Introduction
The excitatory neurotransmitter glutamate is
involved in the pathophysiology of certain neurological disorders,
as well as in neuron loss, by increasing cell component damage,
including mitochondrial dysfunction (1). Glutamate excitotoxicity is also
considered a major mechanism underlying neuronal death, leading to
neurodegeneration, as occurring in hypoxia, ischemia, traumas and
chronic neurodegenerative disorders (2). It has been indicated that glutamate
mediates excitotoxicity in primary cultured neurons by stimulating
the N-methyl-d-aspartate receptor, which leads to increased calcium
permeability and formation of reactive oxygen species (ROS), and
the release of lysosomal enzymes (3,4).
Oxidative stress is known to be involved in several human
neuropathologies, including acute hypoxia-ischemia/reperfusion and
chronic neurodegenerative disorders, e.g. Parkinson's and
Alzheimer's diseases (5,6). Following acute glutamate
administration, increased intracellular ROS accumulation,
Ca+2 levels, production of peroxynitrite and depletion
of glutathione were observed in the cerebral cortex of rats
(7). ROS generation is involved in
the pathophysiology of several neuropsychiatric disorders, since
20% of the total amount of oxygen in the body is metabolized by the
brain, and the brain has a limited anti-oxidant capacity (8,9). High
levels of extracellular glutamate, resulting in glutathione
depletion and cell injury, in addition to the inhibition of
glutamate toxicity by several anti-oxidants, including α-tocopherol
and superoxide dismutase (SOD), indicate oxidative glutamate
toxicity. Cultured cortical neurons from mice overexpressing the
free radical-scavenging enzyme SOD were indicated to be resistant
to glutamate toxicity and the involvement of glutathione in
neuro-degeneration was demonstrated (10,11).
Excitotoxic neuronal injury is linked to the generation of free
radicals and glutathione is involved in neurodegeneration (10,11). It
has also been speculated that the principal function of ascorbate
and α-tocopherol as anti-oxidants may be based on a synergistic
effect with glutathione in the central nervous system (10,11).
Valproic acid (VPA), widely used as an
anti-convulsant agent and efficient mood stabilizer, has been
reported to have a utility in the treatment of excitotoxicity in
the hippocampus (12). The
neuroprotective effect and anti-oxidant activity of VPA were
studied in primary cultured rat cerebral cortical cells and chronic
VPA treatment was suggested to inhibit glutamate-induced cell
death, DNA fragmentation, intracellular free calcium imbalance,
lipid peroxidation and protein oxidation (13). VPA directly inhibits histone
deacetylase (HDAC), causing histone hyperacetylation and heat shock
protein (HSP) induction, where HSP induction is correlated with
damage resistance. VPA was indicated to exert a neuroprotective
effect through these mechanisms in the cerebral ischemia model
(14), it was also indicated that
VPA has neuroprotective effects against brain ischemia due to its
anti-inflammatory and anti-oxidant activities, as well as against
HDAC and glycogen synthase kinase 3 (GSK3) inhibition (15,16).
In the present study, the neuroprotective effect of
VPA against oxidative glutamate toxicity was studied in the SH-SY5Y
cell line, which is frequently used as a model for the study of
oxidative stress associated with neuronal death and its
anti-oxidant capacity, by measuring oxidative and anti-oxidant
biochemical parameters.
Materials and methods
Cell culture
The study was performed using SH-SY5Y human
neuroblastoma cells originally obtained from the American Type
Culture Collection and kindly supplied by Dr İbrahim Akalın
(Department of Medical Genetics, İstanbul Medeniyet University,
İstanbul, Turkey). Every effort was made for the cell lines to be
kept pure and free from contamination in the laboratory. The
preventions were taken to avoid contamination; the sampling area,
incubator and water bath was kept clean, the laboratory enviroment
was sanitized, the biological safety cabinet was used for all
procedures, all cell culture equipment, including reagents and
media were sterile, antibiotics were included and sterile water was
used to prevent contamination.
The SH-SY5Y cells were grown in Dulbecco's modified
Eagle's medium (Thermo Fisher Scientific, Inc.) containing 10%
fetal bovine serum (Thermo Fisher Scientific, Inc.), penicillin
(100 unit/ml) and streptomycin (100 µg/ml). The cells were
incubated at 37˚C with 5% CO2. SH-SY5Y cells were seeded
into 96-well plates (1x104 cells/well). To investigate
the effects of L-glutamate and VPA, SH-SY5Y cells were incubated in
complete culture medium for 24 h prior to the addition of
L-glutamate or VPA.
Drug concentrations
Cells were treated with 9 different concentrations
of glutamate (1, 5, 10, 15, 20, 25, 30, 40 and 50 mM; L-glutamate;
cat. no. G1251; Sigma-Aldrich; Merck KGaA) to determine the
glutamate toxicity in the cultured SH-SY5Y cells. The glutamate
concentrations that caused a significant reduction in cell
viability were determined by drawing dose-cell viability curves.
The glutamate concentration of 15 mM that caused a ~20% decrease in
cell viability after 24 h was then used for subsequent experiments.
The cells were challenged with this concentration calculated from
the dose-response experiment for 2 different time intervals (3 and
24 h). Cell viability was determined by MTT assays, as described
below.
SH-SY5Y cells were treated with 1, 5 and 10 mM VPA
(Depakin 400 mg/4 ml; lyophilized powder; Sanofi S.A.) for 2 h
prior to exposure to 15 mM glutamate. The effect of VPA treatment
was tested following 2 different time intervals (3 and 24 h).
Cell viability assay
An MTT (Thermo Fisher Scientific, Inc.) assay was
used to evaluate cell viability. After adding MTT solution (5
mg/ml) to each well, cells were incubated for 3 h with 5%
CO2 at 37˚C. Following the removal of the culture
medium, 200 µl dimethyl sulfoxide was used to dissolve the formazan
product. Absorbance values were measured at 560 nm using a
microplate reader (Multiskan™ GO microplate spectrophotometer;
Thermo Fisher Scientific, Inc.). Cell viability was calculated by
considering the controls as 100%.
