B-D). The phosphorylation level of p70 S6k, which is a
substrate of mTOR, was significantly increased by RANKL induction.
RANKL/M-CSF-induced activation of p70 S6k in STIM1mut
BMDMs was consistent with the activation of Akt/mTOR signaling
(Fig. 3E). Transcription factor
NFATC2 is highly phosphorylated in resting cells, while
dephosphorylation occurs with intracellular calcium-dependent
calcineurin activation, followed by translocation to the nucleus
(32). RANKL/M-CSF may induce
dephosphorylation of STIM1wt BMDMs at
NFATC2Ser172, and the p-NFATC2 levels were significantly
downregulated in the STIM1p.R304W and upregulated in the
STIM1p.R429C BMDMs compared with those in the
STIM1wt BMDMs, while the level of NFATC2 phosphorylation
in STIM1p.E136X BMDMs was not altered following
treatment with RANKL/M-CSF (Fig. 3F
and G). To verify the effect of
calcineurin/Akt/mTOR/NFATC2 signalling on RANKL/M-CSF-induced
proliferation, the STIM1wt BMDMs were treated with the
calcineurin inhibitor CsA, the mTOR inhibitor Rap and
NFATC2-interfering RNA. The interference efficiency of NFATC2 siRNA
is provided in Supplementary Fig.
S1A-C. The results indicated that CsA, Rap and NFATC2 siRNA
inhibited cell division and cell cycle entry, and reduced the
expression of Ki67, cathepsin K and MMP9 (Fig. 3H). Similarly, CsA, Rap and NFATC2
siRNA inhibited the RANKL/M-CSF-induced proliferative activity and
osteoclastogenesis (Fig. 3I and
J). These results indicate that the
calcineurin/Akt/mTOR/NFATC2 pathway is involved in SOCE-controlled
cell proliferation, differentiation and osteoclastogenesis.
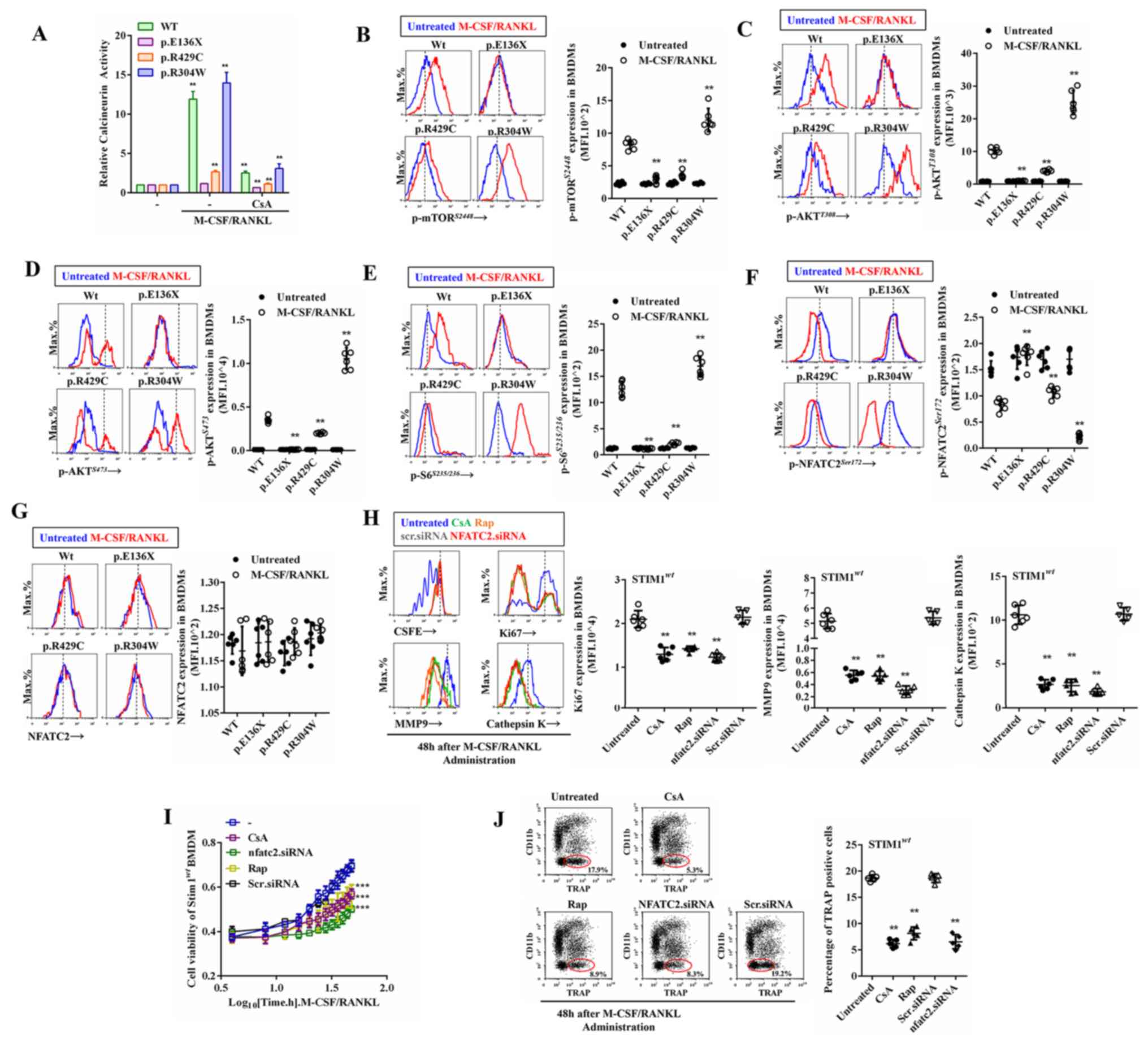 | Figure 3Calcineurin/Akt/mTOR/NFATC2
signalling controlled by store-operated Ca2+ entry. (A)
M-CSF/RANKL-induced calcineurin activity in BMDMs in the presence
or absence of CsA. (B-G) Phosphorylation levels of (B)
mTORS2448, (C) AktT308, (D)
AktS473, (E)
p70S6S235/236 and
dephosphorylation levels of (F) NFATC2 and (G) total NFATC2 were
detected by flow cytometry prior to and after treatment of
STIM1wt or STIM1mut BMDMs with M-CSF/RANKL.
(H) The CSFE fluorescence intensity, as well as the expression of
ki67, cathepsin K and MMP9 in STIM1wt or
STIM1mut BMDMs were detected prior to and after
treatment with M-CSF/RANKL in the presence or absence of 7 nM CsA,
0.1 nM Rap or NFATC2 siRNA. (I) Proliferation and (J)
differentiation of STIM1wt or STIM1mut BMDMs
detected prior to and after treatment with M-CSF/RANKL in the
presence or absence of 7 nM CsA, 0.1 nM Rap or NFATC2 siRNA. Values