Cell lysate preparation
SH-SY5Y cells were harvested by trypsin-EDTA 0.25%
(Thermo Fisher Scientific, Inc.) and collected by centrifugation at
1,000-2,000 x g for 10 min at 4˚C. Cells were then harvested in
ice-cold buffer (0.05 M potassium phosphate pH 7.0, 1 mM EDTA) and
homogenized by sonication on ice. The solution was centrifuged at
12,000 x g at 4˚C for 20 min to remove cell debris. The supernatant
was used for determining the quantity of total protein and for the
enzyme activity assay. The protein concentration was determined
using the bicinchoninic acid assay kit (Thermo Fisher Scientific,
Inc). All spectrophotometric measurements were made using an Epoch
microplate spectrophotometer (BioTek Instruments, Inc.).
Catalase (CAT) activity assay
CAT activity in cell lysates was tested by
spectrophotometrically monitoring the degradation of hydrogen
peroxide (H2O2) for 3 min at 240 nm at 25˚C
in the presence of 3% H2O2 and 0.1 mM EDTA in
0.05 M potassium phosphate buffer with a pH of 7.0.
SOD activity assay
The SOD assay was performed by quantifying the
inhibition of nitro blue tetrazolium (NBT) at 560 nm. The assay
mixture (200 µl) comprised 0.0033 mM riboflavin, 10 mM
L-methionine, 0.033 mM NBT and 0.66 mM EDTA-Na2 in 0.05
M potassium phosphate buffer (pH 7.8). The 96-well plates
containing the assay mixture were incubated for 20 min with 300
nmol/m2/sec at 560 nm excitation at 25˚C. One unit of
SOD activity was defined as the amount of protein (in mg) causing
50% inhibition of photoreduction, following which specific enzyme
activity was expressed as units/mg protein.
Malondialdehyde (MDA) activity
assay
Lipid peroxidation products, including MDA, react
with thiobarbituric acid to form a colored product with an
absorption maximum of 532 nm. The results are expressed as the
molar equivalent of MDA (calculated from the standard curve
prepared with tetraethoxypropane) per mg of protein.
H2O2 activity
assay
H2O2 in cells was quantified
using a H2O2 assay kit (cat no. ab102500;
Abcam). In brief, at 24 h after drug administration, cells were
harvested, homogenized and centrifuged. The supernatant was used
for the assay. The absorbance was detected at 570 nm using a
microplate reader and the optical density was used for
quantification of H2O2 levels. Distilled
water was used as a negative control instead of cell lysate
sample.
DAPI staining
At 24 h after glutamate exposure and/or
pre-treatment with VPA, the cells were fixed with methanol/acetic
acid at a ratio of 3:1 at room temperature for 10 min (17). Cells were then washed twice with PBS,
stained with DAPI (BioShop Canada, Inc.) for 5 min and examined by
fluorescence microscopy (Axio Vert.A1; Carl Zeiss AG).
Apoptosis-associated changes in cellular and nuclear morphology
were examined and a reduced nuclear size, chromatin condensation,
nuclear fragmentation and intense fluorescence were considered to
indicate apoptosis. Cells were imaged using Zen 2,6 Blue Edition
software (Carl Zeiss, AG).
Statistical analysis
Values are expressed as the mean ± standard error of
the mean and analyzed by one-way analysis of variance, followed by
a Bonferroni's multiple-comparisons post-hoc test by Grahpad Prism
8 software (GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Dose-response curve of glutamate and
VPA
A dose-response curve of glutamate was plotted to
determine the toxic concentration of glutamate in SH-SY5Y cells at
3 and at 24 h of exposure. The decrease in cell viability after
exposure to glutamate for 3 h was not significant at the glutamate
concentrations of 1, 5 and 10 mM. The significant decrease in cell
viability started with 15 mM glutamate and decreased progressively
with increasing glutamate concentrations. The progressive decline
in cell viability was also observed at all concentrations of
glutamate (1-50 mM) at 24 h of exposure (Fig. 1A). The glutamate concentration of 15
mM that caused a ~20% decrease in cell viability after 24 h was
then used for the subsequent experiments.
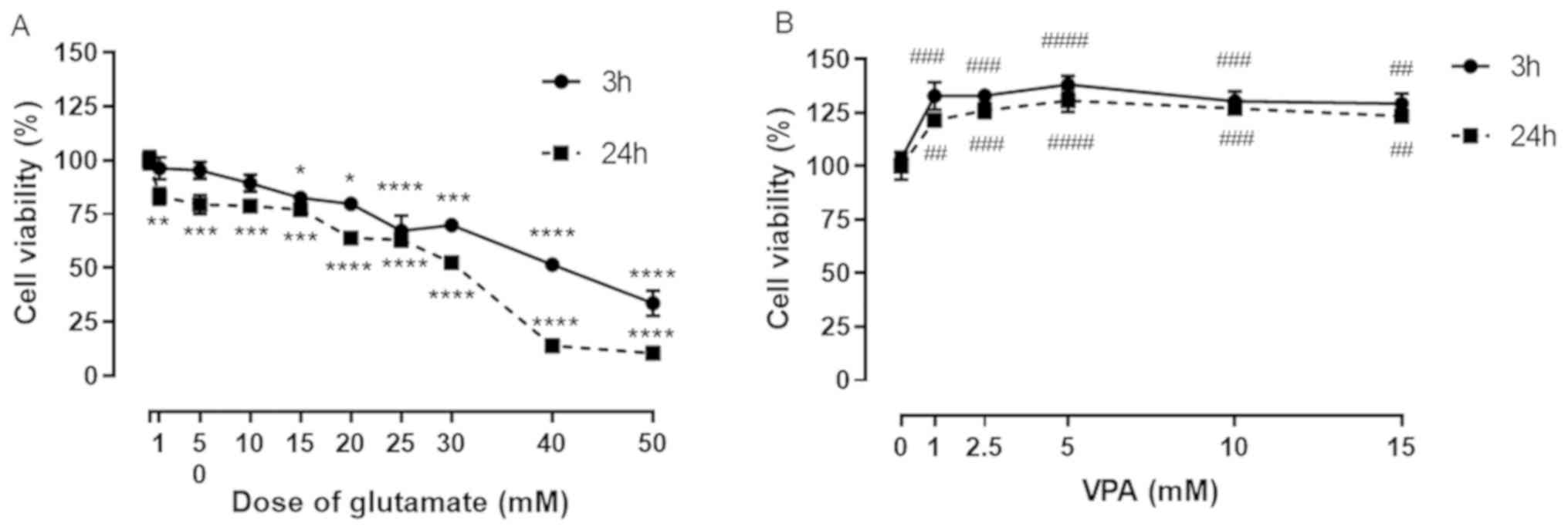 | Figure 1Effect of L-glutamate on SH-SY5Y cell