are expressed as the mean ± standard deviation (n=6).
**P<0.01 and ***P<0.001 vs. Untreated
BMDMs. BMDMs, bone marrow-derived mononuclear macrophages; CSFE,
carboxyfluorescein succinimidyl ester; STIM1, stromal interaction
molecule 1; wt, wild-type; mut, mutant; MMP, matrix
metallopeptidase; TRAP, tartrate-resistant acid phosphatase; RANKL,
receptor activator of NF-κB ligand; M-CSF, macrophage
colony-stimulating factor; siRNA, small interfering RNA; CsA,
cyclosporin A; Rap, rapamycin; p-Akt, phosphorylated Akt. |
Lentiviral transfection of STIM1 is
not always able to restore disorders in osteoclastogenesis caused
by its defects in vitro
Lentiviral overexpression of plasmid expressing wild
type STIM1 (ca.STIM1) was performed in an attempt to restore the
RANKL/M-CSF-induced disorders of osteoclastogenesis caused by of
STIM1 mutation (Fig. 4A,
Supplementary Fig. S1C-D and
F). Transfection of
STIM1wt into BMDMs with caSTIM1 did not affect the
amplitude and threshold of the calcium influx. This is because the
upper limit of the calcium influx peak depends on the concentration
of calcium stores in the ER and not the level of STIM1 expression
(27). Transfection of ca.STIM1
restored calcium influx in STIM1p.R429C and
STIM1p.E136X BMDMs in response to
RANKL/M-CSF, but was not able to reverse the constitutive
activation of SOCE in STIM1p.R304W BMDMs (Fig. 4B). This was most probably due to the
residual constitutively active STIM1p.R304W protein.
Regarding cell viability, caSTIM1 did not affect the proliferative
activity of wild-type cells but restored the RANLK/M-CSF-induced
proliferative disorder caused by the STIM1p.R429C and
STIM1p.E136X mutations; however, caSTIM1 did not restore
the proliferative disorder caused by the STIM1p.R304W
mutation (Fig. 4C). In terms of cell
differentiation, caSTIM1 did not affect
STIM1p.R304W BMDMs but restored the impaired
capacity of the STIM1p.R429C and STIM1p.E136X
mutant cells to differentiate upon induction by RANLK/M-CSF
(Fig. 4D). Although lentiviral
transfection of caSTIM1 failed to restore the osteoclastogenesis
capacity of STIM1p.R304W cells,
inhibitors of calcineurin/Akt/mTOR signaling inhibited excessive
differentiation and proliferation induced by RANLK/M-CSF in
vitro (Fig. 4E and F). In addition, NFATC2 siRNA inhibited
excessive differentiation and proliferation of
STIM1p.R304W BMDMs induced by RANLK/M-CSF (Fig. 4E and F). These results confirm the important role
of SOCE in osteoclastogenesis and suggest the possibility of gene
therapy for the treatment of congenital bone diseases.