viability. (A) Graph indicating the glutamate-induced
dose-dependent decrease in cell viability in SH-SY5Y cells as % of
control. Cells were treated with 9 different concentrations of
glutamate (1, 5, 10, 15, 20, 25, 30, 40 and 50 mM). The cell
viability determined after 3 and 24 h of treatment. (B) Graph of
the effect of different VPA concentrations (1-15 mM) on cell
viability at 3 and 24 h after treatment. Cell viability (% of
control) is expressed as the mean value of four separate
experiments (n=8 per condition). *P<0.05,
**P<0.01,
***P<0.001 and
****P<0.0001 vs.
control in A and ##P<0.01, ###P<0.001
and ####P<0.0001 vs. control in B. VPA, valproic
acid. |
VPA treatment alone at concentrations of 1, 2.5, 5,
10 and 15 mM significantly increased the cell viability of SH-SY5Y
cells, when compared to the control, at 3 and 24 h. A significant
increase was observed in cells treated with 1 mM VPA compared with
the control (~130% of control; Fig.
1B), but no further increases could be observed with further
increases in VPA concentration (Fig.
1B).
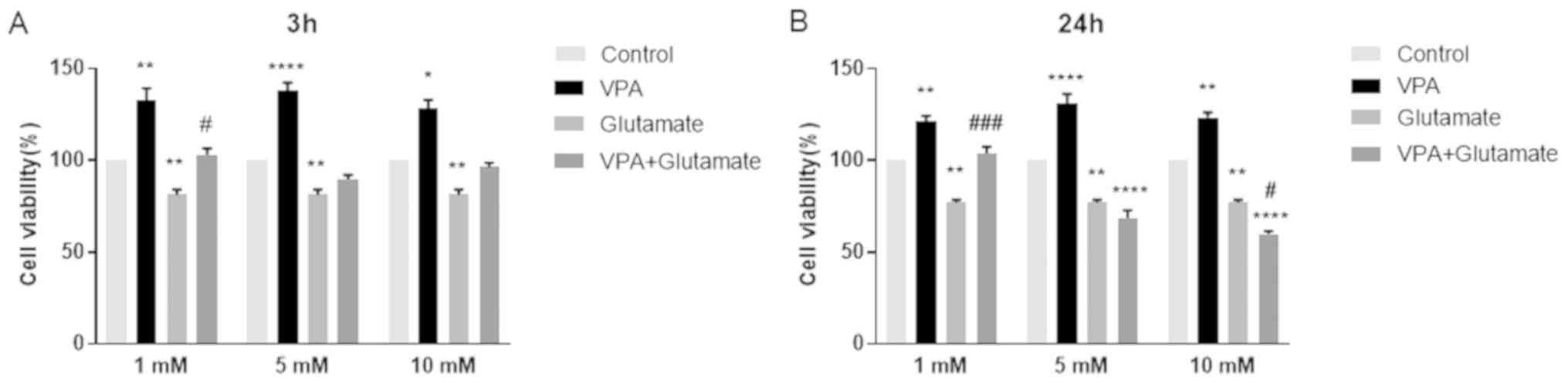
Effect of VPA pre-treatment on the
viability of cells with glutamate-induced excitotoxicity
The effects of different concentrations of VPA (1, 5
and 10 mM) on cell viability, alone and prior to glutamate (15 mM)
exposure for 3 and 24 h, are displayed in Fig. 2.
The viability of glutamate-treated cells was
significantly increased in the group pre-treated with 1 mM VPA when
compared with that of the cells treated with glutamate alone for 3
h. An increase in cell viability at 3 h was also observed after
pre-treatment with 5 and 10 mM VPA; however, these changes were
insignificant (Fig. 2A). Of note,
pre-treatment with 1 mM VPA significantly increased the viability
of cells exposed to glutamate for 24 h (Fig. 2B). However, 5 and 10 mM VPA
concentrations were ineffective in reducing glutamate-induced
excitotoxicity; conversely, they further decreased the viability of
cells after glutamate exposure for 24 h. Thus, the concentration of
1 mM VPA was used in the further experiments to investigate the
potential anti-oxidant effect of VPA in treating glutamate-injured
neurons (Fig. 2).