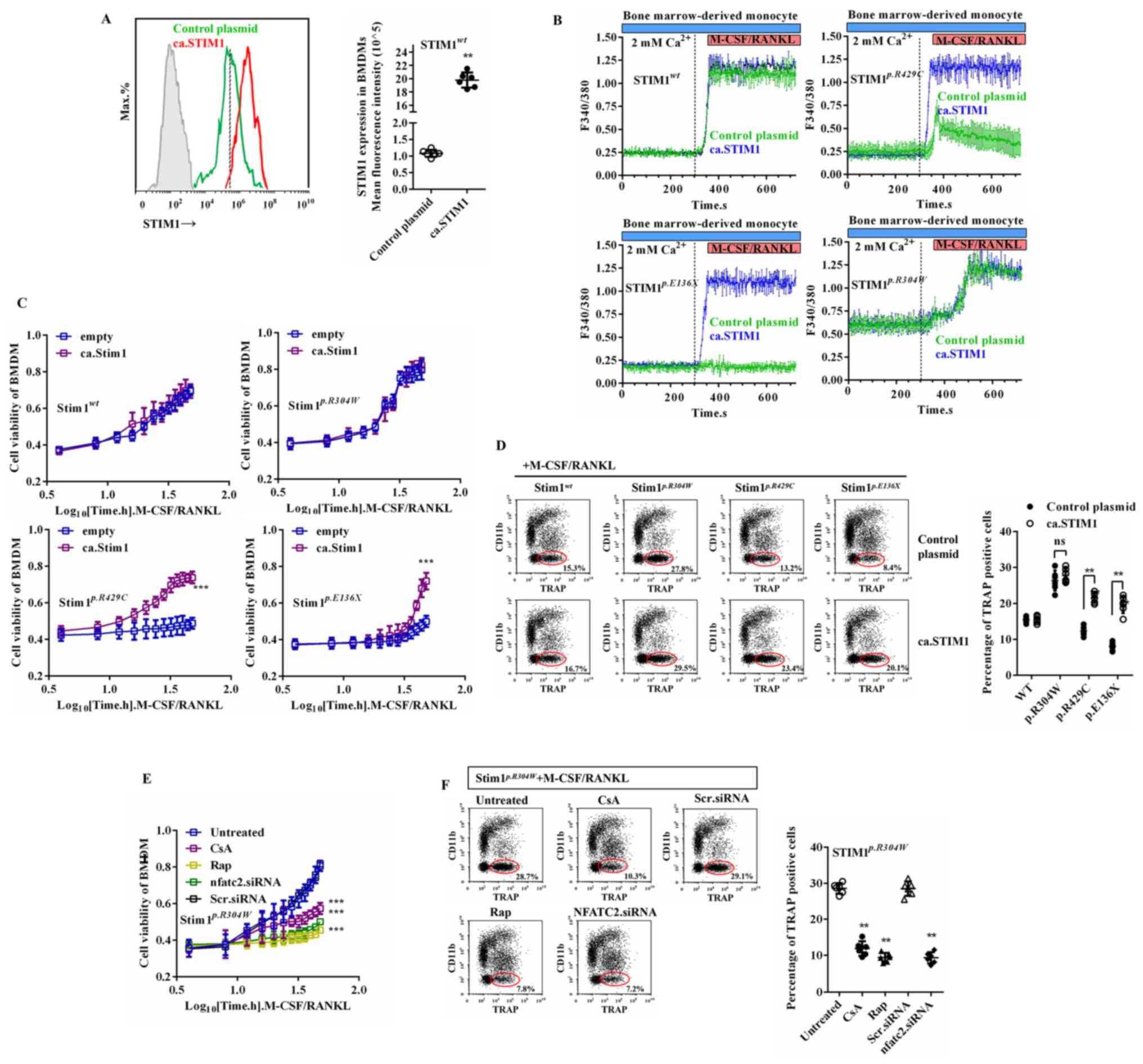 | Figure 4STIM1 overexpression restores
store-operated Ca2+ entry. (A) Reverse transcription
lentiviral transfection efficiency of ca.STIM1 expression vector in
the BMDMs. (B) Ca2+ influx responses in Fura-2-loaded
STIM1wt and STIM1mut BMDMs induced by
M-CSF/RANKL following transfection with an empty plasmid or
ca.STIM1, as measured using the 340/380 nm ratio. (C) Proliferation
and (D) differentiation of the STIM1wt and
STIM1mut BMDMs induced by M-CSF/RANKL following
transfection with an empty plasmid or ca.STIM1. (E) Proliferation
and (F) differentiation of the STIM1p.R304W BMDMs induced by
M-CSF/RANKL following treatment with CsA, Rap and transfection with
NFATC2 siRNA. Values are expressed as the mean ± standard deviation
(n=6). **P<0.01 and ***P<0.001 vs.