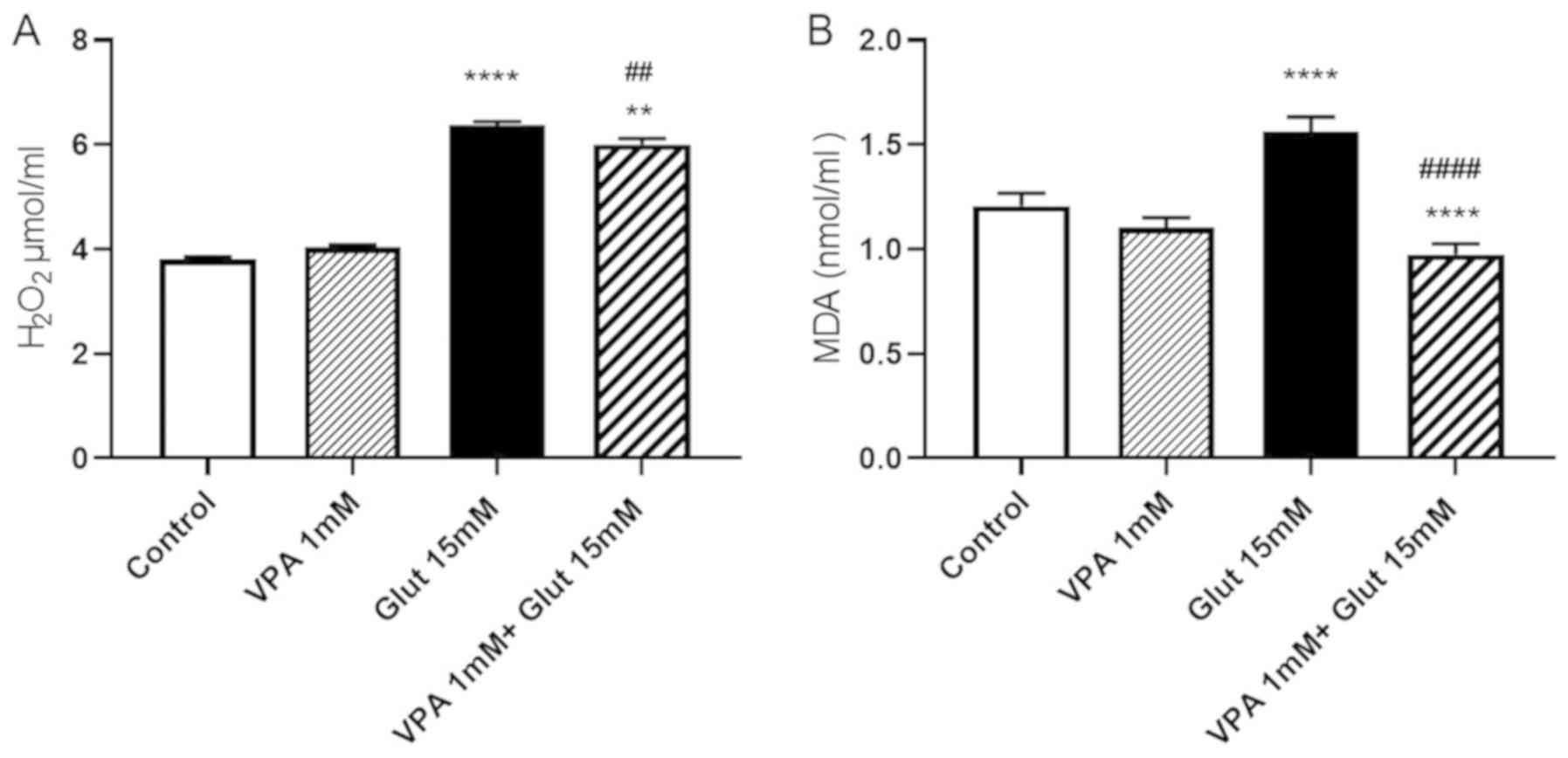
Effects of VPA pre-treatment on
H2O2 and MDA contents in glutamate-induced
excitotoxicity
At 24 h of incubation with 15 mM glutamate,
H2O2 levels were increased as compared with
those in the control group (P<0.0001). VPA treatment alone (1
mM) did not change the H2O2 levels as
compared with those in the control. To explore whether VPA has
protective effects against free radical-induced cell injury by
scavenging free radicals, the effect of 1 mM VPA on
H2O2 levels was evaluated. Treatment with VPA
was indicated to decrease H2O2 levels in
cells exposed to glutamate when compared to treatment with
glutamate alone (P<0.01), but was not able to reduce them to the
control levels (P<0.01 vs. control group; Fig. 3A).
MDA levels were significantly increased after a 24-h
incubation with glutamate (P<0.0001). Pre-treatment with VPA (1
mM) reversed the increase of MDA after glutamate exposure, when
compared to the control and glutamate alone groups (P<0.0001 vs.
control group; P<0.0001 vs. glutamate alone group; Fig. 3B).
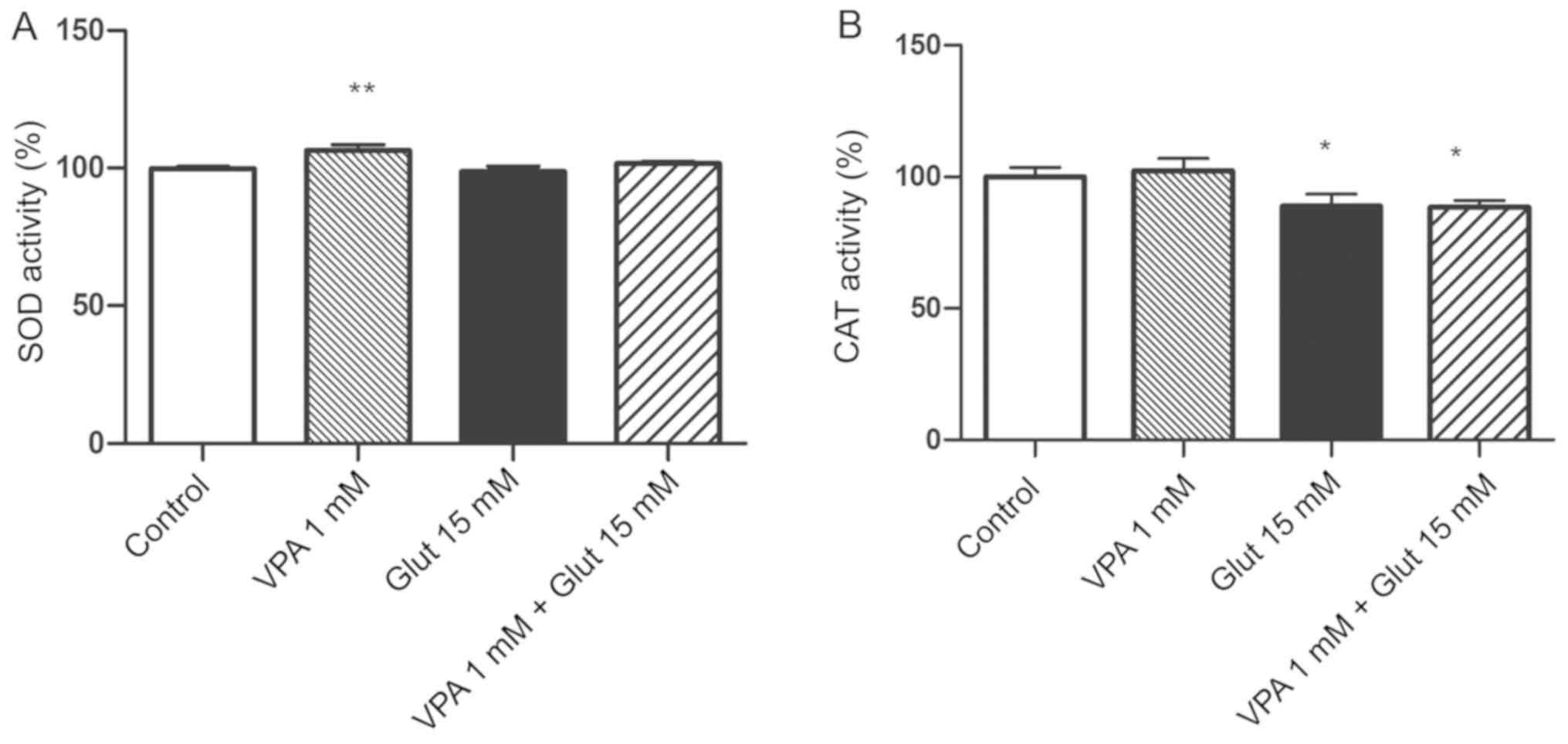
Effects of VPA pre-treatment on
anti-oxidant enzyme activities in cells with glutamate-induced
excitotoxicity
The SOD activity was increased in the 1 mM VPA alone
group as compared with that in the control group (P<0.01;
Fig. 4A). The SOD activity slightly
decreased after glutamate exposure, as compared to the control
group, but this change was not statistically significant. CAT
activity was significantly decreased after exposure to 15 mM
glutamate (Fig. 4B). VPA treatment
alone slightly increased CAT activity, but the decrease was not
significant; furthermore, pre-treatment with VPA had no obvious
effect to inhibit the decrease of CAT in glutamate-exposed cells
(P<0.05 vs. control; Fig.
4B).
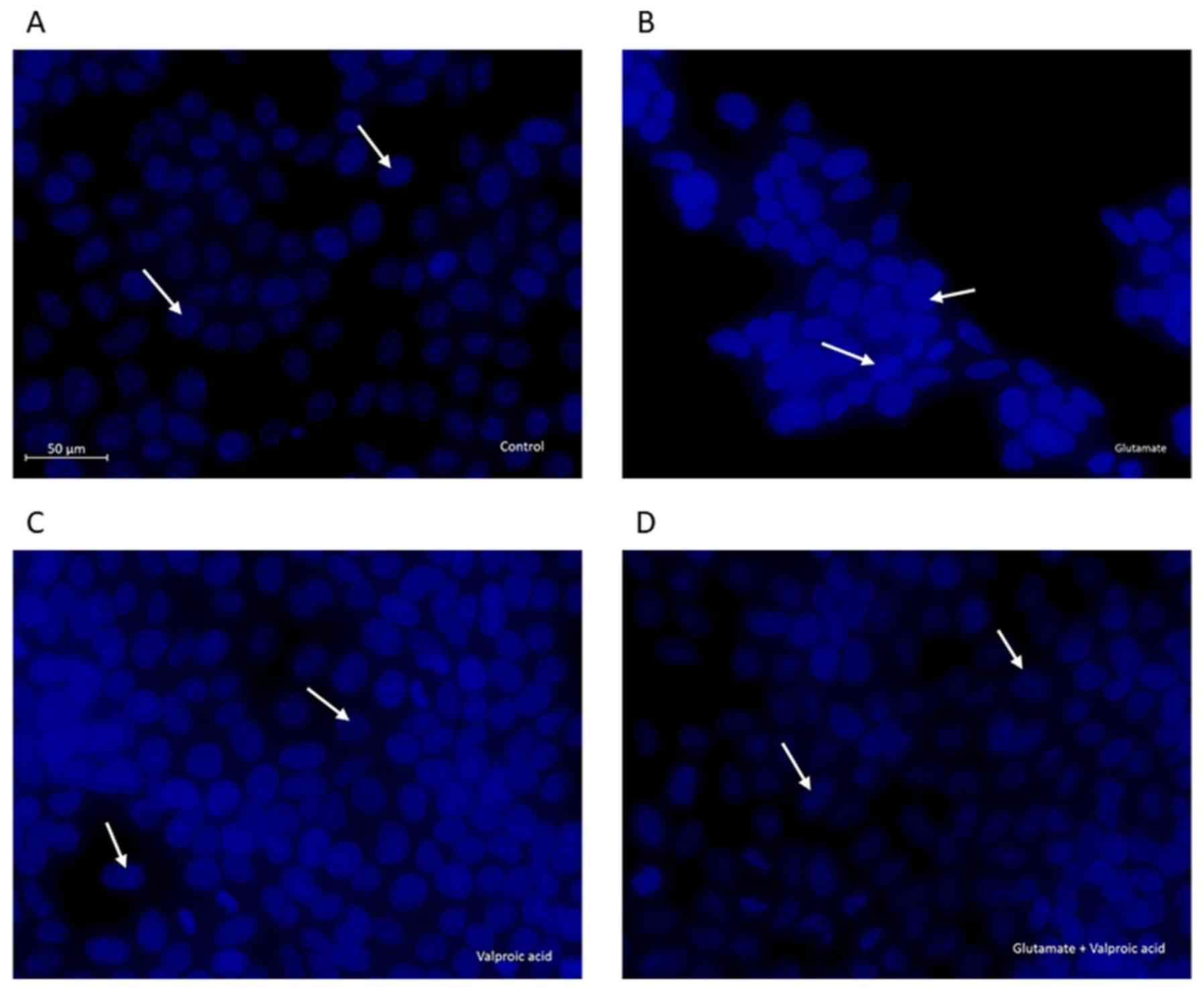
Effects of VPA pre-treatment on
nuclear changes in cells with glutamate-induced excitotoxicity
Changes in cellular and nuclear morphology were
examined in the control, glutamate-exposed, valproic acid-treated
and valproic acid-pre-treated glutamate-exposed cells by DAPI
staining (Fig. 5). The control cell
nuclei were regular in shape. Morphological characteristics of
apoptosis, including reduced nuclear size, chromatin condensation,
nuclear fragmentation and intense fluorescence, were observed in
glutamate-exposed cells. These changes in nuclear morphology were
alleviated by pre-treatment of cells with VPA (1 mM) following
exposure to 15 mM glutamate.