Control plasmid or Untreated BMDMs; ns, no significance. BMDMs,
bone marrow-derived mononuclear macrophages; STIM1, stromal
interaction molecule 1; wt, wild-type; mut, mutant; MMP, matrix
metallopeptidase; TRAP, tartrate-resistant acid phosphatase; RANKL,
receptor activator of NF-κB ligand; M-CSF, macrophage
colony-stimulating factor; siRNA, small interfering RNA; CsA,
cyclosporin A; Rap, rapamycin. |
Discussion
Ca2+ mediates a variety of cellular
physiological functions by transmitting intracellular signals.
Activation of membrane-receptor-coupled phospholipase Cγ produces
inositol-1,4,5-trisphosphate, which binds to receptors on the ER
membrane and stimulates Ca2+ excretion into the ER,
which triggers a wide range of physiological effects. After calcium
storage in the ER is depleted, the cells activate ion channels to
replenish intracellular and ER calcium; this process is called SOCE
(33,34). ORAI and STIM are the two most
important components of the SOCE process. STIM on the ER membrane
pulls the ER membrane to the plasma membrane after Ca2+
storage failure (35); the
Ca2+ release-activated Ca2+ channel (CRAC) is
formed by binding of STIM to the transmembrane channel protein ORAI
on the membrane, and the opening of CRAC leads to SOCE (36-38).
Therefore, SOCE is a calcium influx mode activated by the status of
the ER calcium ion concentration. Ca2+ entry into the
intracellular phase through SOCE may last from several minutes to
several hours and drives a wide range of cellular functions,
including secretion, gene transcription, cell contraction and
phagocytosis (39). The disease
caused by impaired SOCE function is called CRAC channel disease
(40).
STIM1 is a dimeric transmembrane protein of the ER
and the gene encoding STIM1 has 12 exons. A single insertion of an
adenine nucleotide mutation at 380-381 leads to early termination
of translation, resulting in the p.E136X nonsense mutant protein,
which is characterized by protein truncation (26). Substitution mutations at positions
910 and 1,285 produce the p.R304W (27) and p.R429C (26) missense protein mutations, the former
of which is associated with normal STIM1 expression, while the
latter affects STIM1 expression. Due to loss of STIM1 expression or
conformational changes, the normal function of SOCE of BMDMs was
disrupted, including the abolition of SOCE caused by the p.E136X
and p.R429C mutations and the sustained activation of SOCE
associated with the p.R304W mutation. RANKL/M-CSF may induce stable
and continuous calcium influx in BMDMs of healthy subjects. By
contrast, STIM1p.R429C BMDMs only had a small amount of
transient calcium influx, while STIM1p.E136X
BMDMs did not respond to induction of calcium influx by
RANKL/M-CSF. The basal intracellular concentration of quiescent
STIM1p.R304W BMDMs was significantly
increased and peaked rapidly under induction with RANKL/M-CSF. In
spite of having the same STIM1 mutation, these three patients had
opposite bone metabolic diseases, including osteosclerosis in those
with the p.E136X and p.R429C mutations and bone loss in the patient
with the p.R304W mutation. These studies observed the possible
effects of STIM1 mutations on abnormal bone metabolism in CID
patients.
M-CSF promotes the proliferation and survival of
osteoclast precursor cells through its receptor M-CSFR via
phosphoinositide 3-kinase (PI3K)/Akt signalling (41,42). The
RANKL-mediated NF-κB, MAPK and PI3K/Akt signalling pathways are
required to activate NFATC2 in osteoclasts (43). Therefore, RANKL/M-CSF synergistically
promotes osteoclast survival and differentiation. The
differentiation of BMDMs from healthy subjects into
TRAP+ cells increased by about 17% under induction by
RANKL/M-CSF, the p.R429C mutation significantly reduced
TRAP+ cell production, whereas the p.E136X mutation was
associated with only a minor response to RANKL/M-CSF induction. By
contrast, p.R304W mutation led to production of a large amount of
TRAP+ cells under the induction of RANKL/M-CSF.
Following RANKL/M-CSF induction, the cell viability, cell cycle
entry and cell division abilities of the STIM1mut BMDMs
were different from those of the healthy control cells. The
STIM1p.R429C BMDMs had a reduced proliferate tendency,
while that of the STIM1p.R304W BMDMs was enhanced, and
the STIM1p.E136X BMDMs did not proliferate at all.
Cathepsin K and MMP9 are markers of osteoclastogenesis and
functional enzymes that exert osteolysis. RANKL/M-CSF-induced
cathepsin K and MMP9 expression were different between the
STIM1mut and STIM1wt BMDMs, including reduced
expression in STIM1p.R429C BMDMs, increased expression
in STIM1p.R304W BMDMs and unaffected in
STIM1p.E136X BMDMs. These results suggest that the
absence of functional STIM1 affects survival, osteoclastogenesis
and function of BMDMs.
mTOR is an atypical serine/threonine kinase that
exists in two different mTOR complexes. mTOR complex 1, which is
composed of mTOR, raptor, GβL and DEPTOR, is inhibited by rapamycin
(44). Studies of RANKL-induced
osteoclast differentiation have clearly confirmed that these cells
undergo regulation of calcineurin activity and that negative
feedback regulates NFATC2-mediated osteoclastogenesis and bone
resorption (28). In the present
study, the calcineurin activity of the STIM1mut BMDMs
was different from that of the STIM1wt BMDMs in presence
of RANKL/M-CSF: That of STIM1p.R429C BMDMs was weakened,
that of STIM1p.R304W BMDMs was enhanced and that of
STIM1p.E136X was not affected. Similarly, the
RANKL/M-CSF-induced phosphorylation levels of mTORS2448
and the mTOR substrate p70
S6S235/236 were significantly
higher in the STIM1p.R304W BMDMs than those in the
STIM1wt BMDMs, and the phosphorylation levels of
AktS473 and AktT308 were also significantly
higher than those in the STIM1p.R304W BMDMs. Attenuated
Akt/mTOR-S6 signaling was observed in STIM1p.R429C
BMDMs, while no response activation of these signals was observed
in STIM1p.E136X BMDMs. in addition, the
RANKL/M-CSF-induced dephosphorylation levels of NFATC2 were
significantly higher in the STIM1p.R304W
BMDMs and lower in the STIM1p.R429C BMDMs than in the
STIM1wt BMDMs, and no response dephosphorylation of
NFATC2 was observed in STIM1p.E136X BMDMs. Calcineurin
inhibitor CsA and mTOR inhibitors rapamycin and NFATC2 siRNA
inhibited RANKL/M-CSF-induced osteoclastogenesis of BMDMs. This
result directly indicates that NFATC2 is involved in
osteoclastogenesis disorders caused by SOCE deficiency.
Finally, STIM1-deficient BMDMs were transfected with
STIM1 overexpression plasmids. SOCE-dependent calcium influx does
not depend on the expression level of STIM1 or ORAI1; rather, it
depends on whether the ER Ca2+ is ‘storage-depleted’.
M-CSF/RANKL stimulates intracellular signal activation and consumes
ER Ca2+. When the Ca2+ stored in the ER is
consumed, the Ca2+ concentration sensor STIM1 on the ER
and the Ca2+ channel STIM1 on the cytoplasmic membrane
are physically combined, thereby causing Ca2+ influx.
Since STIM1 in patients with p.E136X and p.R429C mutations was
completely inactivated, SOCE-Ca2+ influx did not occur
in response to M-CSF/RANKL-induced activation. Of note,
transfection of caSTIM1 for overexpression was able to restore
normal SOCE in these cells. At the same time, the SOCE channel in
STIM1p.R304W BMDMs is continuously activated, so the
basic intracellular Ca2+ concentration is higher than
that of normal cells. However, as the STIM1 still carried a
conformational mutation, caSTIM1 cannot restore normal SOCE in
STIM1p.R304W BMDMs. In addition, the RANKL/M-CSF-induced
persistent stable calcium influx, and the proliferation and
differentiation into osteoclasts that were reduced due to p.E136X
and p.R429C mutation-associated STIM1 deficiency were restored
in vitro. In turn, CsA, Rap and NFATC2 siRNA were able to
recover the RANKL/M-CSF-induced proliferation and differentiation
that were reduced by p.E136X and p.R429C mutation-associated STIM1
deficiency. These results suggest the potential ability of gene
therapy to treat congenital bone loss or osteopetrosis.