Discussion
The present study focused on an acute excitotoxicity
model of cell culture created by short-term exposure to glutamate
for up to 24 h. Glutamate exposure for 24 h was also indicated to
cause excitotoxicity in cortical neurons in primary culture
(3). In the present study, the
effect of VPA on the early stages of acute glutamate-induced
neurotoxity, including intracellular ROS, were investigated in
SH-SY5Y cells. Exposure to 15 mM glutamate was found to be the
least effective compared with other higher doses of glutamate at
causing toxicity in SH-SY5Y cells, as indicated by an MTT assay
performed after glutamate treatment for 24 h. Among 3
concentrations of VPA (1, 5 and 10 mM), only pre-treatment with 1
mM VPA significantly inhibited glutamate-induced toxicity to
increase the number of viable cells. Therefore, 15 mM glutamate and
1 mM VPA were used in the subsequent experiments to investigate the
potential anti-oxidant effect of VPA to prevent glutamate-induced
neuronal injury.
It was observed that acute exposure of glutamate to
induce neurotoxicity significantly enhanced
H2O2 levels, suggesting that
glutamate-stimulated excitotoxicity in these cells is caused by
oxidative damage. Major detrimental forms of ROS include
superoxide, hydroxyl radicals and H2O2.
H2O2 is one of the key targets for
interventions among various ROS products. As
H2O2 is neither a radical nor an ion, it is
able to readily cross the cell membrane and affect cellular
structures distant from its origin. Oxidative stress lead to
protein dysfunction, DNA damage and lipid peroxidation, resulting
in cell death in a manner that is dependent on the excessive
production of ROS (18). A recent
study revealed that 1-50 mM glutamate affected
H2O2 synthesis by brain mitochondria, and
this effect was associated with complex II, a source of superoxide
formation in mitochondria, and is dependent on the mitochondrial
potential (19). Ha et al
(20) revealed that, following
prolonged exposure to glutamate, extracellular
H2O2 accumulated in a time- and
concentration-dependent manner in HT22 cells.
H2O2 formation due to mitochondrial
superoxide leakage perpetuates oxidative stress in neuronal
injury.
In the present study, increased levels of MDA were
observed in cells exposed to glutamate. MDA, a product of the
breakdown of polyunsaturated fatty acid, commonly known as a marker
of oxidative stress, indicates that glutamate excitotoxicity may be
associated with oxidative stress. MDA also serves as a convenient
indicator of lipid peroxidation (21). In combination, the increased levels
of H2O2 and MDA suggested that
glutamate-induced neurotoxicity in SH-SY5Y cells is mediated by
oxidative damage. This was consistent with the results of Sun et
al (17), who indicated that
glutamate exerted its toxicity through oxidative damage in SH-SY5Y
cells.
The other result of the present study was that
pre-treatment with 1 mM VPA decreased the glutamate-induced
increase in H2O2 and MDA levels, revealing a
neuroprotective effect of VPA by decreasing oxidative stress.
Previous studies also confirmed that the protective effects of VPA
are associated with a reduction of oxidative stress. Chronic
treatment with VPA was reported to exert neuroprotective effects
against excitotoxicity via inhibition of oxidative damage by
decreasing glutamate-induced MDA levels (13). Frey et al (22) also demonstrated that valproate
prevented amphetamine-induced lipid peroxidation in the hippocampus
and in the prefrontal cortex, revealing the neuroprotective effects
of VPA in response to oxidative stress. VPA has also been reported
to inhibit the activation of the JNK pathway by decreasing ROS
production in a model of spinal cord injury (23). It was reported that treatment with
VPA following cerebral ischemia prevented ROS production via the
inhibition of HDAC and induction of HSP (24). Silva et al (15) suggested that VPA exerted
neuroprotective effects by attenuating the increased HDAC and GSK3
immunoreactivity, which are involved in inflammation and brain
function in certain areas of the brain of ischemic animals.
Inhibition of these enzymes was demonstrated to reduce ischemic
cerebral damage by restoring failing mitochondrial bioenergetics
and preventing ROS production (14,25).
The mechanisms through which VPA and other mood
stabilizers decrease ROS generation remain to be fully elucidated,
but it has been suggested that buffering overloaded intracellular
calcium, stabilizing mitochondrial function and increased
expression of endoplasmic reticulum stress protein may have a role
in it (13,26,27). The
inhibition of the GRP78 expression led to an increase in ROS and
intracellular calcium levels following oxidative insult (28).
Oxidative stress occurs when cellular anti-oxidant
defenses are inadequate to maintain the levels of ROS below the
toxic threshold, due to excessive ROS production and/or loss of
anti-oxidant defenses (29,30). CAT is one of the most common
anti-oxidant enzymes in almost all living organisms that are
exposed to oxygen; it catalyzes the reduction of
H2O2 to water and removes organic
hydroperoxides (31). SOD is a
protective enzyme involved in catalyzing the dismutation of
superoxide to less reactive H2O2 and
molecular oxygen (32,33). These anti-oxidants may protect
neuronal cells against oxidative damage by
H2O2 (18,34,35). It
has been reported that anti-oxidant systems in neurodegenerative
disorders have coordinated effects induced by SOD and CAT (36).
In the present study, it was demonstrated that CAT
activity is significantly decreased in cells exposed to glutamate.