In conclusion, the present study confirmed that
SOCE was involved in the survival, differentiation and function of
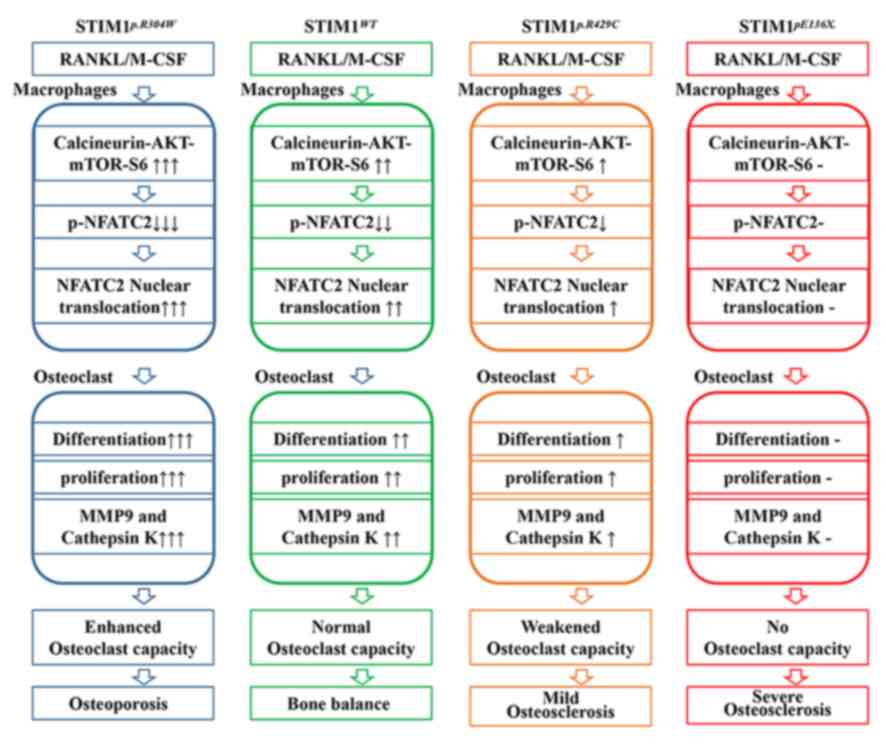
osteoclasts. As presented in Fig. 5,
different mutations in STIM1 affect the function of SOCE in
different ways, which leads to abnormalities of
calcineurin/Akt/mTOR/S6 phosphorylation and NFATC2
dephosphorylation, and results in aberrant differentiation,
proliferation and osteoclast differentiation of BMDMs, which leads
to different types of bone metabolic disorder.
Supplementary Material
Figure S1. (A‑C) Knockdown and
overexpression/transfection efficiency of NFATC2 siRNA and ca.STIM1
expression plasmid, respectively. (A) Expression of NFATC2 mRNA in
293T cells transfected with 1, 2 or 4 μg NFATC2 siRNA. (B)
Expression of NFATC2 protein in 293T cells transfected with 4 μg
NFATC2 siRNA. (C) Fluorescence intensity in 293T cells transfected
with RFP‑labeled ca.STIM1 plasmid (1, 2, 4, 8 or 16 μg). (D)
Relative STIM1 mRNA expression in 293T cells transfected with
ca.STIM1 plasmid (1, 2, 4, 8 or 16 μg). (E) NFATC2 protein
expression after transfection of STIM1wt and
STIM1p.R304W cells with NFATC2 siRNA. (F) STIM1 protein
expression after transfection of STIM1p.R304W,
STIM1p.R429c and STIM1p.E136X cells with
ca.STIM1. Values are expressed as the mean ± standard deviation
(n=3). *P<0.05, **P<0.01 vs. Control plasmid or Scrambled
siRNA. STIM1, stromal interaction molecule 1; siRNA, small
interfering RNA; RFP, red fluorescence protein; ca.STIM1, plasmid
expressing wild type STIM1.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Key Research
Project of Wannan Medical College (grant no. WK2017Z07) and the
National Natural Science Foundation of China (grant no.
81800082).
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
YJH and QL were a major contributor in acquisition
of data, ZYF was a major contributor in analysis and interpretation
of data, LRZ was a major contributor in conception, design and
writing the manuscript. All authors read and approved the final
manuscript.
Ethical approval and consent to
participate
The experiments involving human subjects were based
on the Declaration of Helsinki and the European Declaration of
Human Rights. The study was approved by the Ethical Review
Committee of Yanjishan Hospital of Wannan Medical College (Wuhu,
China; no. 201627).
Patient consent for publication
Informed consent was obtained from the parents of
the patients and the healthy donor.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Boyle WJ, Simonet WS and Lacey DL:
Osteoclast differentiation and activation. Nature. 423:337–342.
2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Harada S and Rodan GA: Control of
osteoblast function and regulation of bone mass. Nature.