However, VPA did not exert any significant effect on CAT activity
in cells cultured with glutamate. These results suggest that CAT
activity has a role in glutamate-induced oxidative damage, but VPA
does not appear to have a sufficient effect on CAT activity in
cells exposed to glutamate. It has also been reported that CAT
activity in the brain is significantly lower than that in other
organs, including the kidney and liver (37). The low CAT activity may be reduced
further by glutamate toxicity, an effect that VPA addition may not
be sufficient to alleviate. VPA alone significantly increased the
levels of this anti-oxidant enzyme, but exerted no significant
effects on glutamate-exposed cells. It was reported that SOD
activity may vary among different cell types, e.g. among adult rat
brain cells; the specific activity of total SOD has been reported
to be ~10-fold higher in glial cells than in neurons (38).
The results of the present study suggested that VPA
exerts a protective effect on glutamate-induced excitotoxicity via
decreasing ROS production without influencing anti-oxidant enzymes.
Studies have reported controversial results regarding the effect of
VPA on anti-oxidant enzymes. It has been indicated that the
activity of glutathione S-transferase, glutathione reductase,
glutathione peroxidase, SOD and CAT was significantly reduced in
the cerebral cortex and cerebellum (39). A study on epileptic children also
demonstrated a significant decrease in the anti-oxidant activity of
SOD and CAT after VPA treatment (40). Glutathione reductase activity was
indicated to be slightly lower in pediatric patients receiving VPA
monotherapy and polytherapy, as compared with that in newly
diagnosed pediatric patients (41).
Other studies have revealed an increase in anti-oxidant enzymes by
VPA treatment. It has been suggested that VPA treatment may alter
the balance between oxidant and anti-oxidant systems (42). In primary cultured rat cerebral
cortical cells, chronic treatment with lithium and valproate at
therapeutic concentrations increased the activity of glutathione
S-transferase (43).
However, the present study was not without its
limitations. The potential contamination of the cells was not
investigated, but the present results are in line with the
literature and they will be confirmed in vivo or in primary
cultured cells in future projects.
In conclusion, the present study demonstrated that
pre-treatment with 1 mM VPA effectively prevented the decline in
neuronal cell viability induced by glutamate exposure in SH-SY5Y
cells via decreasing oxidative stress.
Acknowledgements
The authors would like to thank Miss Ilgın Akpınar
from İstanbul University, Institution of Science, Department of
Botany (Istanbul, Turkey) and Dr İbrahim Akalın from İstanbul
Medeniyet University, Department of Medical Genetics (Istanbul,
Turkey) for their technical contributions.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BTB designed the study. BTB, EO and GA performed the
experiments and analyzed and interpreted the data. All authors
contributed to the writing of the manuscript and read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Wang R and Reddy PH: Role of Glutamate and
NMDA receptors in Alzheimer's disease. J Alzheimers Dis.
57:1041–1048. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bleich S, Römer K, Wiltfang J and
Kornhuber J: Glutamate and the glutamate receptor system: A target
for drug action. Int J Geriatr Psychiatry. 18 (Suppl 1):S33–S40.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Schubert D and Piasecki D: Oxidative
glutamate toxicity can be a component of the excitotoxicity
cascade. J Neurosci. 21:7455–7462. 2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kritis AA, Stamoula EG, Paniskaki KA and
Vavilis TD: Researching glutamate-induced cytotoxicity in different
cell lines: A comparative/collective analysis/study. Front Cell
Neurosci. 9(91)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Andersen JK: Oxidative stress in
neurodegeneration: Cause or consequence? Nat Med. 10 (Supp
l):S18–S25. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Venkateshappa C, Harish G, Mythri RB,
Mahadevan A, Bharath MM and Shankar SK: Increased oxidative damage
and decreased antioxidant function in aging human substantia nigra
compared to striatum: Implications for Parkinson's disease.
Neurochem Res. 37:358–369. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kumar A, Singh RL and Babu GN: Cell death
mechanisms in the early stages of acute glutamate neurotoxicity.
Neurosci Res. 66:271–278. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Floyd RA: Antioxidants, oxidative stress,
and degenerative neurological disorders. Proc Soc Exp Biol Med.
222:236–245. 1999.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Calabrese V, Scapagnini G, Giuffrida
Stella AM, Bates TE and Clark JB: Mitochondrial involvement in
brain function and dysfunction: Relevance to aging,
neurodegenerative disorders and longevity. Neurochem Res.
26:739–764. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Atlante A, Calissano P, Bobba A,
Giannattasio S, Marra E and Passarella S: Glutamate neurotoxicity,
oxidative stress and mitochondria. FEBS Lett. 497:1–5.
2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Aoyama K and Nakaki T: Inhibition of
GTRAP3-18 may increase neuroprotective glutathione (GSH) synthesis.
Int J Mol Sci. 13:12017–12035. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Vajda FJ: Valproate and neuroprotection. J
Clin Neurosci. 9:508–514. 2002.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shao L, Young LT and Wang JF: Chronic
treatment with mood stabilizers lithium and valproate prevents
excitotoxicity by inhibiting oxidative stress in rat cerebral
cortical cells. Biol Psychiatry. 58:879–884. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Pang T, Wang YJ, Gao YX, Xu Y, Li Q, Zhou
YB, Xu L, Huang ZJ, Liao H, Zhang LY, et al: A novel GSK-3β
inhibitor YQ138 prevents neuronal injury induced by glutamate and
brain ischemia through activation of the Nrf2 signaling pathway.