423:349–355. 2003.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zaidi M: Skeletal remodeling in health and
disease. Nat Med. 13:791–801. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Kobayashi E and Setsu N: Osteosclerosis
induced by denosumab. Lancet. 7(539)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G,
Huang J, Dai W, Li C, Zheng C, et al: LGR4 is a receptor for RANKL
and negatively regulates osteoclast differentiation and bone
resorption. Nat Med. 22:539–546. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fu J, Li S, Feng R, Ma H, Sabeh F, Roodman
GD, Wang J, Robinson S, Guo XE, Lund T, et al: Multiple
myeloma-derived MMP-13 mediates osteoclast fusogenesis and
osteolytic disease. J Clin Invest. 126:1759–1772. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Okamoto K, Nakashima T, Shinohara M,
Negishi-Koga T, Komatsu N, Terashima A, Sawa S, Nitta T and
Takayanagi H: Osteoimmunology: The conceptual framework unifying
the immune and skeletal systems. Physiol Rev. 97:1295–1349.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen X, Zhi X, Cao L, Weng W, Pan P, Hu H,
Liu C, Zhao Q, Zhou Q, Cui J and Su J: Matrine derivate MASM
uncovers a novel function for ribosomal protein S5 in
osteoclastogenesis and postmenopausal osteoporosis. Cell Death Dis.
8(e3037)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Suematsu A, Nakashima T, Takemoto-Kimura
S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T,
Kodama T, et al: Regulation of osteoclast differentiation and
function by the CaMK-CREB pathway. Nat Med. 17:1410–1416.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Ortuño MJ, Robinson ST, Subramanyam P,
Paone R, Huang YY, Guo XE, Colecraft HM, Mann JJ and Ducy P:
Serotonin-reuptake inhibitors act centrally to cause bone loss in
mice by counteracting a local anti-resorptive effect. Nat Med.
22:1170–1179. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xu S, Zhang Y, Wang J, Li K, Tan K, Liang
K, Shen J, Cai D, Jin D, Li M, et al: TSC1 regulates osteoclast
podosome organization and bone resorption through mTORC1 and
Rac1/Cdc42. Cell Death Differ. 25:1549–1566. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hoefle G, Holzmueller H and Drexel H:
Alendronate versus calcitriol for prevention of bone loss after
cardiac transplantation. N Engl J Med. 350:2306–2308.
2004.PubMed/NCBI
|
|
13
|
Janssen NM and Genta MS: The effects of
immunosuppressive and anti-inflammatory medications on fertility,
pregnancy, and lactation. Arch Intern Med. 160:610–619.
2000.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Huynh H, Wei W and Wan Y: mTOR inhibition
subdues milk disorder caused by maternal VLDLR loss. Cell Rep.
19:2014–2025. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhou C, You Y, Shen W, Zhu YZ, Peng J,
Feng HT, Wang Y, Li D, Shao WW, Li CX, et al: Deficiency of sorting
nexin 10 prevents bone erosion in collagen-induced mouse arthritis
through promoting NFATc1 degradation. Ann Rheum Dis. 75:1211–1218.
2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Takayanagi H, Kim S, Koga T, Nishina H,
Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al:
Induction and activation of the transcription factor NFATc1 (NFAT2)
integrate RANKL signaling in terminal differentiation of
osteoclasts. Dev Cell. 3:889–901. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kim H, Kim T, Jeong BC, Cho IT, Han D,
Takegahara N, Negishi-Koga T, Takayanagi H, Lee JH, Sul JY, et al:
Tmem64 modulates calcium signaling during RANKL-mediated osteoclast
differentiation. Cell Metab. 17:249–260. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Masuyama R, Vriens J, Voets T, Karashima
Y, Owsianik G, Vennekens R, Lieben L, Torrekens S, Moermans K,
Vanden Bosch A, et al: TRPV4-mediated calcium influx regulates
terminal differentiation of osteoclasts. Cell Metab. 8:257–265.
2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hwang SY and Putney JW: Orai1-mediated
calcium entry plays a critical role in osteoclast differentiation
and function by regulating activation of the transcription factor
NFATc1. FASEB J. 26:1484–1492. 2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Crabtree NJ, Shaw NJ, Bishop NJ, Adams JE,
Mughal MZ, Arundel P, Fewtrell MS, Ahmed SF, Treadgold LA, Högler
W, et al: Amalgamated reference data for size-adjusted bone
densitometry measurements in 3598 children and young adults-the
ALPHABET study. J Bone Miner Res. 32:172–180. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Guo B, Xu Y, Gong J, Tang Y and Xu H: Age
trends of bone mineral density and percentile curves in healthy
Chinese children and adolescents. J Bone Miner Metab. 31:304–314.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Notredame C, Higgins DG and Heringa J:
T-Coffee: A novel method for fast and accurate multiple sequence
alignment. J Mol Biol. 302:205–217. 2000.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Huang Z, Ruan HB, Xian L, Chen W, Jiang S,
Song A, Wang Q, Shi P, Gu X and Gao X: The stem cell factor/Kit
signalling pathway regulates mitochondrial function and energy
expenditure. Nat Commun. 5(4282)2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Misceo D, Holmgren A, Louch WE, Holme PA,
Mizobuchi M, Morales RJ, De Paula AM, Stray-Pedersen A, Lyle R,
Dalhus B, et al: A dominant STIM1 mutation causes Stormorken
syndrome. Hum Mutat. 35:556–564. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Picard C, Mccarl CA, Papolos A, Khalil S,
Lüthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, et
al: STIM1 mutation associated with a syndrome of immunodeficiency
and autoimmunity. N Engl J Med. 360:1971–1980. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Maus M, Jairaman A, Stathopulos PB, Muik
M, Fahrner M, Weidinger C, Benson M, Fuchs S, Ehl S, Romanin C, et
al: Missense mutation in immunodeficient patients shows the
multifunctional roles of coiled-coil domain 3 (CC3) in STIM1
activation. Proc Natl Acad Sci USA. 112:6206–6211. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Morin G, Bruechle NO, Singh AR, Knopp C,
Jedraszak G, Elbracht M, Brémond-Gignac D, Hartmann K, Sevestre H,
Deutz P, et al: Gain-of-function mutation in STIM1 (P.R304W) is
associated with Stormorken syndrome. Hum Mutat. 35:1221–1232.