Acta Pharmacol Sin. 37:741–752. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Silva MR, Correia AO, Dos Santos GCA,
Parente LLT, de Siqueira KP, Lima DGS, Moura JA, da Silva Ribeiro
AE, Costa RO, Lucetti DL, et al: Neuroprotective effects of
valproic acid on brain ischemia are related to its HDAC and GSK3
inhibitions. Pharmacol Biochem Behav. 167:17–28. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen S, Wu H, Klebe D, Hong Y and Zhang J:
Valproic acid: A new candidate of therapeutic application for the
acute central nervous system injuries. Neurochem Res. 39:1621–1633.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sun ZW, Zhang L, Zhu SJ, Chen WC and Mei
B: Excitotoxicity effects of glutamate on human neuroblastoma
SH-SY5Y cells via oxidative damage. Neurosci Bull. 26:8–16.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Uttara B, Singh AV, Zamboni P and Mahajan
RT: Oxidative stress and neurodegenerative diseases: A review of
upstream and downstream antioxidant therapeutic options. Curr
Neuropharmacol. 7:65–74. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lobysheva NV, Selin AA, Vangeli IM,
Byvshev IM, Yaguzhinsky LS and Nartsissov YR: Glutamate induces
H2O2 synthesis in nonsynaptic brain mitochondria. Free Radic Biol
Med. 65:428–435. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ha JS, Lim HM and Park SS: Extracellular
hydrogen peroxide contributes to oxidative glutamate toxicity.
Brain Res. 1359:291–297. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Maes M, Mihaylova I, Kubera M,
Uytterhoeven M, Vrydags N and Bosmans E: Increased plasma peroxides
and serum oxidized low density lipoprotein antibodies in major
depression: Markers that further explain the higher incidence of
neurodegeneration and coronary artery disease. J Affect Disord.
125:287–294. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Frey BN, Valvassori SS, Réus GZ, Martins
MR, Petronilho FC, Bardini K, Dal-Pizzol F, Kapczinski F and
Quevedo J: Effects of lithium and valproate on amphetamine-induced
oxidative stress generation in an animal model of mania. J
Psychiatry Neurosci. 31:326–332. 2006.PubMed/NCBI
|
|
23
|
Lee JY, Maeng S, Kang SR, Choi HY, Oh TH,
Ju BG and Yune TY: Valproic acid protects motor neuron death by
inhibiting oxidative stress and endoplasmic reticulum
stress-mediated cytochrome C release after spinal cord injury. J
Neurotrauma. 31:582–594. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xuan A, Long D, Li J, Ji W, Hong L, Zhang
M and Zhang W: Neuroprotective effects of valproic acid following
transient global ischemia in rats. Life Sci. 90:463–468.
2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Valerio A, Bertolotti P, Delbarba A,
Perego C, Dossena M, Ragni M, Spano P, Carruba MO, De Simoni MG and
Nisoli E: Glycogen synthase kinase-3 inhibition reduces ischemic
cerebral damage, restores impaired mitochondrial biogenesis and
prevents ROS production. J Neurochem. 116:1148–1159.
2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang JF, Azzam JE and Young LT: Valproate
inhibits oxidative damage to lipid and protein in primary cultured
rat cerebrocortical cells. Neuroscience. 116:485–489.
2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu H, Bowes RC, van de Water B, Sillence
C, Nagelkerke JF and Stevens JL: Endoplasmic reticulum chaperones
GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances,
and cell death in renal epithelial cells. J Biol Chem.
272:21751–21759. 1997.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Halliwell B: Free radicals and
antioxidants-quo vadis? Trends Pharmacol Sci. 32:125–130.
2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ďuračková Z: Free radicals and
antioxidants for Non-experts. In: Systems Biology of Free Radicals
and Antioxidants. Laher I (ed). Springer, Berlin, Heidelberg,
pp3-38, 2014.
|
|
31
|
Jornada LK, Valvassori SS, Steckert AV,
Moretti M, Mina F, Ferreira CL, Arent CO, Dal-Pizzol F and Quevedo
J: Lithium and valproate modulate antioxidant enzymes and prevent
ouabain-induced oxidative damage in an animal model of mania. J
Psychiatr Res. 45:162–168. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dringen R, Pawlowski PG and Hirrlinger J:
Peroxide detoxification by brain cells. J Neurosci Res. 79:157–165.
2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yasui K and Baba A: Therapeutic potential
of superoxide dismutase (SOD) for resolution of inflammation.
Inflamm Res. 55:359–363. 2006.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chadwick W, Zhou Y, Park SS, Wang L,
Mitchell N, Stone MD, Becker KG, Martin B and Maudsley S: Minimal
peroxide exposure of neuronal cells induces multifaceted adaptive
responses. PLoS One. 5(e14352)2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Halliwell B: Oxidative stress and
neurodegeneration: Where are we now? J Neurochem. 97:1634–1658.
2006.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kapczinski F, Frey BN, Andreazza AC,
Kauer-Sant'Anna M, Cunha AB and Post RM: Increased oxidative stress
as a mechanism for decreased BDNF levels in acute manic episodes.
Br J Psychiatry. 30:243–245. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wilson JX: Antioxidant defense of the
brain: A role for astrocytes. Can J Physiol Pharmacol.
75:1149–1163. 1997.PubMed/NCBI
|
|
38
|
Savolainen H: Superoxide dismutase and
glutathione peroxidase activities in rat brain. Res Commun Chem
Pathol Pharmacol. 21:173–176. 1978.PubMed/NCBI
|
|
39
|
Chaudhary S and Parvez S: An in vitro
approach to assess the neurotoxicity of valproic acid-induced
oxidative stress in cerebellum and cerebral cortex of young rats.
Neuroscience. 225:258–268. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhang Y, Wu JY, Weng LH, Li XX, Yu LJ and
Xu Y: Valproic acid protects against MPP+-mediated
neurotoxicity in SH-SY5Y Cells through autophagy. Neurosci Lett.
638:60–68. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sołowiej E and Sobaniec W: The effect of
antiepileptic drug therapy on antioxidant enzyme activity and serum
lipid peroxidation in young patients with epilepsy. Neurol
Neurochir Pol. 37:991–1003. 2003.PubMed/NCBI(In Polish).
|
|
42
|
Yiş U, Seçkin E, Kurul SH, Kuralay F and
Dirik E: Effects of epilepsy and valproic acid on oxidant status in
children with idiopathic epilepsy. Epilepsy Res. 84:232–237.
2009.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wang JF, Shao L, Sun X and Young LT:
Glutathione S-transferase is a novel target for mood stabilizing
drugs in primary cultured neurons. J Neurochem. 88:1477–1484.
2004.PubMed/NCBI View Article : Google Scholar
|