2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vaeth M, Maus M, Klein-Hessling S,
Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E,
Berberich-Siebelt F, et al: Store-operated Ca2+ entry
controls clonal expansion of T cells through metabolic
reprogramming. Immunity. 47:664–679.e6. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li CH, Yang ZF, Li ZX, Ma Y, Zhang L,
Zheng C, Qiu W, Wu X, Wang X, Li H, et al: Maslinic acid suppresses
osteoclastogenesis and prevents ovariectomy-induced bone loss by
regulating RANKL-mediated NF-κB and MAPK signaling pathways. J Bone
Miner Res. 26:644–656. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ohta K, Naruse T, Ishida Y, Shigeishi H,
Nakagawa T, Fukui A, Nishi H, Sasaki K, Ogawa I and Takechi M:
TNF-α-induced IL-6 and MMP-9 expression in immortalized
ameloblastoma cell line established by hTERT. Oral Dis. 23:199–209.
2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Huynh H and Wan Y: mTORC1 impedes
osteoclast differentiation via calcineurin and NFATc1. Commun Biol.
1(29)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hogan PG, Chen L, Nardone J and Rao A:
Transcriptional regulation by calcium, calcineurin, and NFAT. Genes
Dev. 15:2205–2232. 2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Cahalan MD: STIMulating store-operated
Ca(2+) entry. Nat Cell Biol. 11:669–677. 2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lewis RS: The molecular choreography of a
store-operated calcium channel. Nature. 446:284–287.
2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Roos J, DiGregorio PJ, Yeromin AV, Ohlsen
K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD,
et al: STIM1, an essential and conserved component of
store-operated Ca2+ channel function. J Cell Biol. 169:435–445.
2005.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Feske S, Gwack Y, Prakriya M, Srikanth S,
Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M and Rao A: A
mutation in Orai1 causes immune deficiency by abrogating CRAC
channel function. Nature. 441:179–185. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Vig M, Peinelt C, Beck A, Koomoa DL, Rabah
D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R and
Kinet JP: CRACM1 is a plasma membrane protein essential for
store-operated Ca2+ entry. Science. 312:1220–1223. 2006.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhang SL, Yeromin AV, Zhang XH, Yu Y,
Safrina O, Penna A, Roos J, Stauderman KA and Cahalan MD:
Genome-wide RNAi screen of Ca(2+) influx identifies genes that
regulate Ca(2+) release-activated Ca(2+) channel activity. Proc
Natl Acad Sci USA. 103:9357–9362. 2006.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Feske S, Skolnik EY and Prakriya M: Ion
channels and transporters in lymphocyte function and immunity. Nat
Rev Immunol. 12:532–547. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Parekh AB: Store-operated CRAC channels:
Function in health and disease. Nat Rev Drug Discov. 9:399–410.
2010.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wang L, Iorio C, Yan K, Yang H, Takeshita
S, Kang S, Neel BG and Yang W: A ERK/RSK-mediated negative feedback
loop regulates M-CSF-evoked PI3K/AKT activation in macrophages.
FASEB J. 32:875–887. 2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pacitto R, Gaeta I, Swanson JA and Yoshida
S: CXCL12-induced macropinocytosis modulates two distinct pathways
to activate mTORC1 in macrophages. J Leukoc Biol. 101:683–692.
2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Walsh MC and Choi Y: Biology of the
RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol.
5(511)2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Dowling RJ, Topisirovic I, Fonseca BD and
Sonenberg N: Dissecting the role of mTOR: Lessons from mTOR
inhibitors. Biochim Biophys Acta. 1804:433–439. 2010.PubMed/NCBI View Article : Google Scholar
|